
Seeking for Non-Zinc-Binding MMP-2 Inhibitors: Synthesis, Biological Evaluation and Molecular Modelling Studies

 ,
, 
 , , ,
, , ,  ,
, 
Abstract
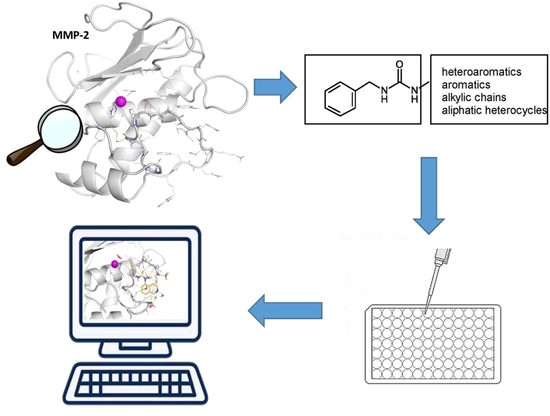
:
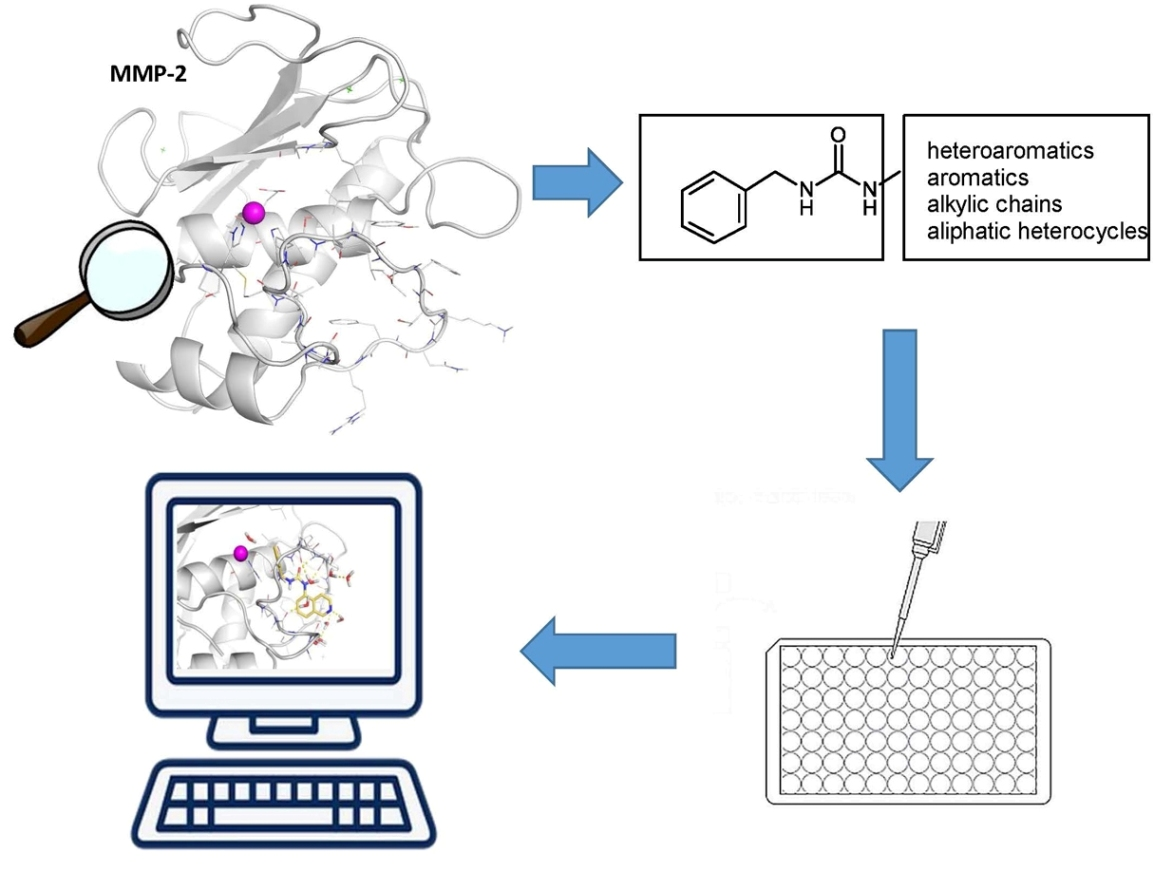
1. Introduction
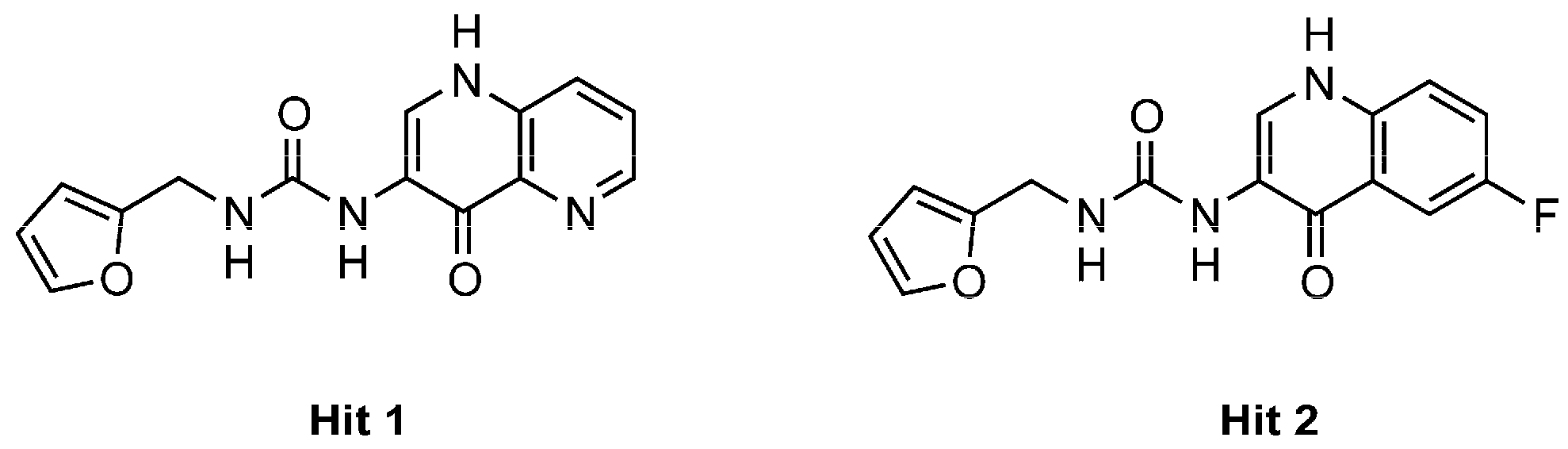
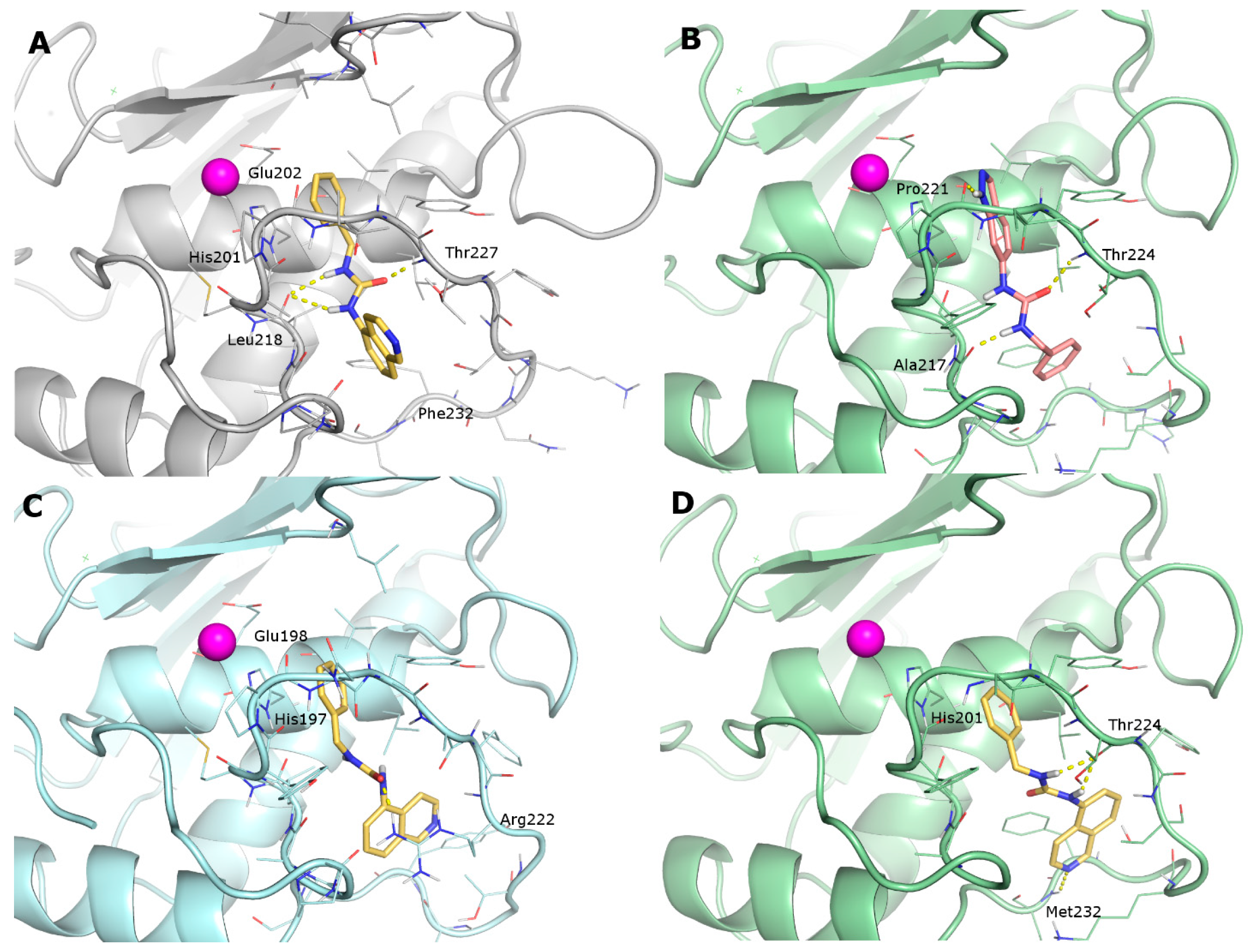
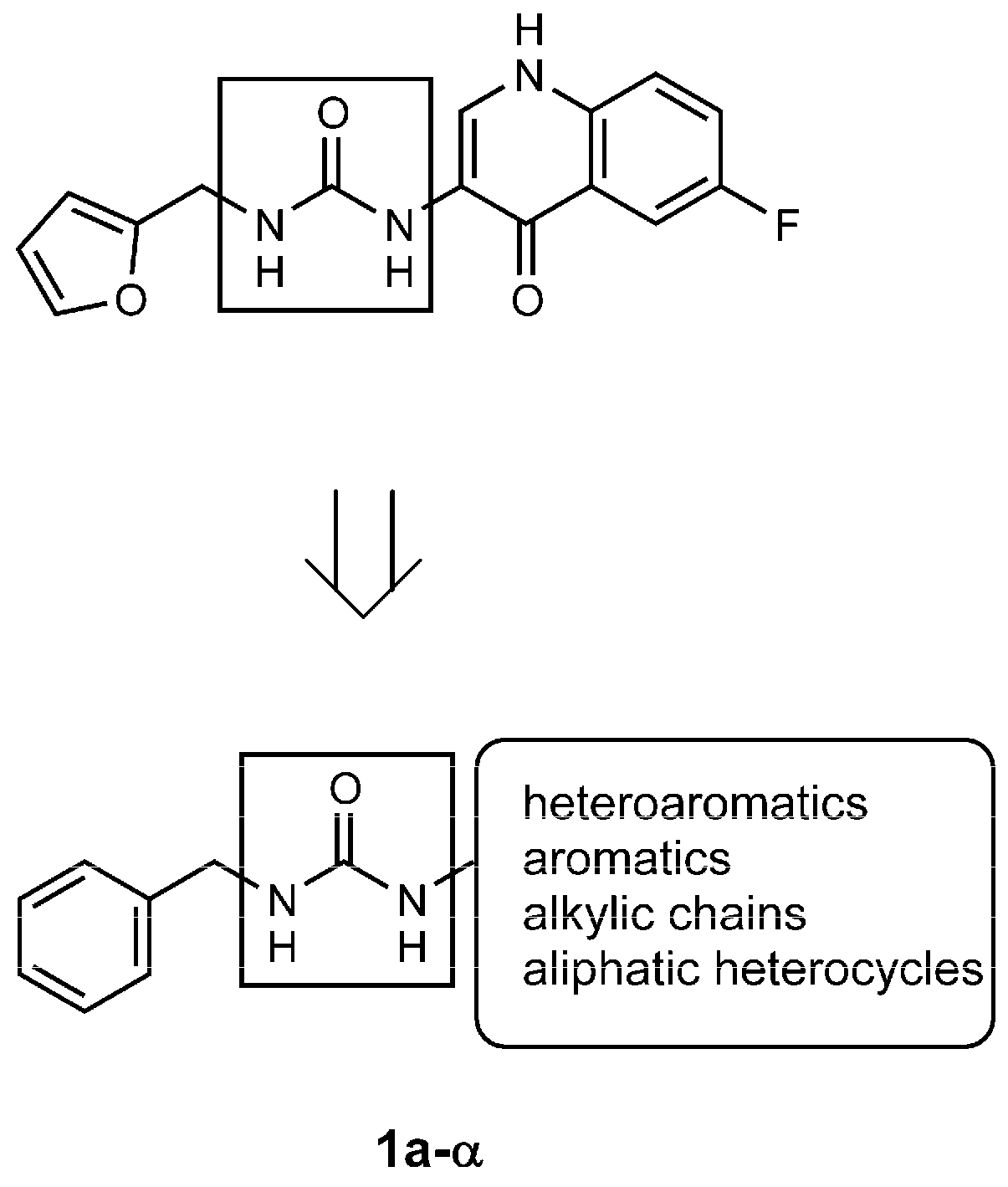
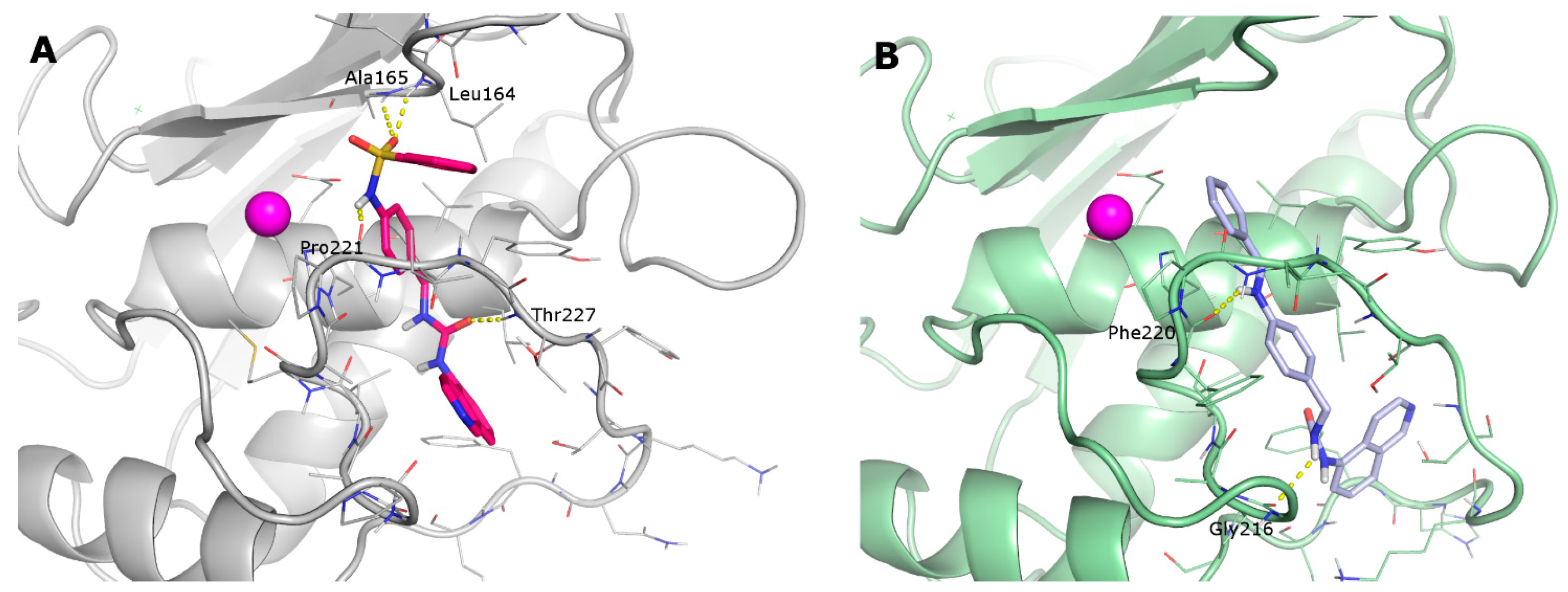
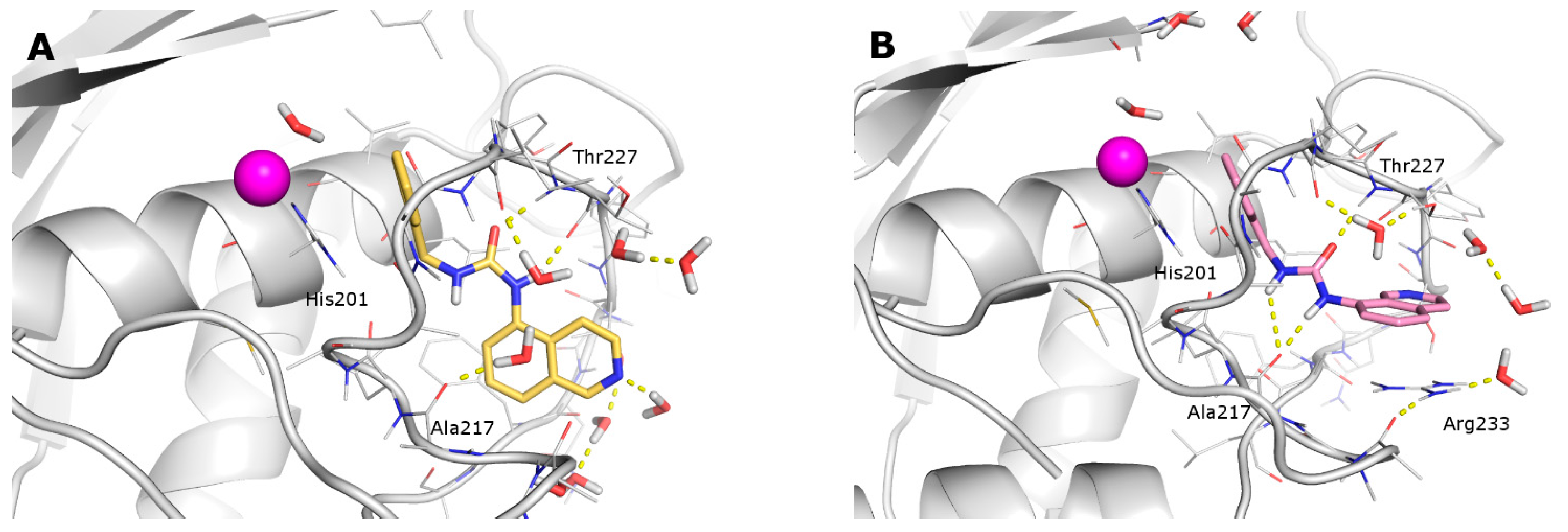
- the furyl/aryl group that provides the π–π stacking with His201 (MMP-2 numbering);
- the CH2 spacer between the furyl/aryl group and the urea function;
- the urea function that provides H-bonds in the S1’ site with NH of Thr227 and CO of Leu218.
2. Results
2.1. Chemistry
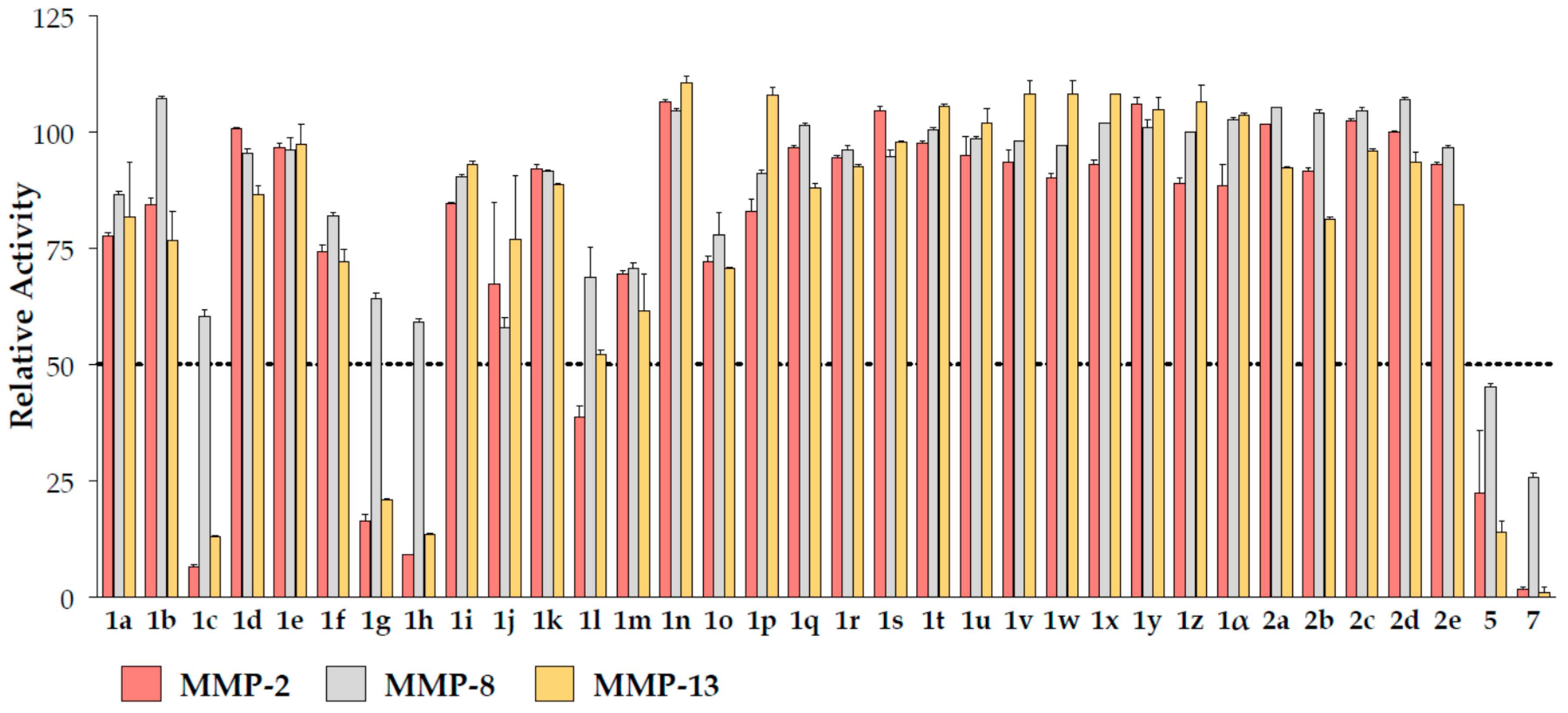
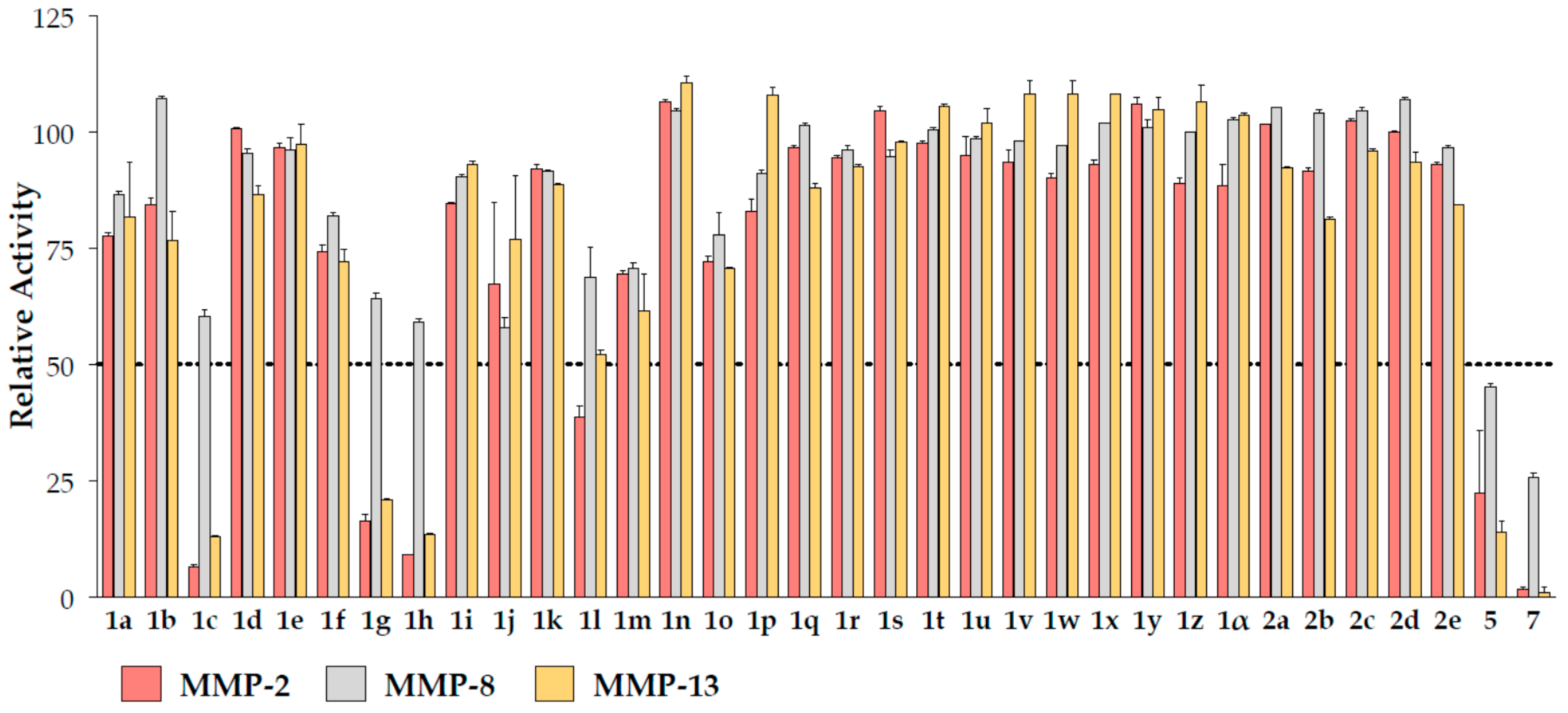
2.2. Biochemical Assays
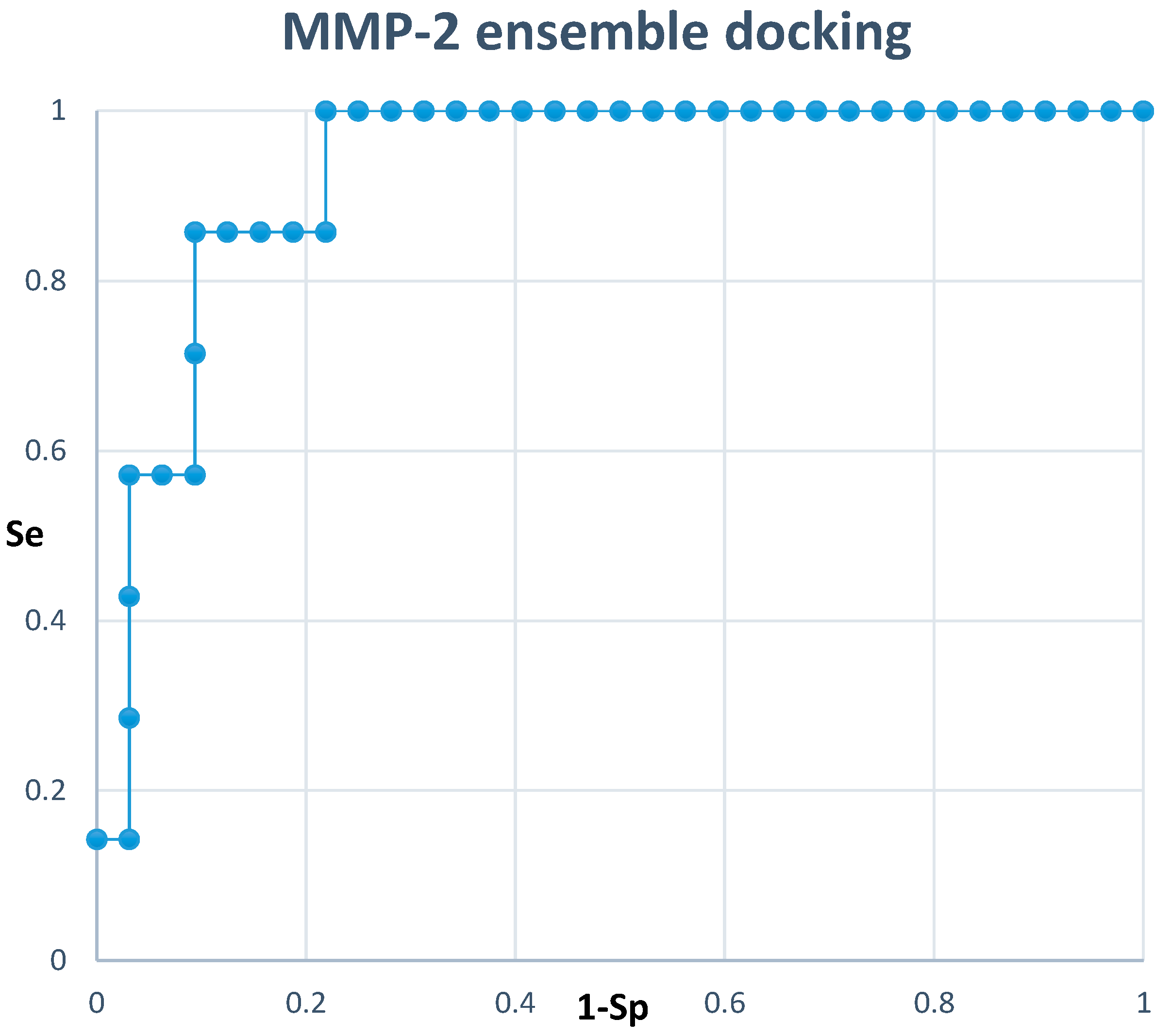
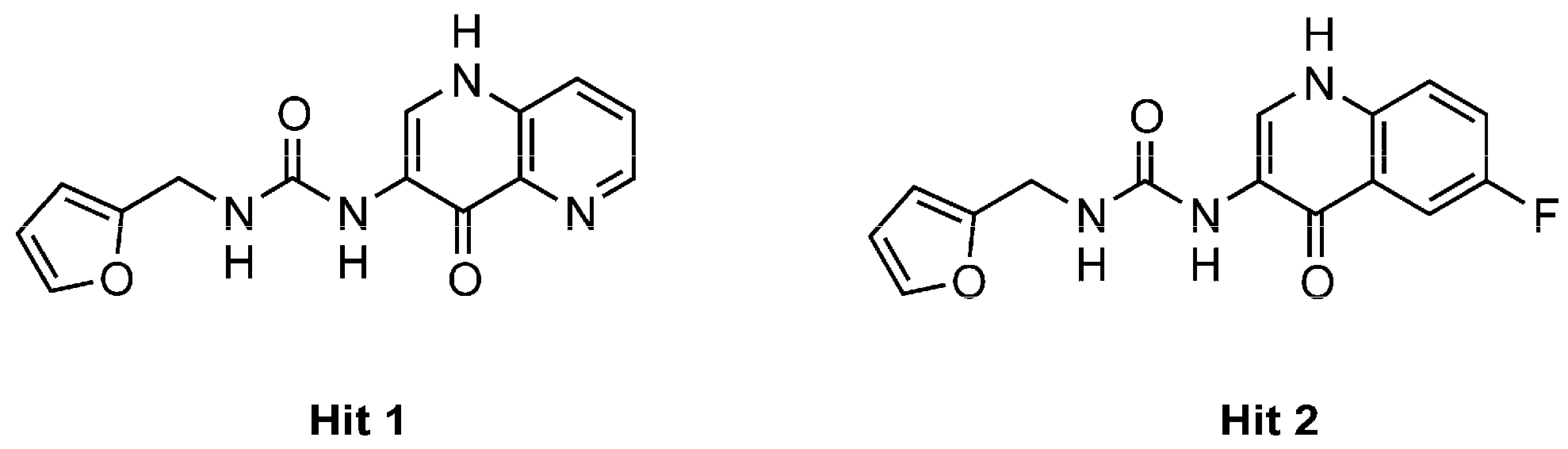
2.3. Docking
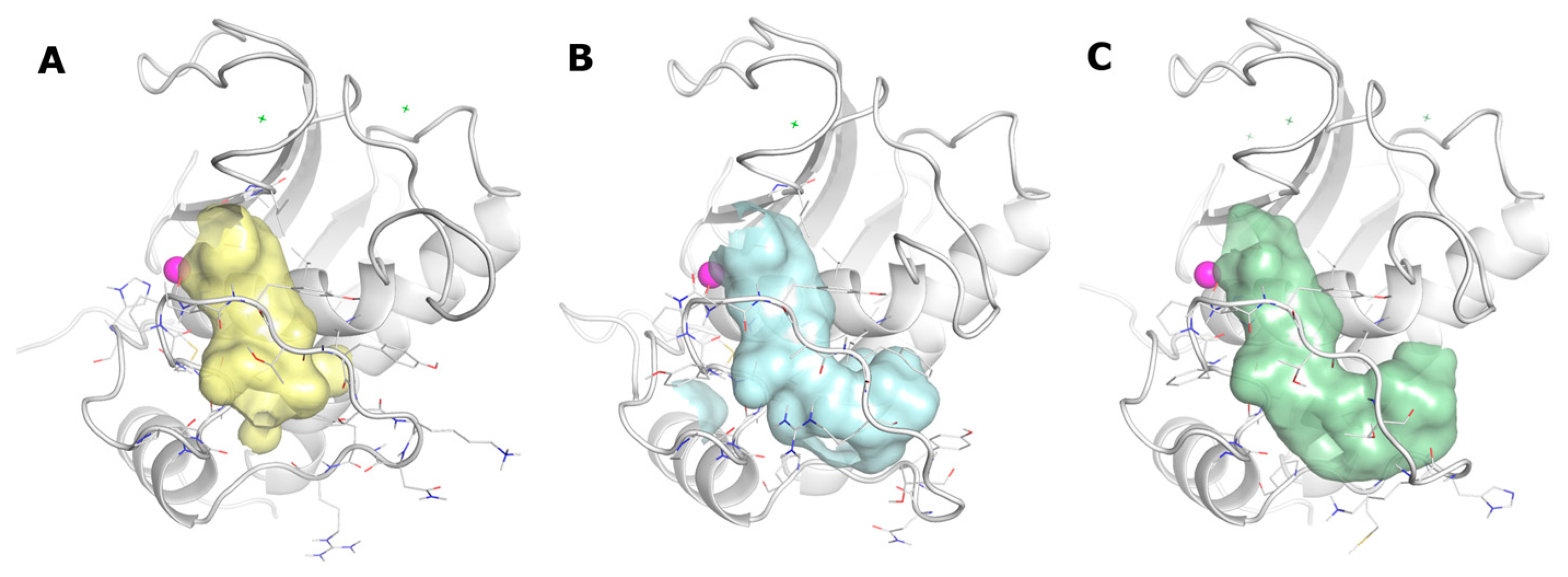
2.4. Molecular Dynamics
3. Discussion
4. Materials and Methods
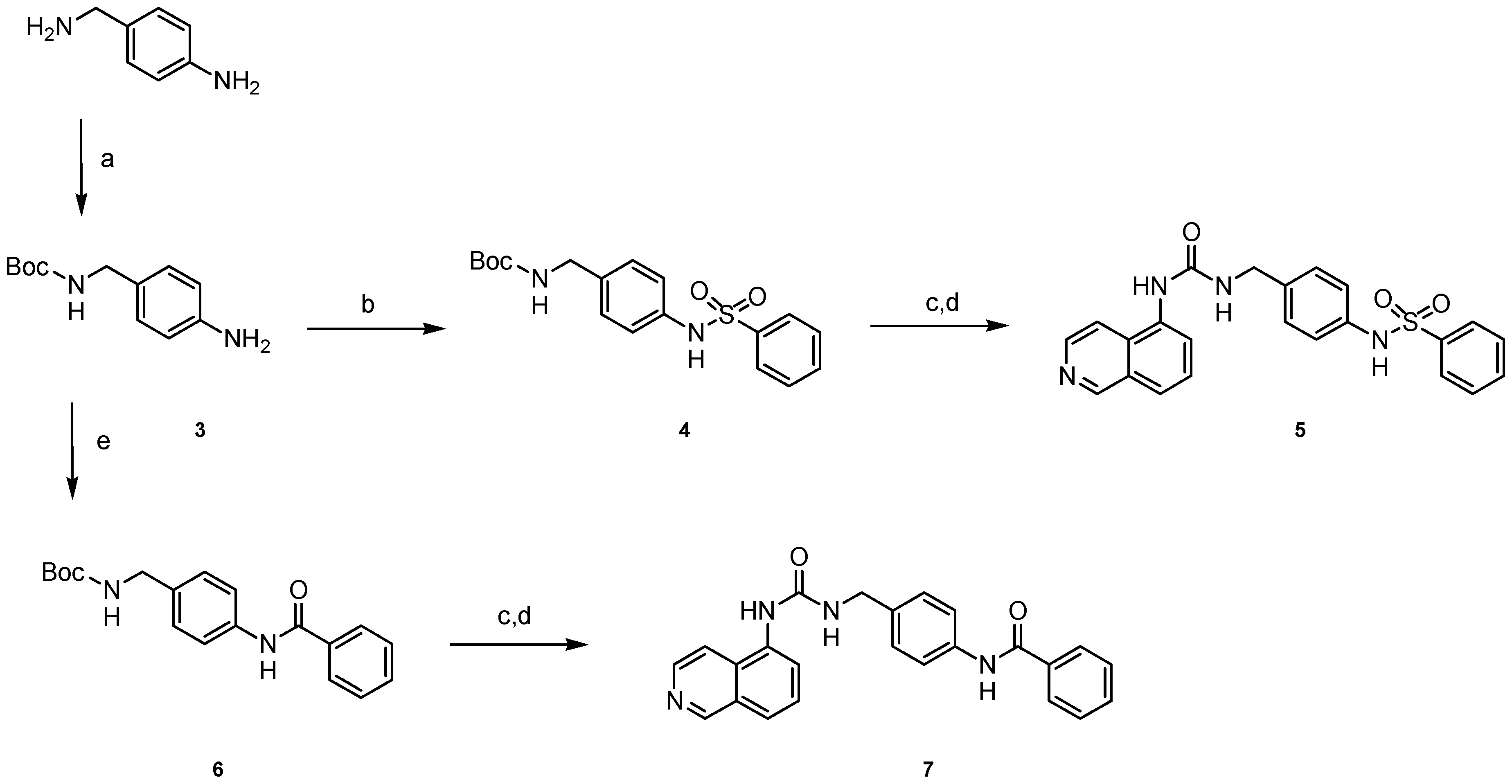
4.1. Chemistry
4.1.1. General
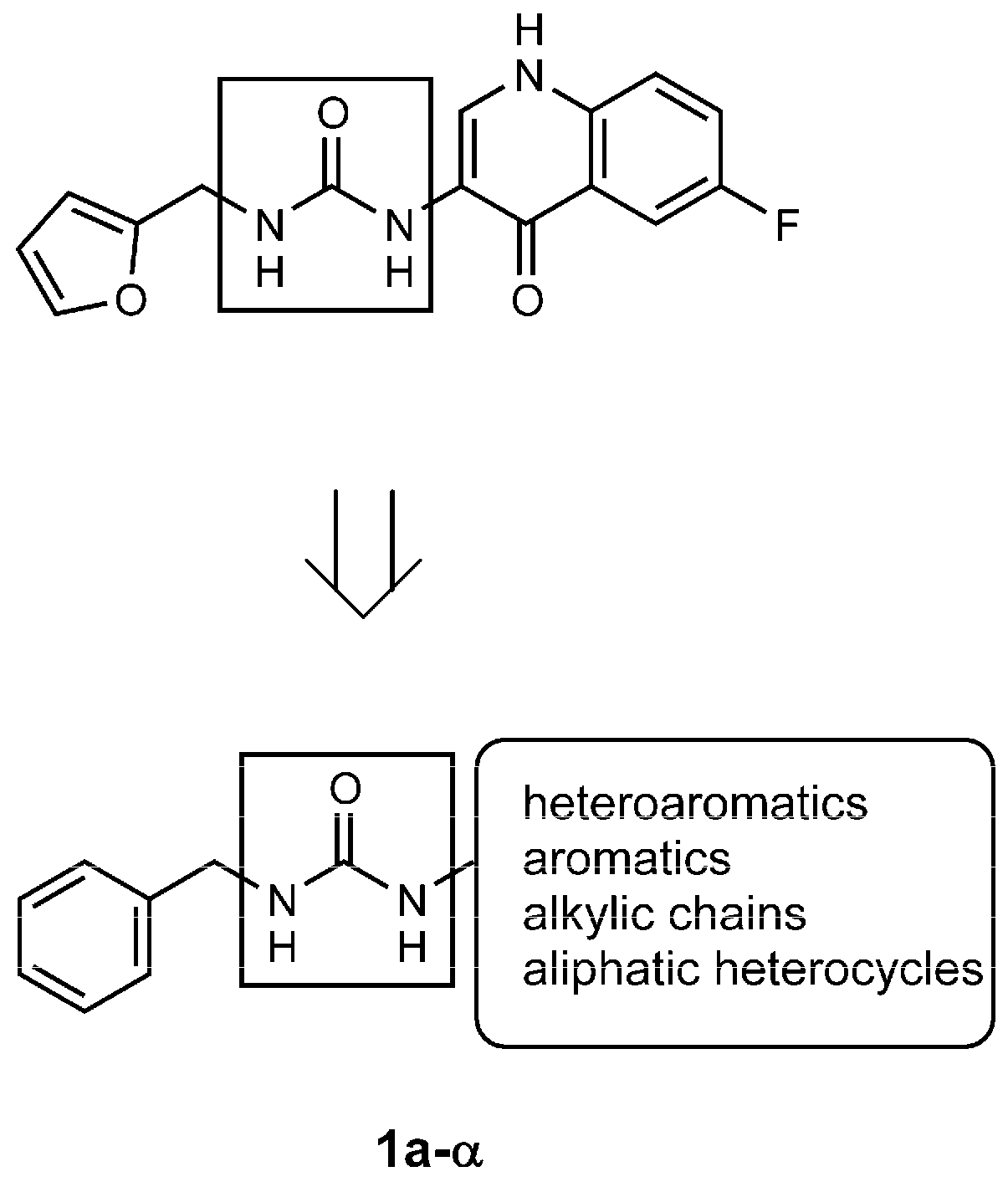
4.1.2. General Procedure for the Synthesis of Ureas 1a–α
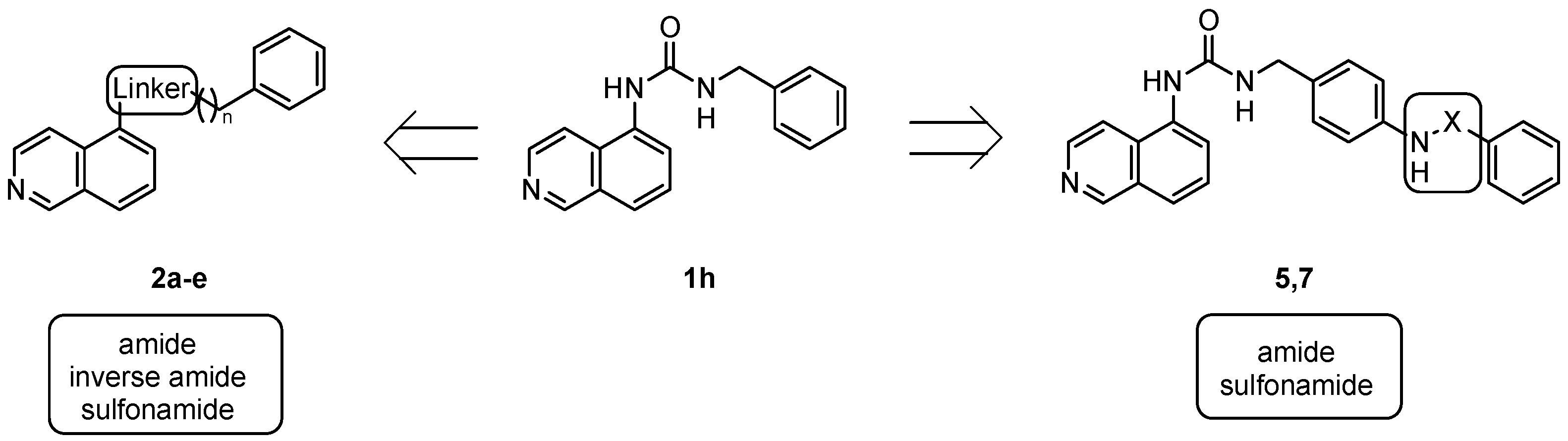
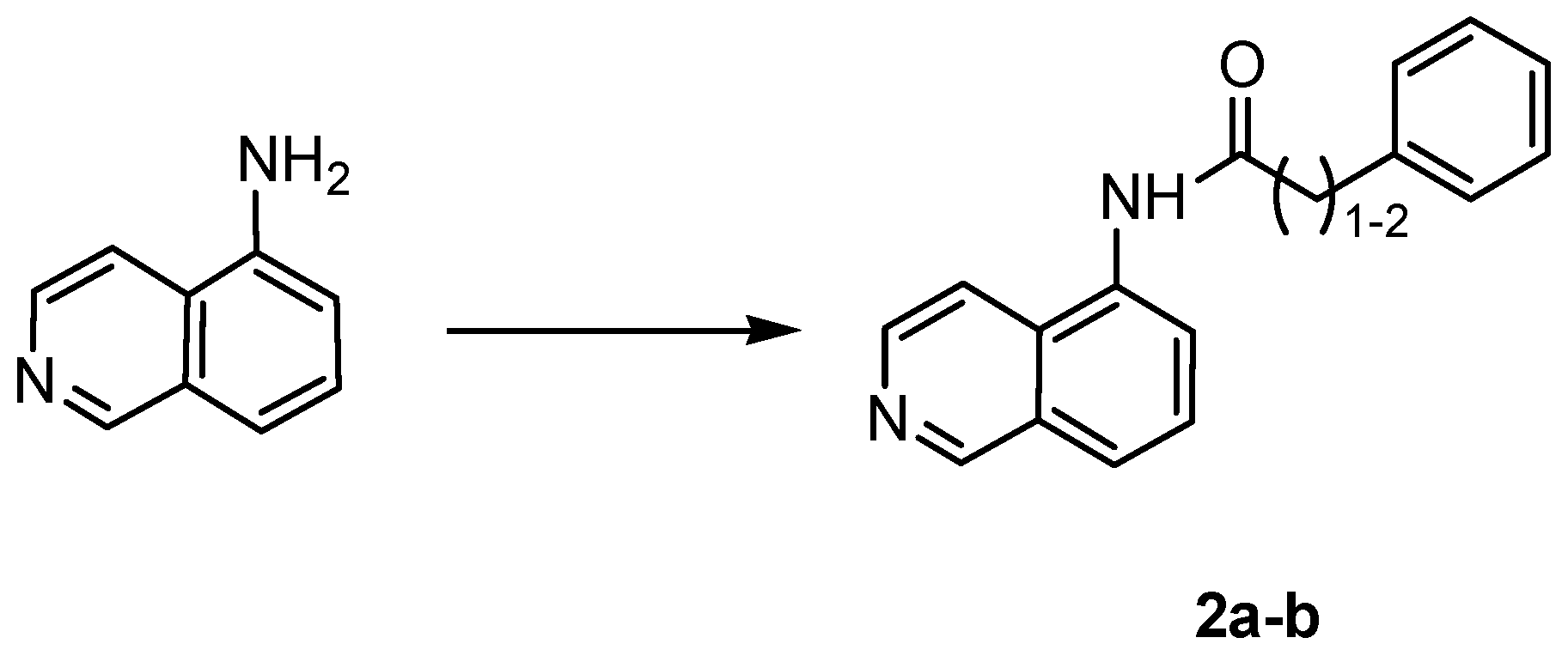
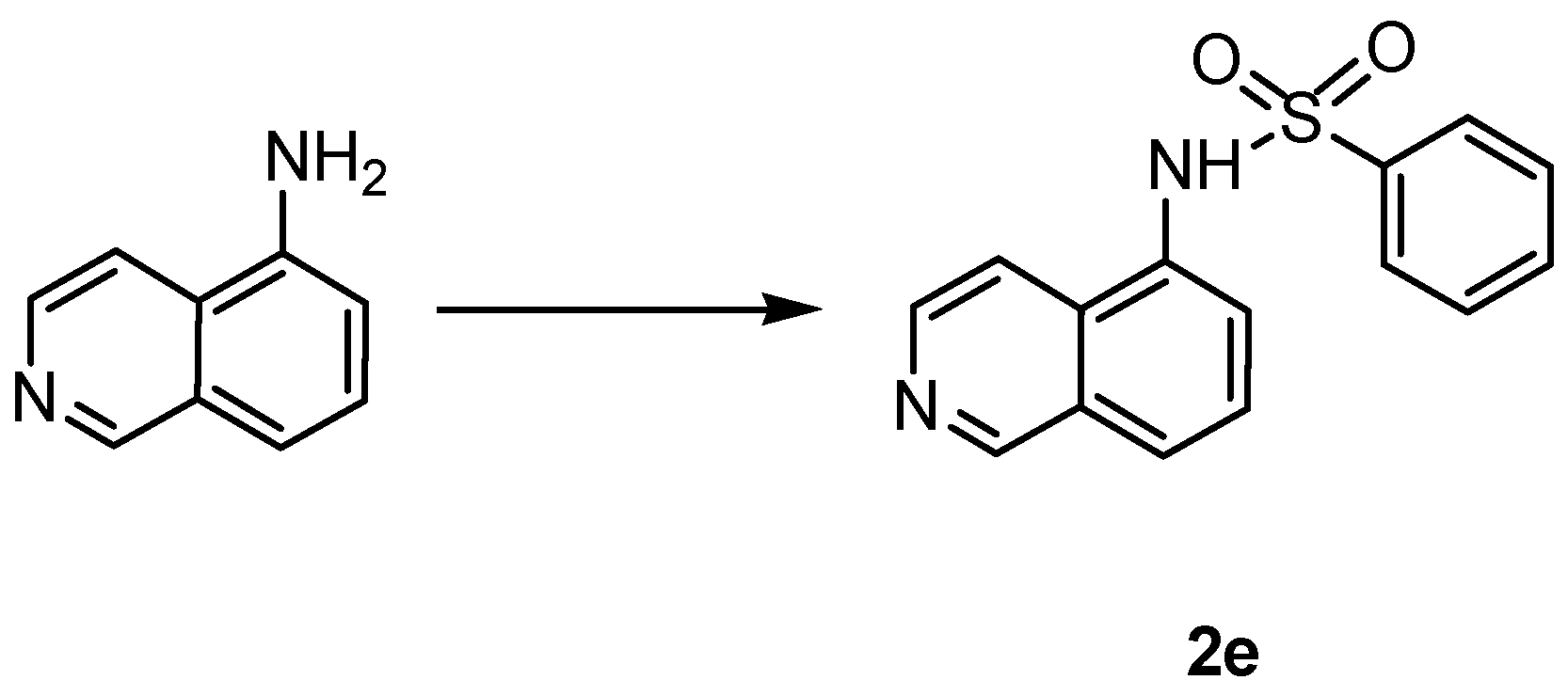
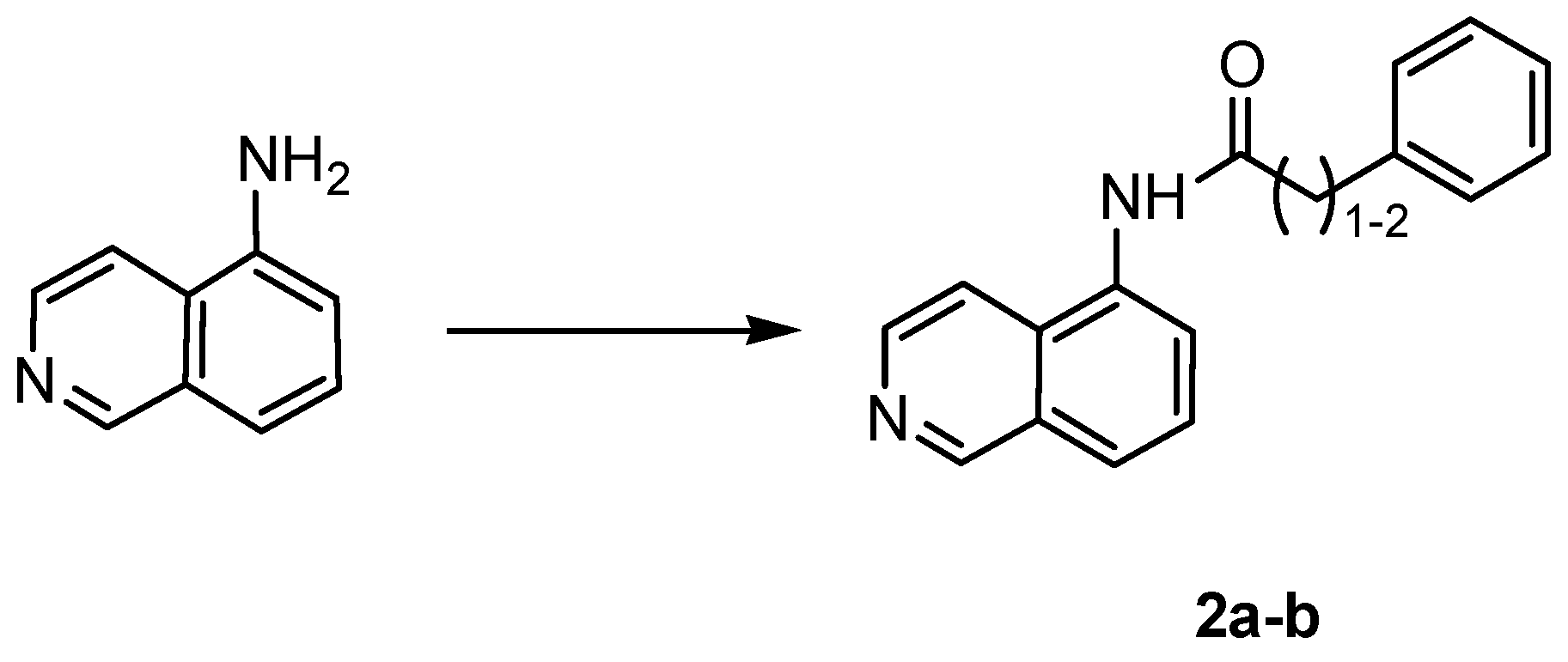
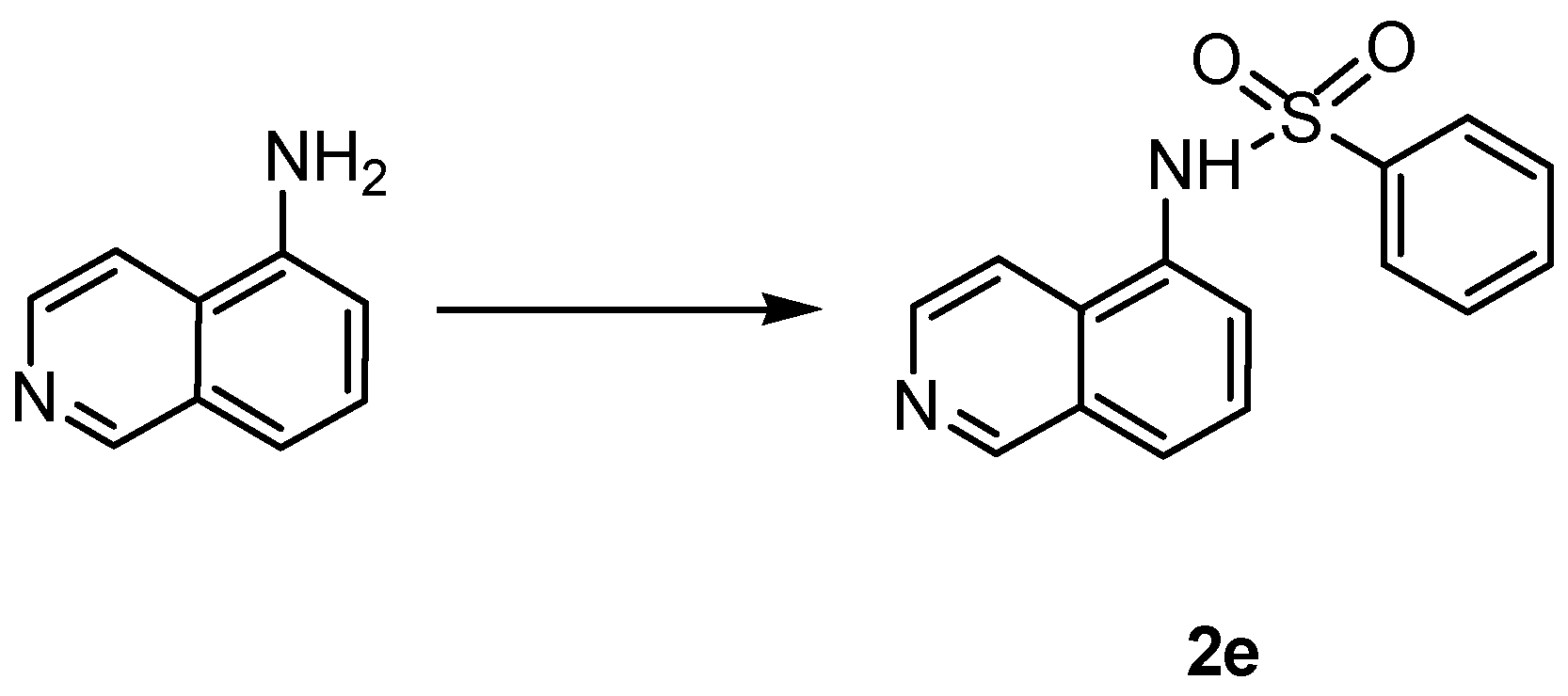
4.1.3. General Procedure for the Synthesis of Amides 2a,b
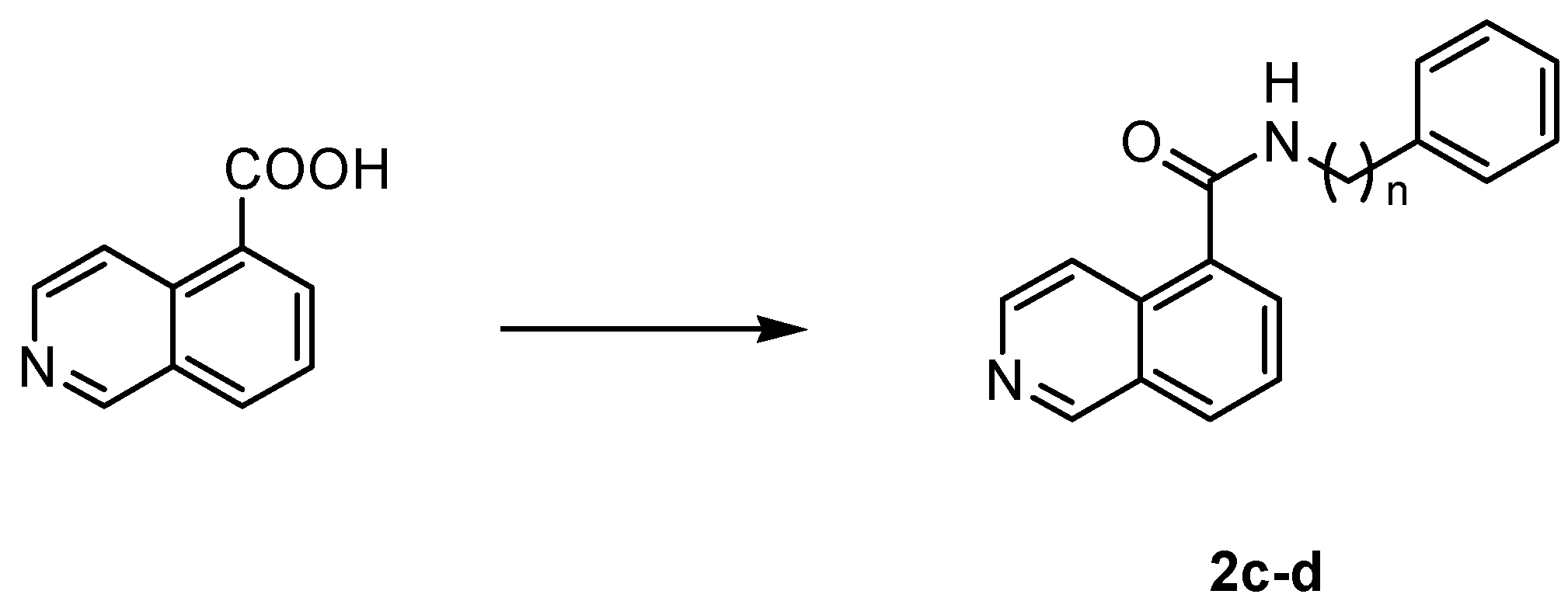
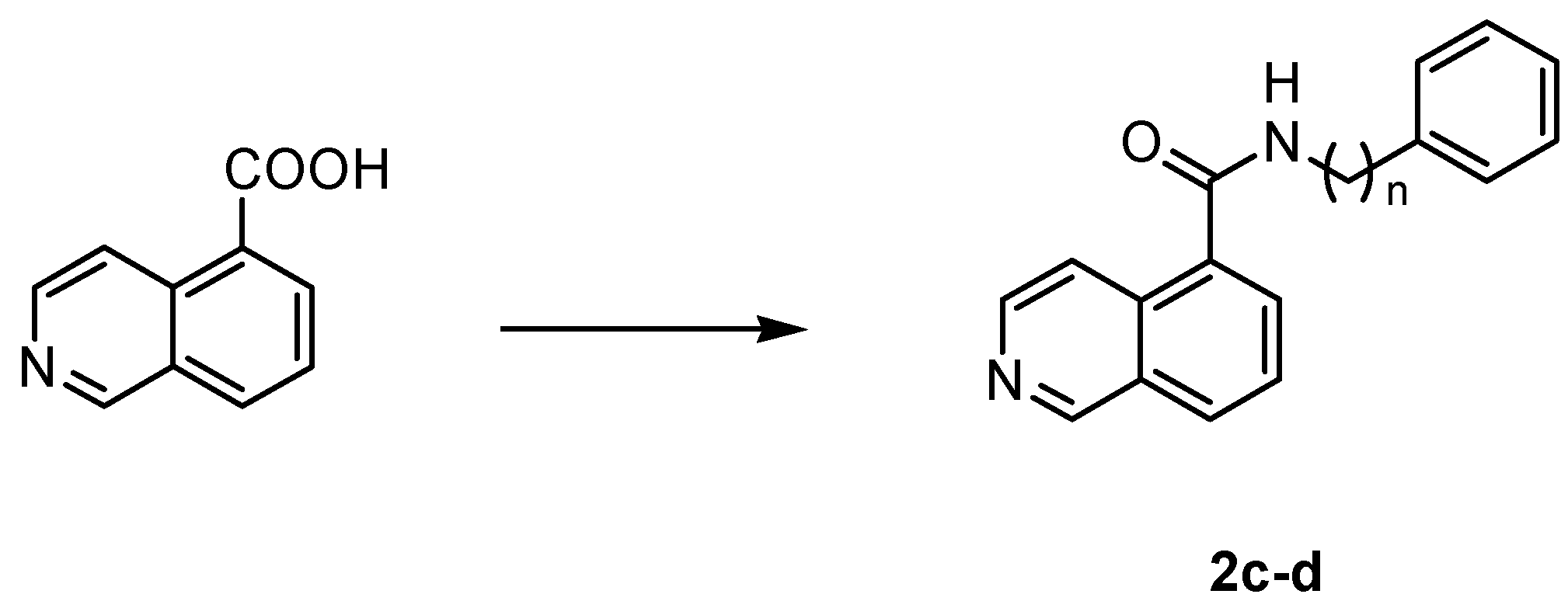
4.1.4. General Procedure for the Synthesis of Amides 2c,d
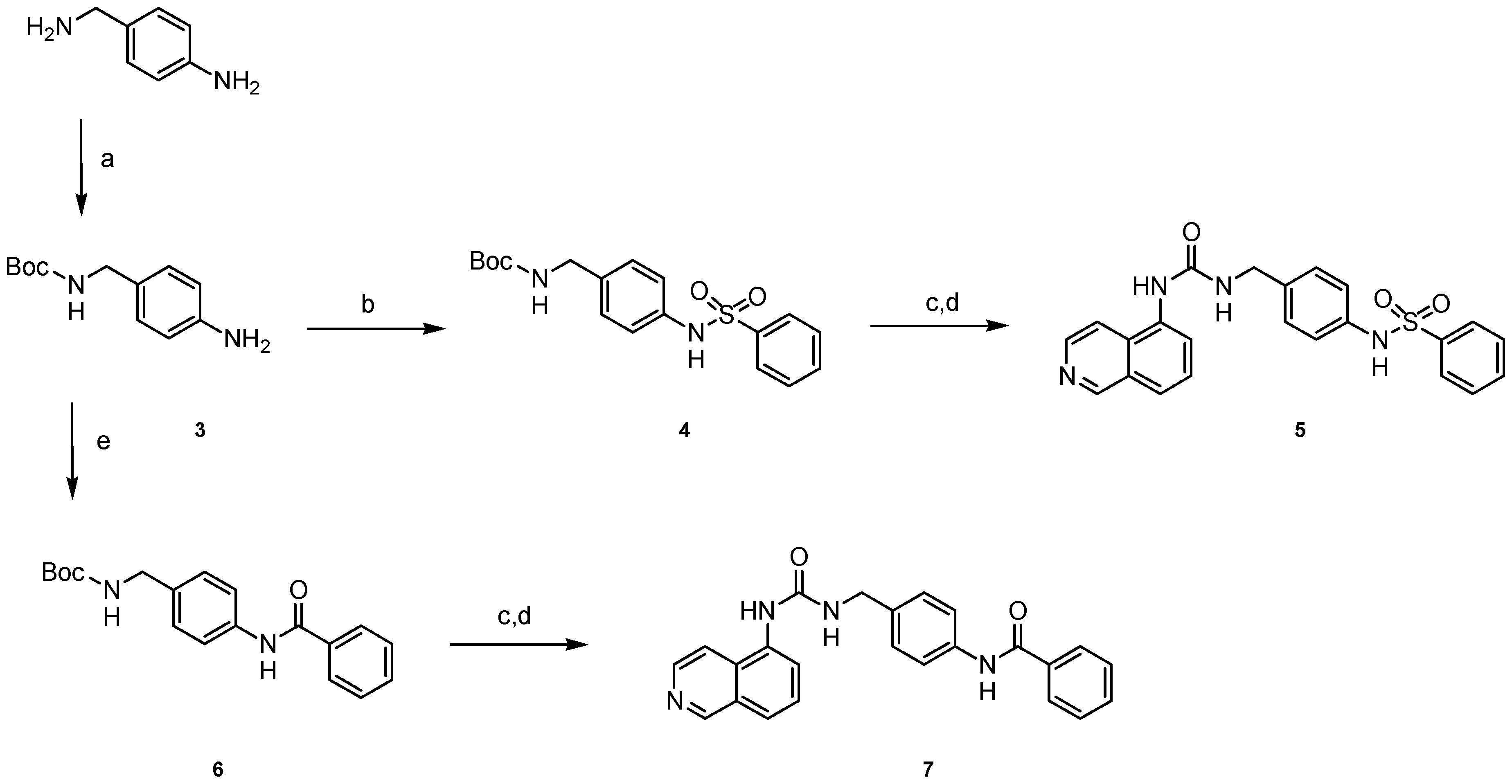
4.1.5. General Procedure for the Synthesis of Carbamates 4 and 6
4.1.6. General Procedure for the Synthesis of Ureas 5 and 7
4.2. Enzyme Inhibition Assay
4.3. Computational Methods
4.3.1. Ligand Preparation
4.3.2. Docking
4.3.3. Molecular Dynamics
- (i)
- Local energy minimization, heating up to 300 K in six MD runs (100 ps) with protein temperature set at 0, 100, 150, 200, 250 and 300 K;
- (ii)
- Production runs of 100 ns at 300 K in an isothermal–isobaric (NPT) ensemble, by using the velocity rescaling scheme (temperature) and the isotropic Berendsen coupling scheme (pressure) [45];
- (iii)
- (i)
- Local energy minimization, heating up to 300 K in six runs with protein temperature set at 0 (100 ps), 100 (200 ps), 150 (200 ps), 200 (200 ps), 250 (200 ps) and 300 K (200 ps);
- (ii)
- Production runs of 200 ns at 300 K in an NPT ensemble, by using the velocity rescaling scheme (temperature) and the isotropic Berendsen coupling scheme (pressure);
- (iii)
- Same setting of MMP-2:1i simulation.
4.3.4. DFT Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Apte, S.S.; Parks, W.C. Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Bio. 2015, 44–46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix metalloproteinases: Role in arthritis. Front Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Lohmander, L.S.; Hoerrner, L.A.; Lark, M.W. Metalloproteinases, tissue inhibitor, and proteoglycan fragments in knee synovial fluid in human osteoarthritis. Arthritis Rheum. 1993, 36, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.R. MMPs: Role in cardiovascular development and disease. Front Biosci. 2006, 11, 447–478. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Van den Steen, P.E.; Sang, Q.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W. Metalloproteinases: Mediators of pathology and regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Peress, N.; Perillo, E.; Zucker, S. Localization of tissue inhibitor of matrix metalloproteinases in Alzheimer's disease and normal brain. J. Neuropathol. Exp. Neurol. 1995, 54, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Chandler, S.; Coates, R.; Gearing, A.; Lury, J.; Wells, G.; Bone, E. Matrix metalloproteinases degrade myelin basic protein. Neurosci. Lett. 1995, 201, 223–226. [Google Scholar] [CrossRef]
- Demers, M.; Couillard, J.; Bélanger, S.; St-Pierre, Y. New roles for matrix metalloproteinases in metastasis. Crit. Rev. Immunol. 2005, 25, 493–523. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell. Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- Cox, J.H.; Overall, C.M. Cytokine substrates: MMP regulation of inflammatory signaling molecules. In The Cancer Degradome; Edwards, D., Hoyer-Hansen, G., Blasi, F., Sloane, B.F., Eds.; Springer: New York, NY, USA, 2008; pp. 519–539. [Google Scholar]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Overall, C. M.; Kleifeld, O. Tumour microenvironment-opinion: Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. New strategies for targeting matrix metalloproteinases. Matrix Biology 2015, 44–46, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Campestre, C.; Agamennone, M.; Tauro, M.; Tortorella, P. Phosphonate emerging zinc binding group in matrix metalloproteinase inhibitors. Curr. Drug Targets 2015, 16, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Romero-Perez, D.; Jacobsen, J.A.; Villarreal, F.J.; Cohen, S.M. Zinc-binding groups modulate selective inhibition of MMPs. ChemMedChem 2008, 3, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Tanakit, A.; Rouffet, M.; Martin, D.P.; Cohen, S.M. Investigating chelating sulfonamides and their use in metalloproteinase inhibitors. Dalton Trans. 2012, 41, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Campestre, C.; Tortorella, P.; Agamennone, M.; Preziuso, S.; Biasone, A.; Nuti, E.; Rossello, A.; Gallina, C. Peptidyl 3-substituted 1-hydroxyureas as isosteric analogues of succinylhydroxamate MMP inhibitors. Eur. J. Med. Chem. 2008, 43, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Rubino, M.T.; Agamennone, M.; Campestre, C.; Fracchiolla, G.; Laghezza, A.; Loiodice, F.; Nuti, E.; Rossello, A.; Tortorella, P. Synthesis, SAR, and biological evaluation of α-sulfonylphosphonic acids as selective matrix metalloproteinase inhibitors. ChemMedChem 2009, 4, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Tauro, M.; Laghezza, A.; Loiodice, F.; Agamennone, M.; Campestre, C.; Tortorella, P. Arylamino methylene bisphosphonate derivatives as bone seeking matrix metalloproteinase inhibitors. Bioorg. Med. Chem. 2013, 21, 6456–6465. [Google Scholar] [CrossRef] [PubMed]
- Devy, L.; Dransfield, D.T. New strategies for the next generation of matrix-metalloproteinase inhibitors: Selectively targeting membrane-anchored MMPs with therapeutic antibodies. Biochem. Res. Int. 2011, 2011, 191670. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, J.A.; Major Jourden, J.L.; Miller, M.T.; Cohen, M.T. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Biophys. Acta 2010, 1803, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Di Pizio, A.; Agamennone, M.; Tortorella, P. Non-zinc-binding inhibitors of MMP-13: GRID-based approaches to rationalize the binding process. Curr. Top. Med. Chem. 2016, 16, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Pochetti, G.; Montanari, R.; Gege, C.; Chevrier, C.; Taveras, A.G.; Mazza, F. Extra binding region induced by non-zinc chelating inhibitors into the S1 subsite of matrix metalloproteinase 8 (MMP-8). J. Med. Chem. 2009, 52, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Dublanchet, A.C.; Ducrot, P.; Andrianjara, C.; O’Gara, M.; Morales, R.; Compère, D.; Denis, A.; Blais, S.; Cluzeau, P.; Courté, K.; et al. Structure-based design and synthesis of novel non-zinc chelating MMP-12 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3787–3790. [Google Scholar] [CrossRef] [PubMed]
- Di Pizio, A.; Laghezza, A.; Tortorella, P.; Agamennone, M. Probing the S1′ Site for the Identification of Non-Zinc-Binding MMP-2 Inhibitor. ChemMedChem 2013, 8, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, D.; Fasciglione, G.F.; Gioia, M.; Ciaccio, C.; Tundo, G.R.; Marini, S.; Coletta, M. Human matrix metalloproteinases: An ubiquitarian class of enzymes involved in several pathological processes. Mol. Asp. Med. 2012, 33, 119–208. [Google Scholar] [CrossRef] [PubMed]
- Blanc, M.; Cussac, M.; Boucherle, A.; Leclerc, G. Synthesis and immunomodulating activity of 1-amino-2-thiohydantoin derivatives. Eur. J. Med. Chem. 1992, 27, 839–843. [Google Scholar] [CrossRef]
- Albrecht, M.; Witt, K.; Frohlich, R.; Kataeva, O. Inter- and intramolecular hydrogen bonding in amide- and urea-substituted 8-hydroxyquinoline derivatives. Tetrahedron 2002, 58, 561–567. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly regioselective arylation of sp3 C–H bonds catalyzed by palladium acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef] [PubMed]
- Dea-Ayuela, A.M.; Castillo, E.; Gonzalez-Alvarez, M.; Vega, C.; Rolón, M.; Bolás-Fernández, F.; Borrás, J.; González-Rosende, M.E. In vivo and in vitro anti-leishmanial activities of 4-nitro-N-pyrimidinand N-pyrazin-2-ylbenzenesulfonamides, and N2-(4-nitrophenyl)-N1-propylglycinamide. Bioorg. Med. Chem. 2009, 17, 7449–7456. [Google Scholar] [CrossRef] [PubMed]
- Acero-Alarcón, A.; Armero-Alarte, T.; Jordá-Gregori, J.M.; Rojas-Argudo, C.; Zaballos-García, E.; Server-Carrio, J.; Ahjyaje, F.Z.; Sepúlveda-Arques, J. Unusual ring closure reaction of amides with pyrimidines: Novel stereoselective synthesis of hexahydroimidazo[1,2-c]pyrimidines. Synthesis 1999, 12, 2124–2130. [Google Scholar] [CrossRef]
- Jain, P.; Saravanan, C.; Singh, S.K. Sulphonamides: Deserving class as MMP inhibitors? Eur. J. Med. Chem. 2013, 60, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Nanjan, P.; Nambiar, J.; Nair, B.G.; Banerji, A. Synthesis and discovery of (I-3,II-3)-biacacetin as a novel non-zinc binding inhibitor of MMP-2 and MMP-9. Bioorg. Med. Chem. 2015, 23, 3781–3787. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Peng, P.; Chang, J.; Liu, M.M.; Yu, J.M.; Zhou, L.; Sun, X. Selective non-zinc binding MMP-2 inhibitors: Novel benzamide ilomastat analogs with anti-tumor metastasis. Bioorg. Med. Chem. 2016, 26, 2174–2178. [Google Scholar] [CrossRef] [PubMed]
- Di Pizio, A.; Agamennone, M.; Aschi, M. An integrated computational approach to rationalize the activity of non-zinc-binding MMP-2 inhibitors. PLoS ONE 2012, 7, e47774. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. Maestro Version 10.2, LigPrep Version 3.4, Epik, Version 3.2, MacroModel, Version 10.8, Jaguar, Version 8.8, Glide Version 6.7; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullmann, B., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comp. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.J. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Frajie, J.C.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.A.; York, D.M.; Pedersen, L.G. Particle mesh ewald: An N log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Lin, F.; Wang, R. Systematic derivation of AMBER force field parameters applicable to zinc-containing systems. J. Chem. Theory Comput. 2010, 6, 1852–1870. [Google Scholar] [CrossRef] [PubMed]
- Gaussian Inc. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5452. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular orbital methods. IX. extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; DeFrees, D.J.; Pople, J.A.; Gordon, M.S. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for 2nd-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Li, X.; Frisch, M.J. Energy-represented DIIS within a hybrid geometry optimization method. J. Comput. Chem. 2006, 2, 835–839. [Google Scholar]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
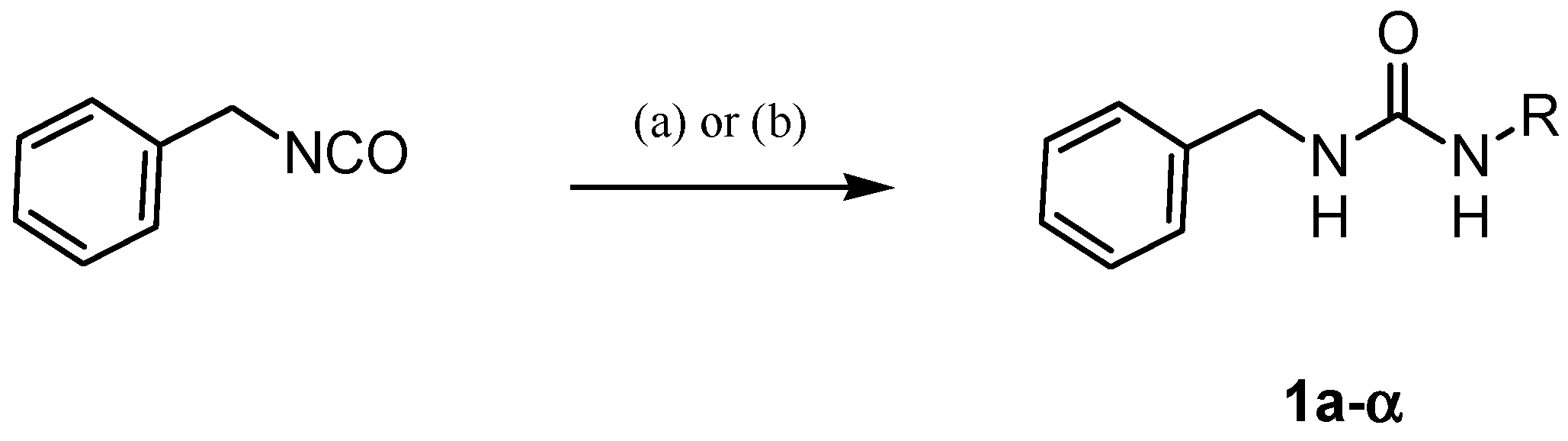



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
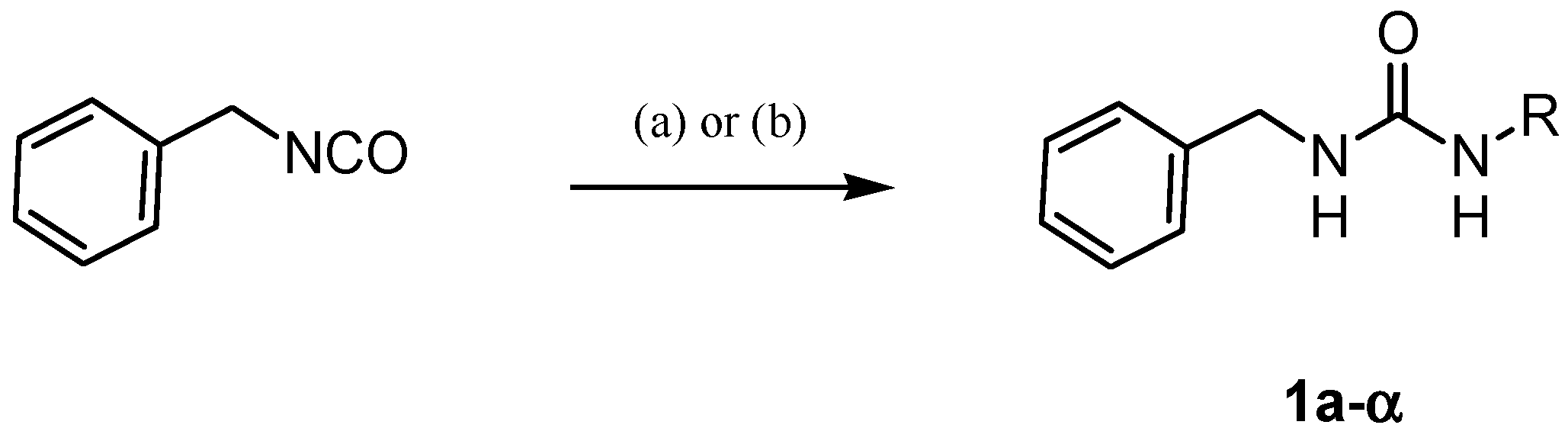{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
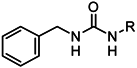 | |||
| ID | R | ID | R |
| 1a | 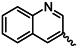 | 1n |  |
| 1b |  | 1o | 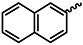 |
| 1c |  | 1p | 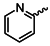 |
| 1d |  | 1q | 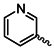 |
| 1e | 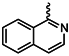 | 1r | 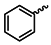 |
| 1f |  | 1s | 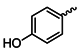 |
| 1g | 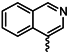 | 1t | 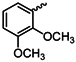 |
| 1h | 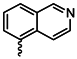 | 1u | 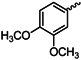 |
| 1i | 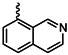 | 1v | 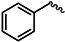 |
| 1j | 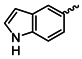 | 1w | 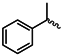 |
| 1k | 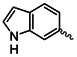 | 1x |  |
| 1l | 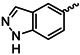 | 1y | 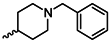 |
| 1m | 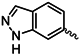 | 1z | 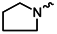 |
| 1α | 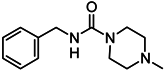 | ||
 | ||
| Cpd | Linker | n |
| 2a | NHCO | 1 |
| 2b | NHCO | 2 |
| 2c | CONH | 1 |
| 2d | CONH | 2 |
| 2e | NHSO2 | - |
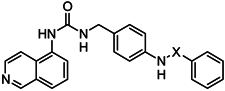 | |
| Cpd | X |
| 5 | SO2 |
| 7 | CO |
| ID | MMP-2 | MMP-8 | MMP-13 |
|---|---|---|---|
| 1c | 55 ± 1 | >100 | 58 ± 1 |
| 1g | 74 ± 3 | >100 | 73 ± 11 |
| 1h | 15 ± 4 | >100 | 14 ± 4 |
| 1l | 86 ± 2 | >100 | 98 ± 2 |
| 5 | 54 ± 7 | 88 ± 5 | 55 ± 7 |
| 7 | 7.4 ± 0.8 | 48.5 ± 1.6 | 6.6 ± 0.4 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammazzalorso, A.; De Filippis, B.; Campestre, C.; Laghezza, A.; Marrone, A.; Amoroso, R.; Tortorella, P.; Agamennone, M. Seeking for Non-Zinc-Binding MMP-2 Inhibitors: Synthesis, Biological Evaluation and Molecular Modelling Studies. Int. J. Mol. Sci. 2016, 17, 1768. https://doi.org/10.3390/ijms17101768
Ammazzalorso A, De Filippis B, Campestre C, Laghezza A, Marrone A, Amoroso R, Tortorella P, Agamennone M. Seeking for Non-Zinc-Binding MMP-2 Inhibitors: Synthesis, Biological Evaluation and Molecular Modelling Studies. International Journal of Molecular Sciences. 2016; 17(10):1768. https://doi.org/10.3390/ijms17101768
Chicago/Turabian StyleAmmazzalorso, Alessandra, Barbara De Filippis, Cristina Campestre, Antonio Laghezza, Alessandro Marrone, Rosa Amoroso, Paolo Tortorella, and Mariangela Agamennone. 2016. "Seeking for Non-Zinc-Binding MMP-2 Inhibitors: Synthesis, Biological Evaluation and Molecular Modelling Studies" International Journal of Molecular Sciences 17, no. 10: 1768. https://doi.org/10.3390/ijms17101768
APA StyleAmmazzalorso, A., De Filippis, B., Campestre, C., Laghezza, A., Marrone, A., Amoroso, R., Tortorella, P., & Agamennone, M. (2016). Seeking for Non-Zinc-Binding MMP-2 Inhibitors: Synthesis, Biological Evaluation and Molecular Modelling Studies. International Journal of Molecular Sciences, 17(10), 1768. https://doi.org/10.3390/ijms17101768