
Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2

Abstract
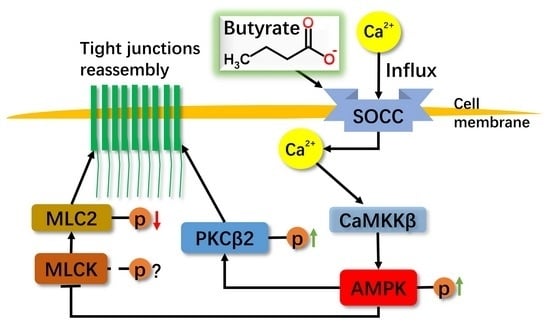
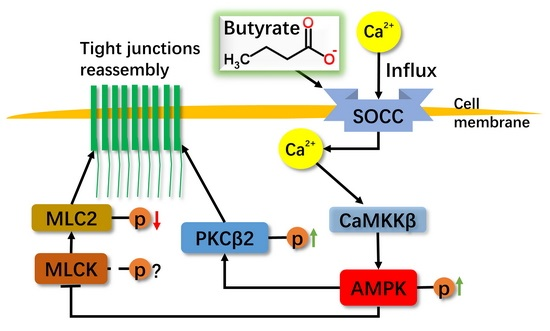
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
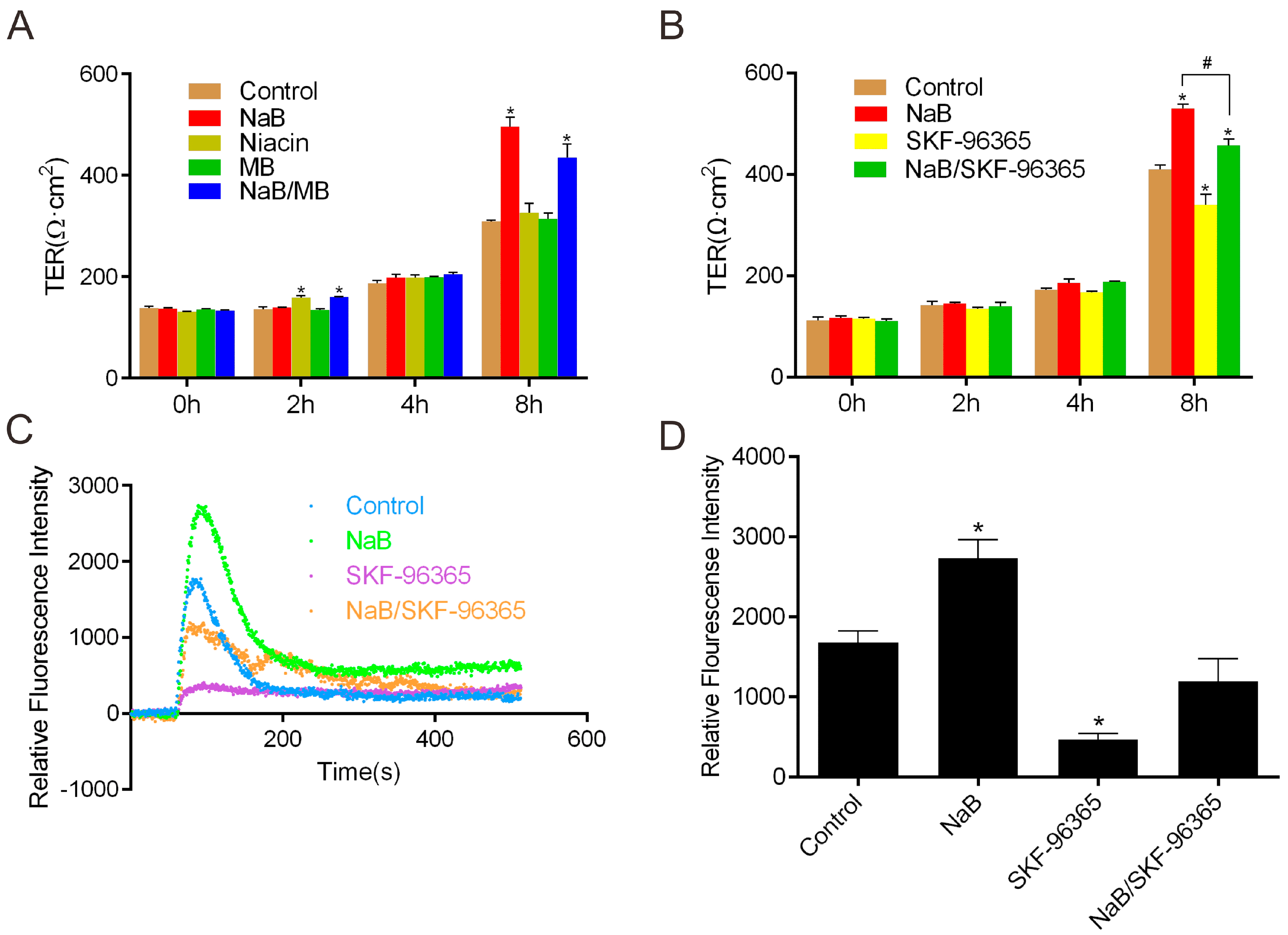
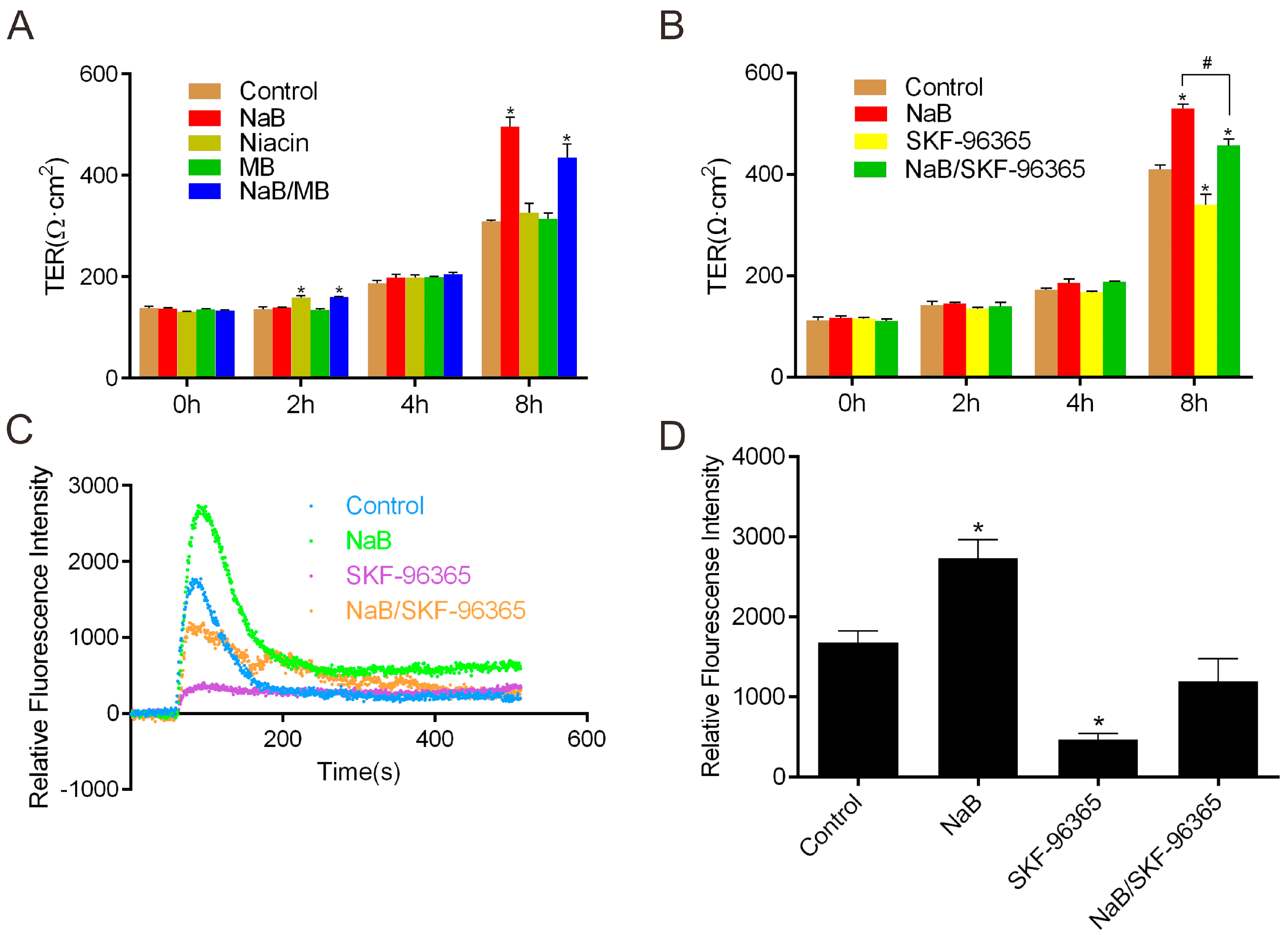
2.1. Effect of Sodium Butyrate (NaB) on GPR109A and Store-Operated Calcium Entry (SOCE) during Reassembly of Tight Junctions (TJs)
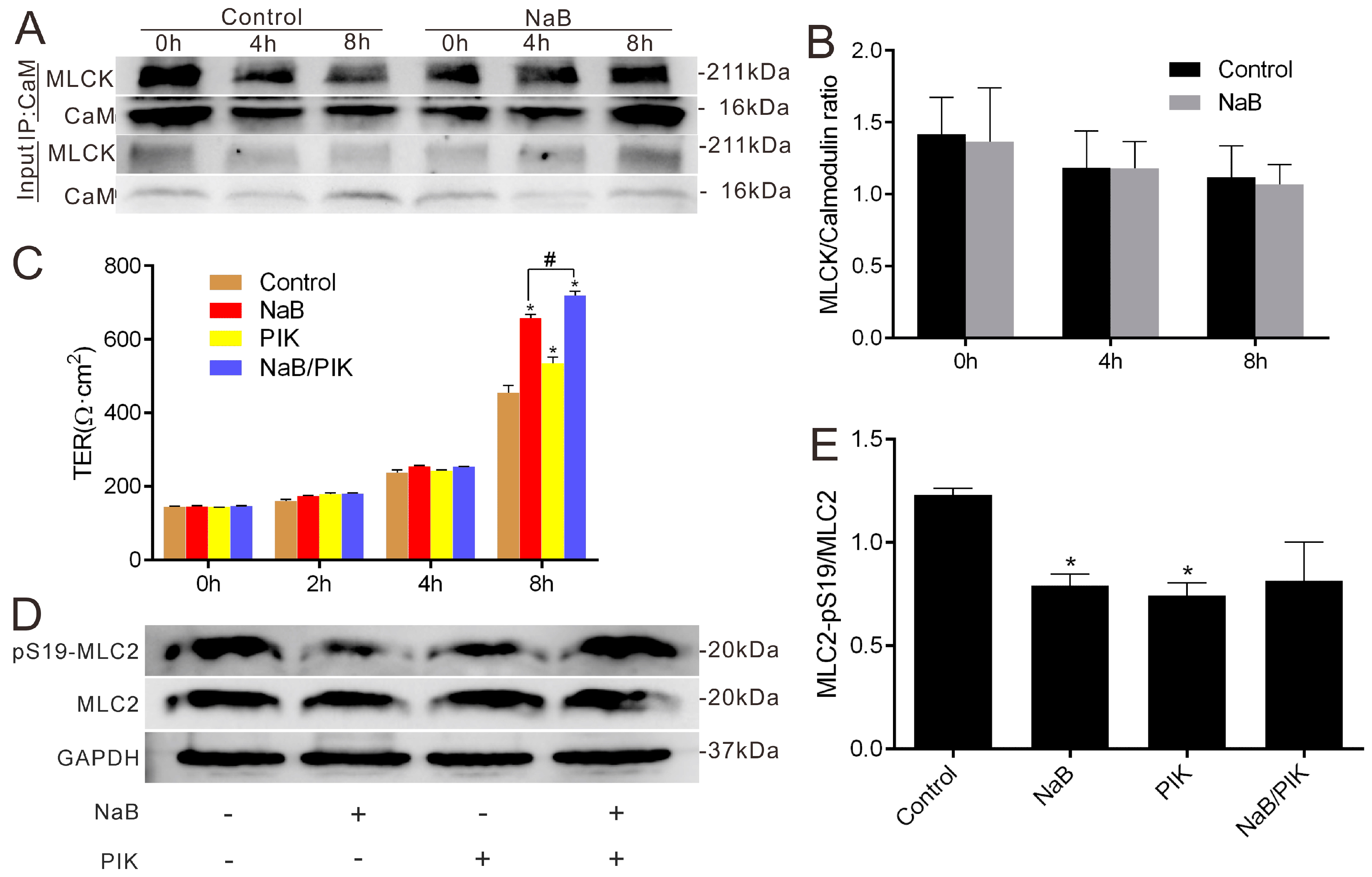
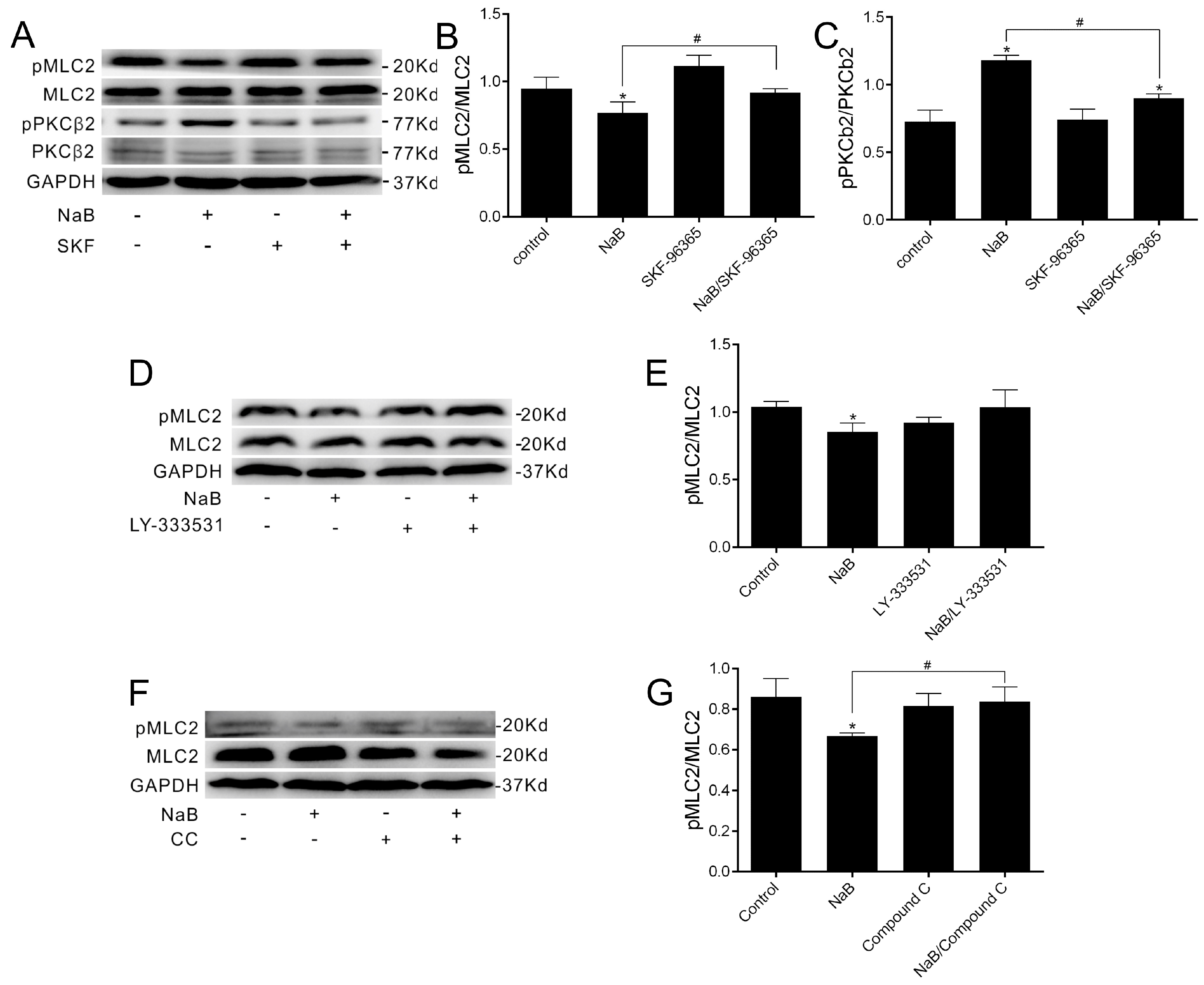
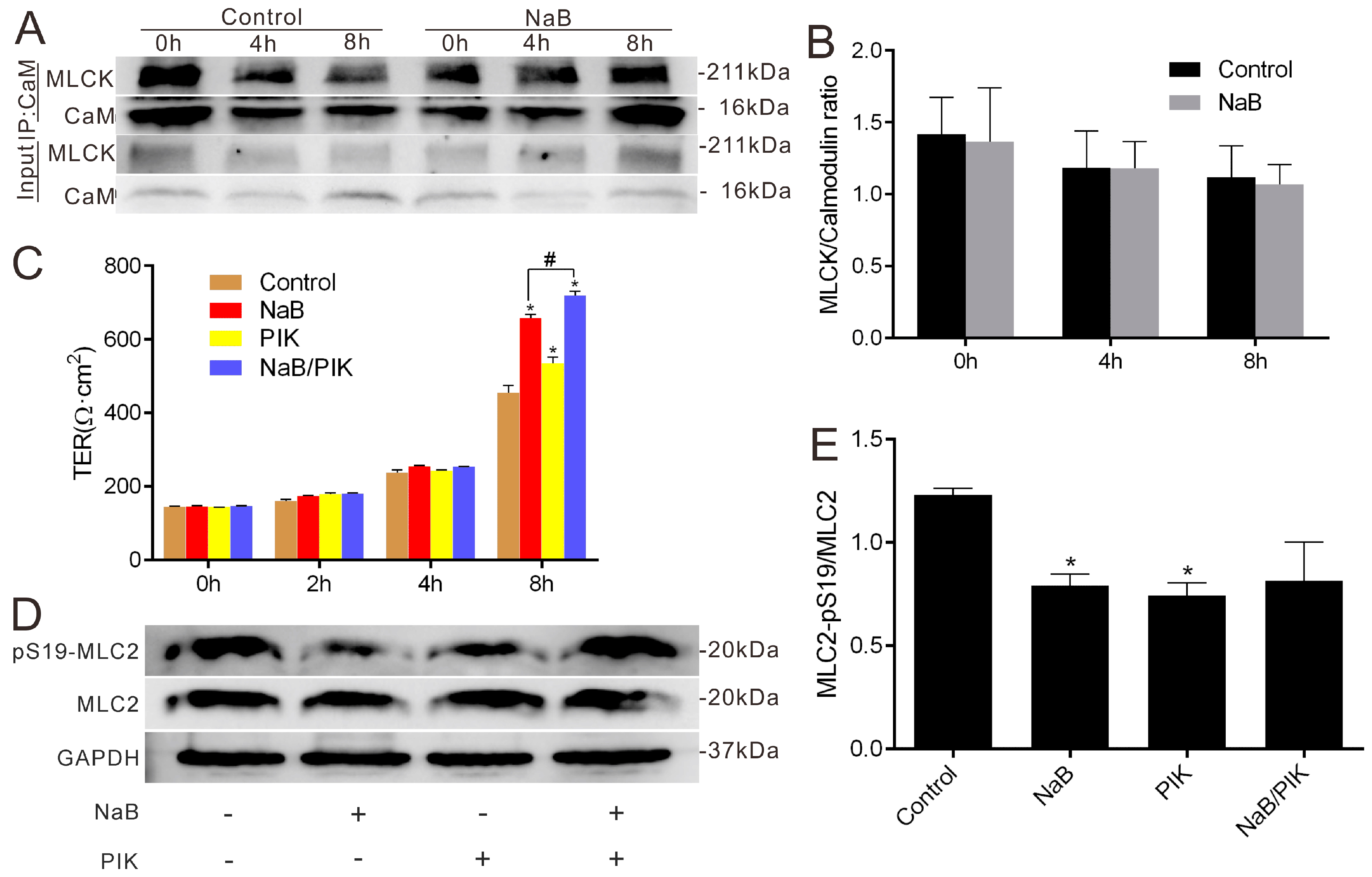
2.2. Effect of NaB on Myosin Light Chain Kinase (MLCK) and Myosin II Regulatory Light Chain (MLC2) during the Reassembly of TJs
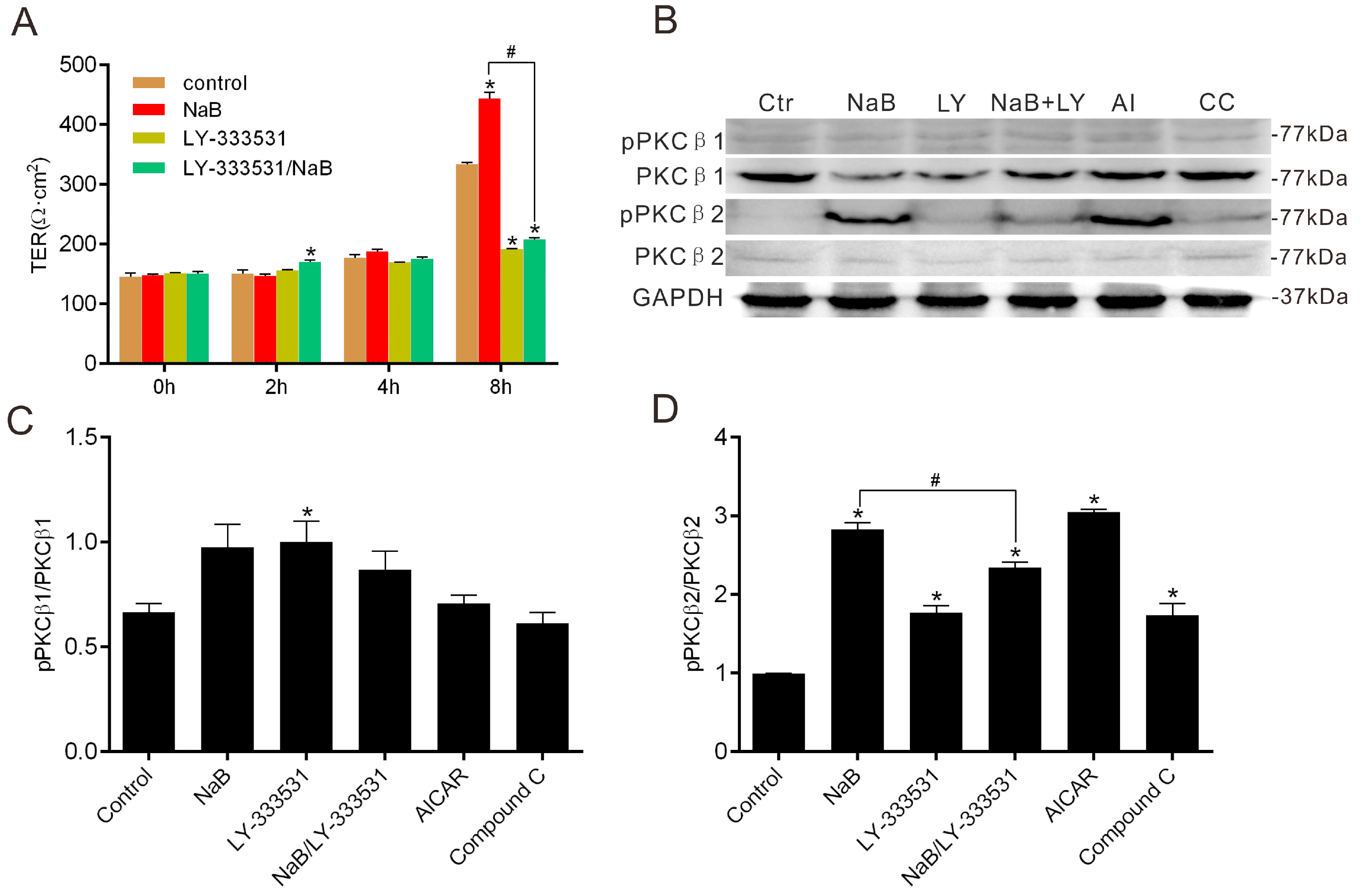
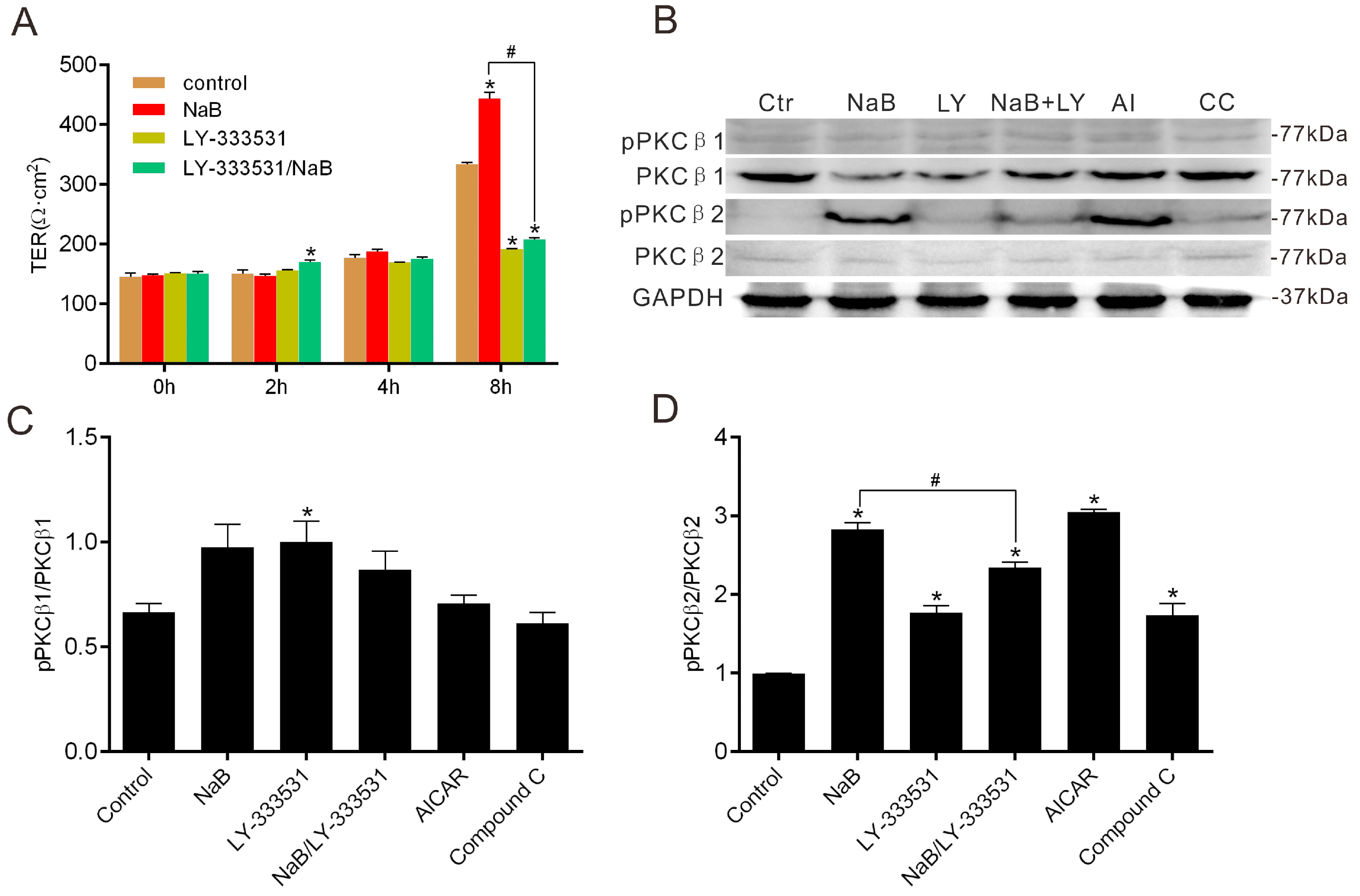
2.3. Effect of NaB on the Phosphorylation of Protein Kinase C β (PKCβ) during Reassembly of TJs
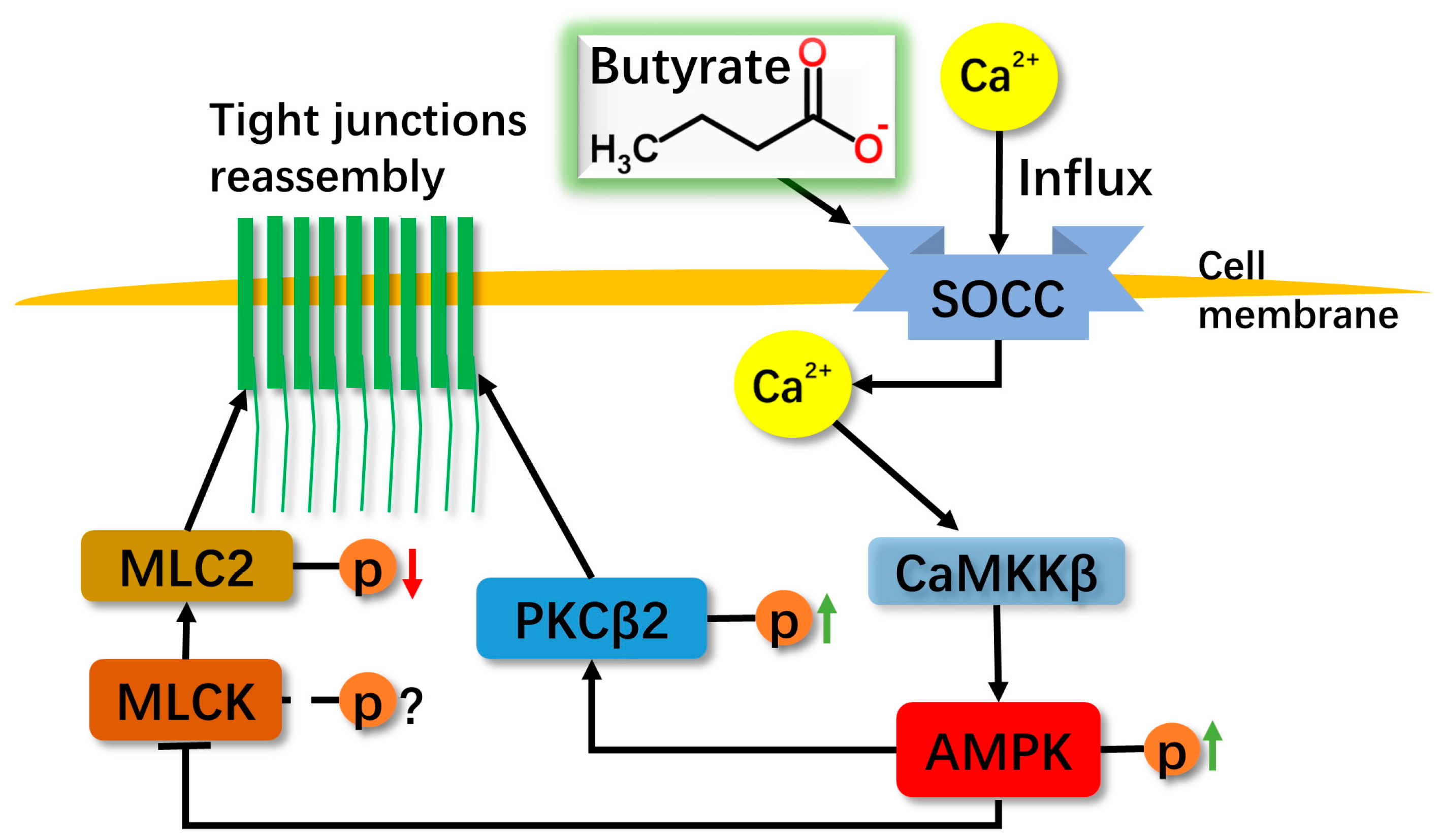
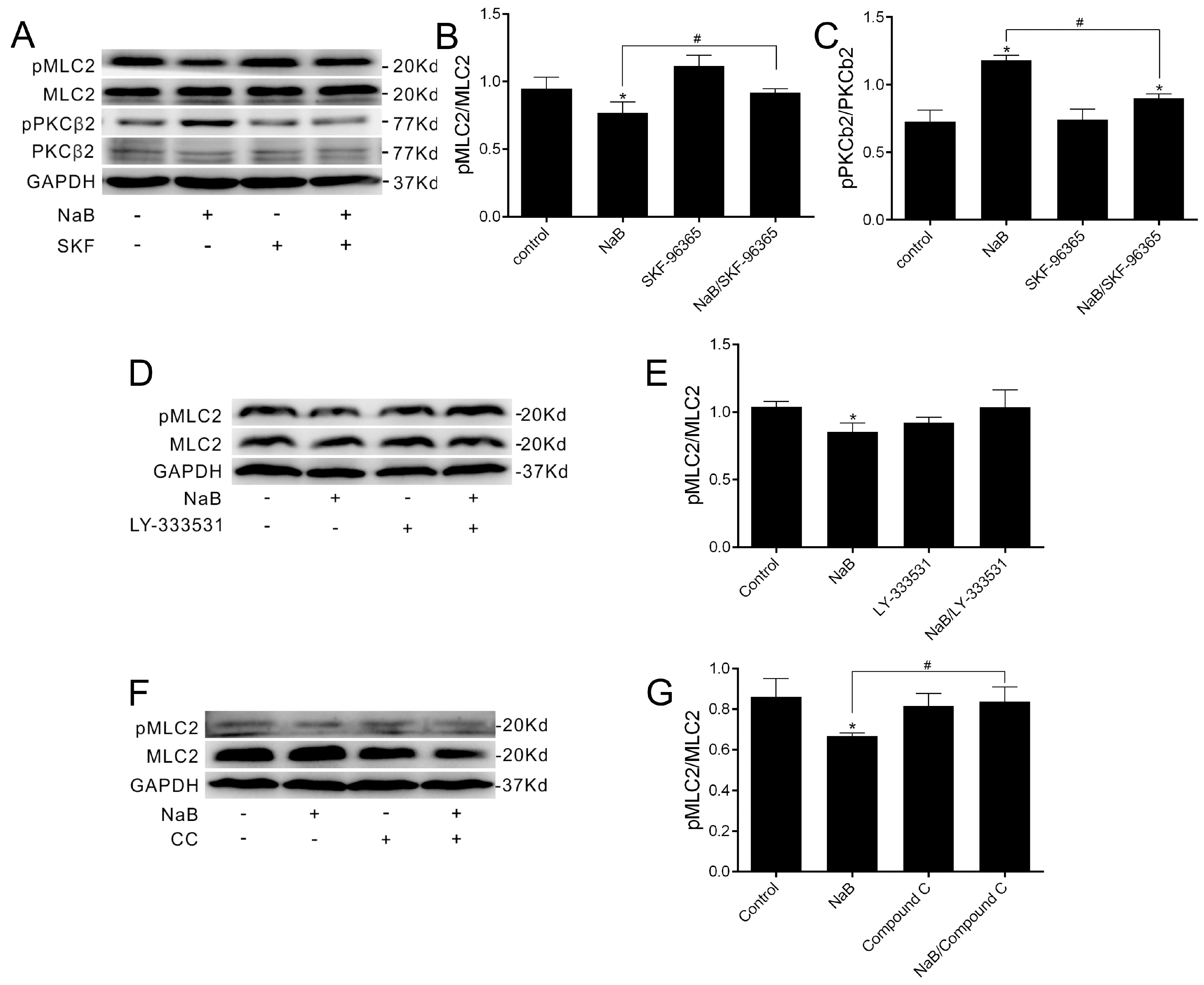
2.4. The Relationship among MLC2, PKCβ2 and AMPK during TJs Reassembly Induced by NaB
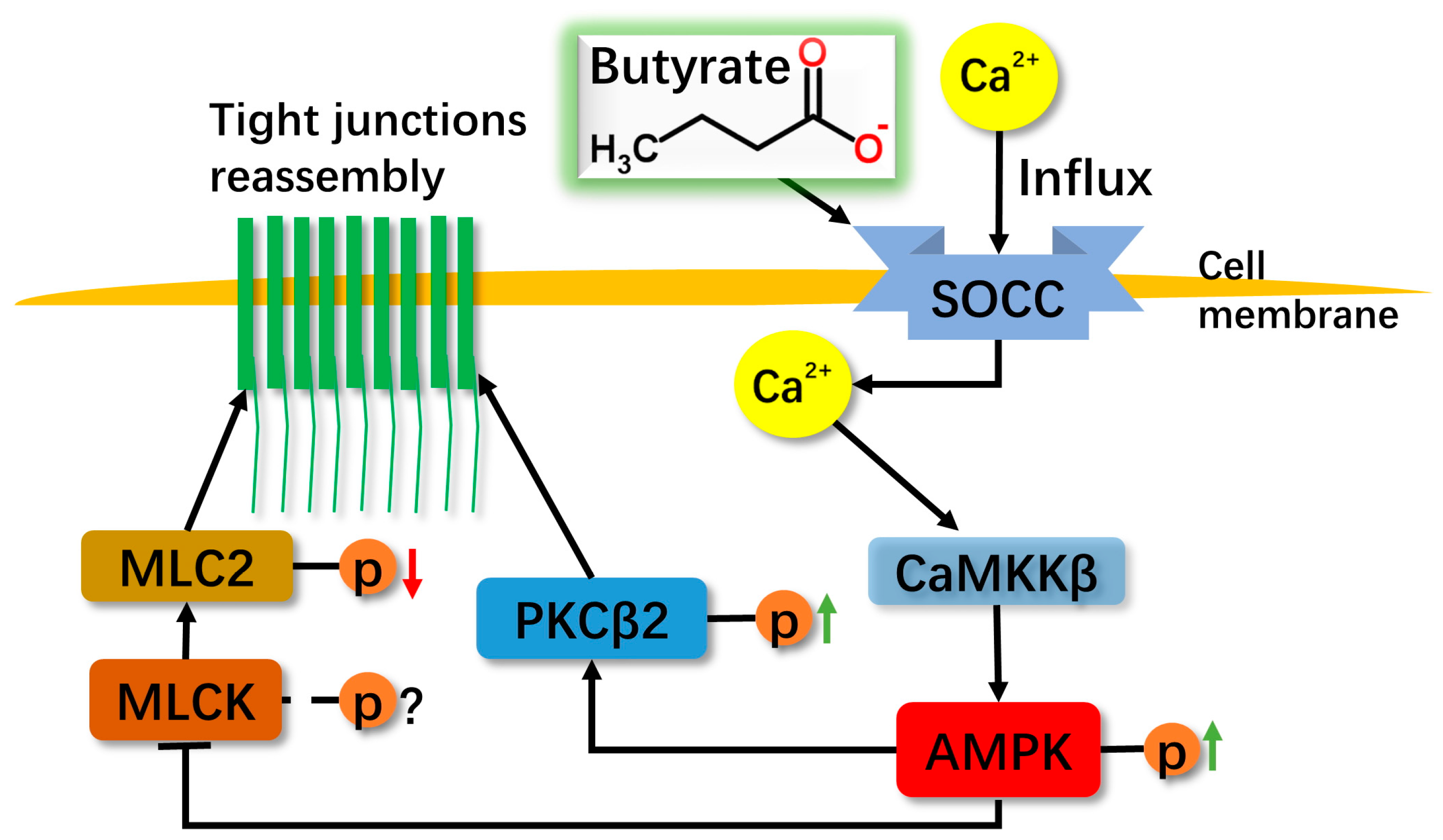
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Calcium Switch Assay
4.4. Measurement of TER
4.5. Intracellular Ca2+ Measurements
4.6. Western Blotting
4.7. Immunoprecipitation
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [PubMed]
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. Part B Comp. Biochem. 1987, 86, 439–472. [Google Scholar] [CrossRef]
- Bugaut, M.; Bentejac, M. Biological effects of short-chain fatty acids in nonruminant mammals. Annu. Rev. Nutr. 1993, 13, 217–241. [Google Scholar] [CrossRef] [PubMed]
- Gill, R.K.; Dudeja, P.K. A novel facet to consider for the effects of butyrate on its target cells. Focus on “the short-chain fatty acid butyrate is a substrate of breast cancer resistance protein”. Am. J. Physiol. Cell Physiol. 2011, 301, C977–C979. [Google Scholar] [CrossRef] [PubMed]
- Harten, S.K.; Shukla, D.; Barod, R.; Hergovich, A.; Balda, M.S.; Matter, K.; Esteban, M.A.; Maxwell, P.H. Regulation of renal epithelial tight junctions by the von Hippel-Lindau tumor suppressor gene involves occludin and claudin 1 and is independent of E-cadherin. Mol. Biol. Cell 2009, 20, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Matloob, A.F.; Du, J.; Pan, H.; Dong, Z.; Zhao, J.; Feng, Y.; Zhong, Y.; Huang, B.; Lu, J. Vitamin D stimulates apoptosis in gastric cancer cells in synergy with trichostatin A /sodium butyrate-induced and 5-aza-2’-deoxycytidine-induced PTEN upregulation. FEBS J. 2010, 277, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Hawley, S.A.; Scott, J.W. AMP-activated protein kinase—Development of the energy sensor concept. J. Physiol. 2006, 574, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Selbert, M.A.; Goldstein, E.G.; Edelman, A.M.; Carling, D.; Hardie, D.G. 5’-AMP activates the amp-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J. Biol. Chem. 1995, 270, 27186–27191. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase β. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Miao, W.; Liu, C.; Kang, Z.; Li, L.; Peng, L. Butyrate facilitates tight junction reassembly via activation of Ca2+/calmodulin-dependent protein kinase. J. Tongji Univ. Med. Sci. 2015, 8–12. [Google Scholar]
- Sun, S.; Li, W.; Zhang, H.; Zha, L.; Xue, Y.; Wu, X.; Zou, F. Requirement for store-operated calcium entry in sodium butyrate-induced apoptosis in human colon cancer cells. Biosci. Rep. 2012, 32, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Van Itallie, C.M. Tight junctions and the molecular basis for regulation of paracellular permeability. Am. J. Physiol. 1995, 269, G467–G475. [Google Scholar] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Jameson, B.J.; Jesaitis, L.A.; Anderson, J.M. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J. Biol. Chem. 1998, 273, 29745–29753. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Ma, T.Y.; Anderson, J.M. Isolation and functional characterization of the actin-binding region in the tight junction protein ZO-1. FASEB J. 2002, 16, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.E.; Turner, J.R. Myosin light chain kinase: Pulling the strings of epithelial tight junction function. Ann. N. Y. Acad. Sci. 2012, 1258, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Turner, J.R. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol. Biol. Cell 2005, 16, 3919–3936. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Wang, P.; Su, Q.; Wang, S.L.; Wang, F.J. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE 2012, 7, e34946. [Google Scholar] [CrossRef] [PubMed]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J.; Mrsny, R.J.; Turner, J.R. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Srivastava, K.; Bayraktutan, U. Small GTPase RhoA and its effector rho kinase mediate oxygen glucose deprivation-evoked in vitro cerebral barrier dysfunction. Stroke 2010, 41, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Banan, A.; Fields, J.Z.; Talmage, D.A.; Zhang, Y.; Keshavarzian, A. PKC-β1 mediates EGF protection of microtubules and barrier of intestinal monolayers against oxidants. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G833–G847. [Google Scholar] [PubMed]
- Seth, A.; Yan, F.; Polk, D.B.; Rao, R.K. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a pkc- and map kinase-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1060–G1069. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. The molecular choreography of a store-operated calcium channel. Nature 2007, 446, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Rigor, R.R.; Hawkins, B.T.; Miller, D.S. Activation of PKC isoform β(I) at the blood-brain barrier rapidly decreases P-glycoprotein activity and enhances drug delivery to the brain. J. Cereb. Blood Flow Metab. 2010, 30, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of GPR109A, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, A.; Priyamvada, S.; Kumar, A.; Natarajan, A.A.; Gill, R.K.; Alrefai, W.A.; Dudeja, P.K. A novel nutrient sensing mechanism underlies substrate-induced regulation of monocarboxylate transporter-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1126–G1133. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.; Morel, N.; Vertommen, D.; Hussain, N.; Neumann, D.; Beauloye, C.; El Najjar, N.; Forcet, C.; Viollet, B.; Walsh, M.P.; et al. AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J. Biol. Chem. 2008, 283, 18505–18512. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.J.; Ferro, T.J.; Blumenstock, F.A.; Brockenauer, A.M.; Malik, A.B. Increased endothelial albumin permeability mediated by protein kinase C activation. J. Clin. Investig. 1990, 85, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ze’ev, A. Tumor promoter-induced disruption of junctional complexes in cultured epithelial cells is followed by the inhibition of cytokeratin and desmoplakin synthesis. Exp. Cell Res. 1986, 164, 335–352. [Google Scholar] [CrossRef]
- Siflinger-Birnboim, A.; Goligorsky, M.S.; del Vecchio, P.J.; Malik, A.B. Activation of protein kinase C pathway contributes to hydrogen peroxide-induced increase in endothelial permeability. Lab Investig. J. Tech. Methods Pathol. 1992, 67, 24–30. [Google Scholar]
- Garcia, J.G.N.; Aschner, J.L.; Malik, A.B. Regulation of thrombin-induced endothelial barrier dysfunction and prostaglandin synthesis. In Thrombin: Structure and Function; Berliner, L.J., Ed.; Springer US: Boston, MA, USA, 1992; pp. 397–430. [Google Scholar]
- Johnson, A.; Phillips, P.; Hocking, D.; Tsan, M.F.; Ferro, T. Protein kinase inhibitor prevents pulmonary edema in response to H2O2. Am. J. Physiol. 1989, 256, H1012–H1022. [Google Scholar] [PubMed]
- Spyridopoulos, I.; Luedemann, C.; Chen, D.; Kearney, M.; Chen, D.; Murohara, T.; Principe, N.; Isner, J.M.; Losordo, D.W. Divergence of angiogenic and vascular permeability signaling by VEGF: Inhibition of protein kinase C suppresses VEGF-induced angiogenesis, but promotes VEGF-induced, no-dependent vascular permeability. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Valenzano, M.C.; DiGuilio, K.; Mercado, J.; Teter, M.; To, J.; Ferraro, B.; Mixson, B.; Manley, I.; Baker, V.; Moore, B.A.; et al. Remodeling of tight junctions and enhancement of barrier integrity of the Caco-2 intestinal epithelial cell layer by micronutrients. PLoS ONE 2015, 10, e0133926. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, W.; Wu, X.; Wang, K.; Wang, W.; Wang, Y.; Li, Z.; Liu, J.; Li, L.; Peng, L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. Int. J. Mol. Sci. 2016, 17, 1696. https://doi.org/10.3390/ijms17101696
Miao W, Wu X, Wang K, Wang W, Wang Y, Li Z, Liu J, Li L, Peng L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. International Journal of Molecular Sciences. 2016; 17(10):1696. https://doi.org/10.3390/ijms17101696
Chicago/Turabian StyleMiao, Wei, Xiujuan Wu, Kang Wang, Wenjing Wang, Yumei Wang, Zhigang Li, Jingjing Liu, Li Li, and Luying Peng. 2016. "Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2" International Journal of Molecular Sciences 17, no. 10: 1696. https://doi.org/10.3390/ijms17101696
APA StyleMiao, W., Wu, X., Wang, K., Wang, W., Wang, Y., Li, Z., Liu, J., Li, L., & Peng, L. (2016). Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. International Journal of Molecular Sciences, 17(10), 1696. https://doi.org/10.3390/ijms17101696