
Time in Redox Adaptation Processes: From Evolution to Hormesis



Abstract
:
1. Adaptation or Hormesis in General
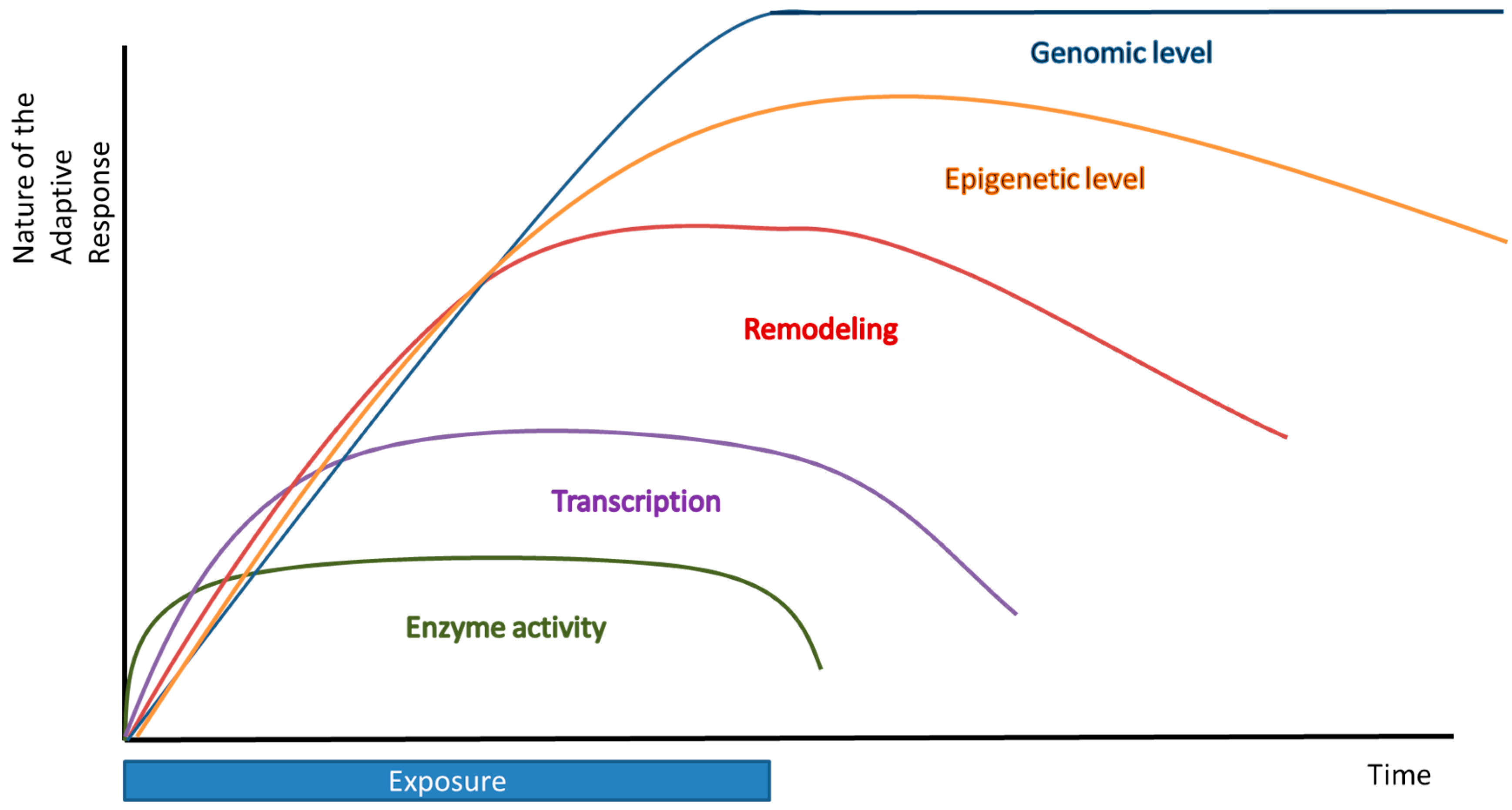
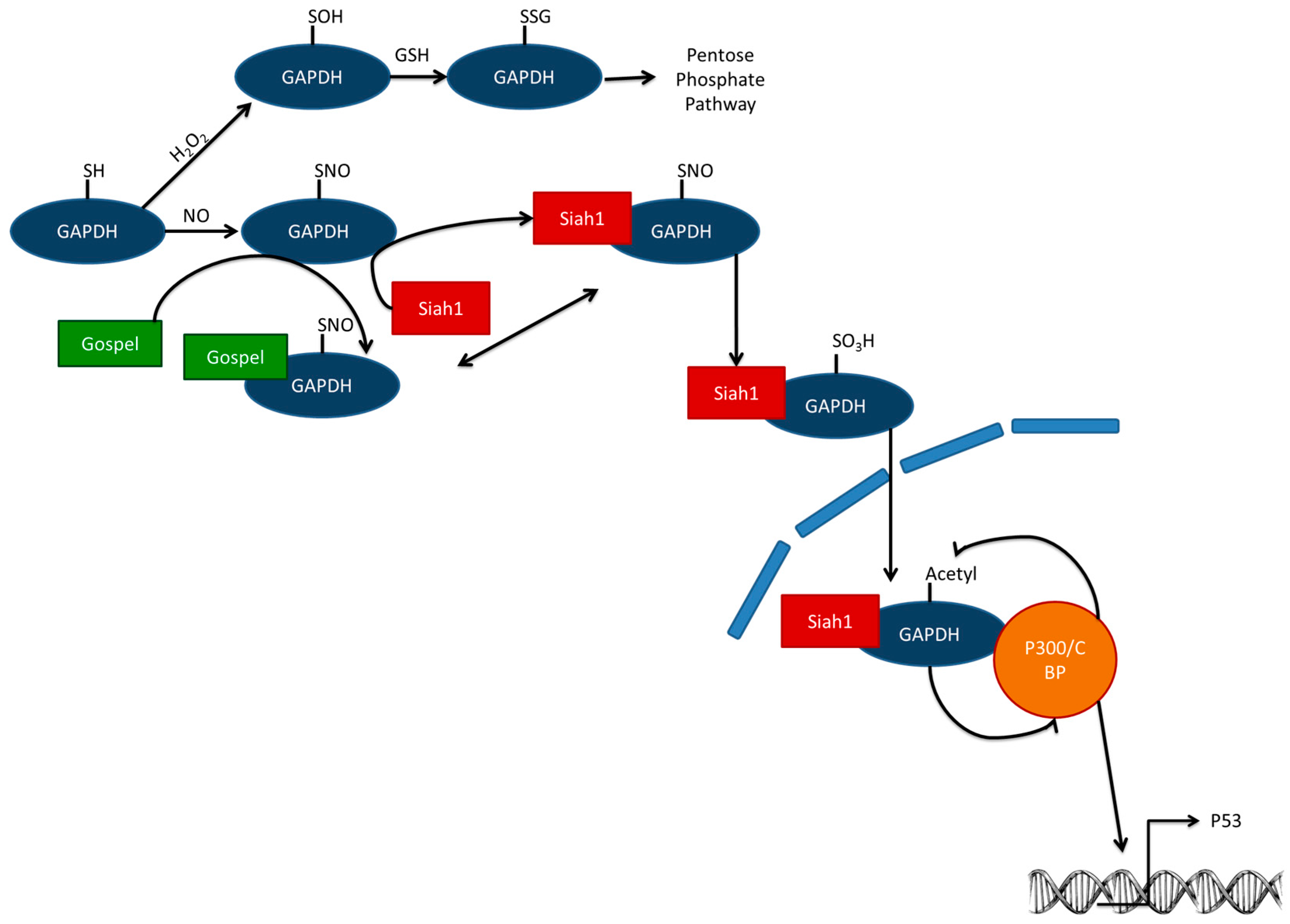
2. Short Term—Enzymatic Level
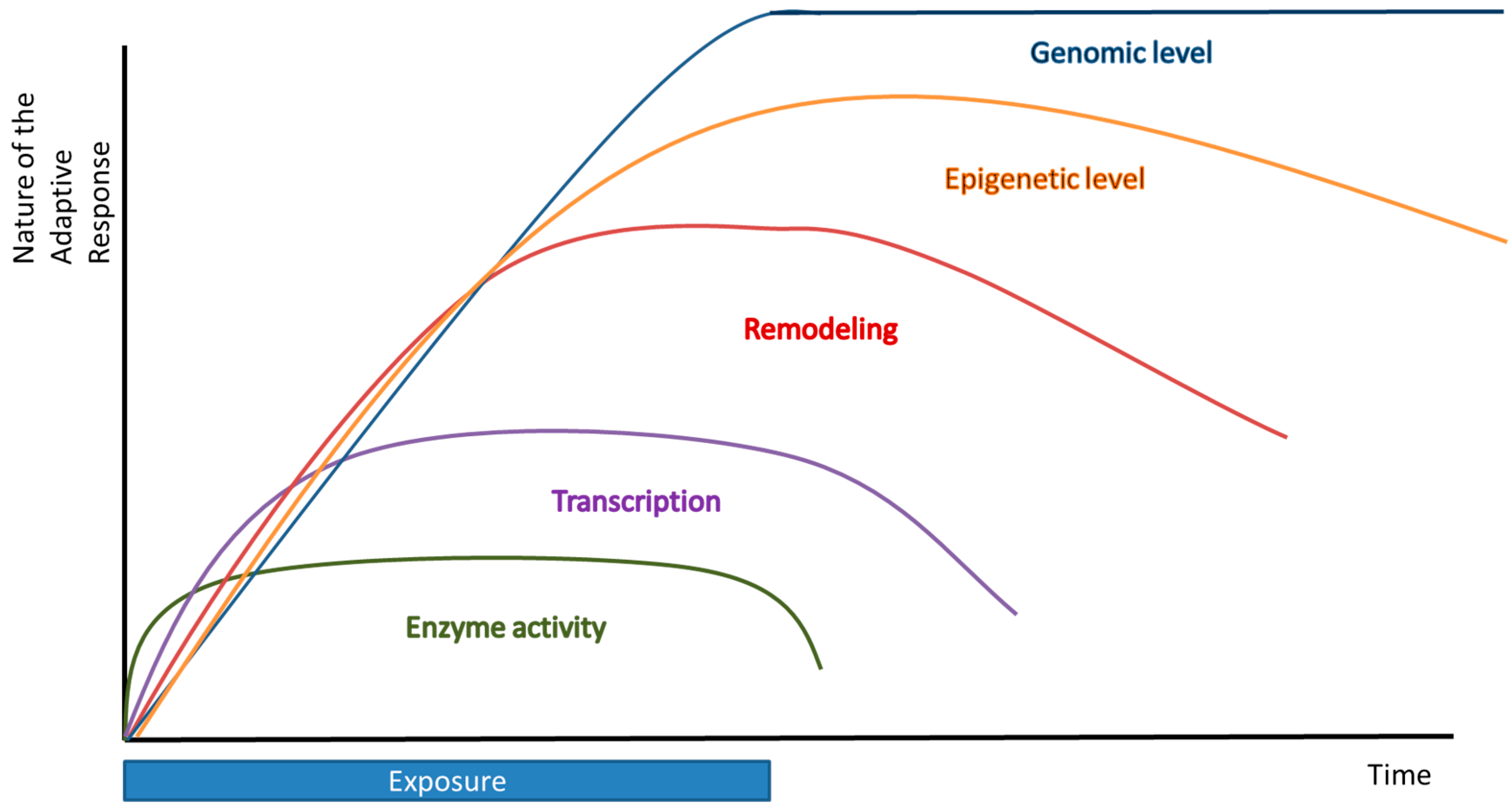
3. Long Term Adaptation—Transcriptional, Epigenetic, and Genomic Level
3.1. Transcriptional Level
3.2. Epigenetic Level
3.3. Genomic Level
4. Factors Influencing Hormesis
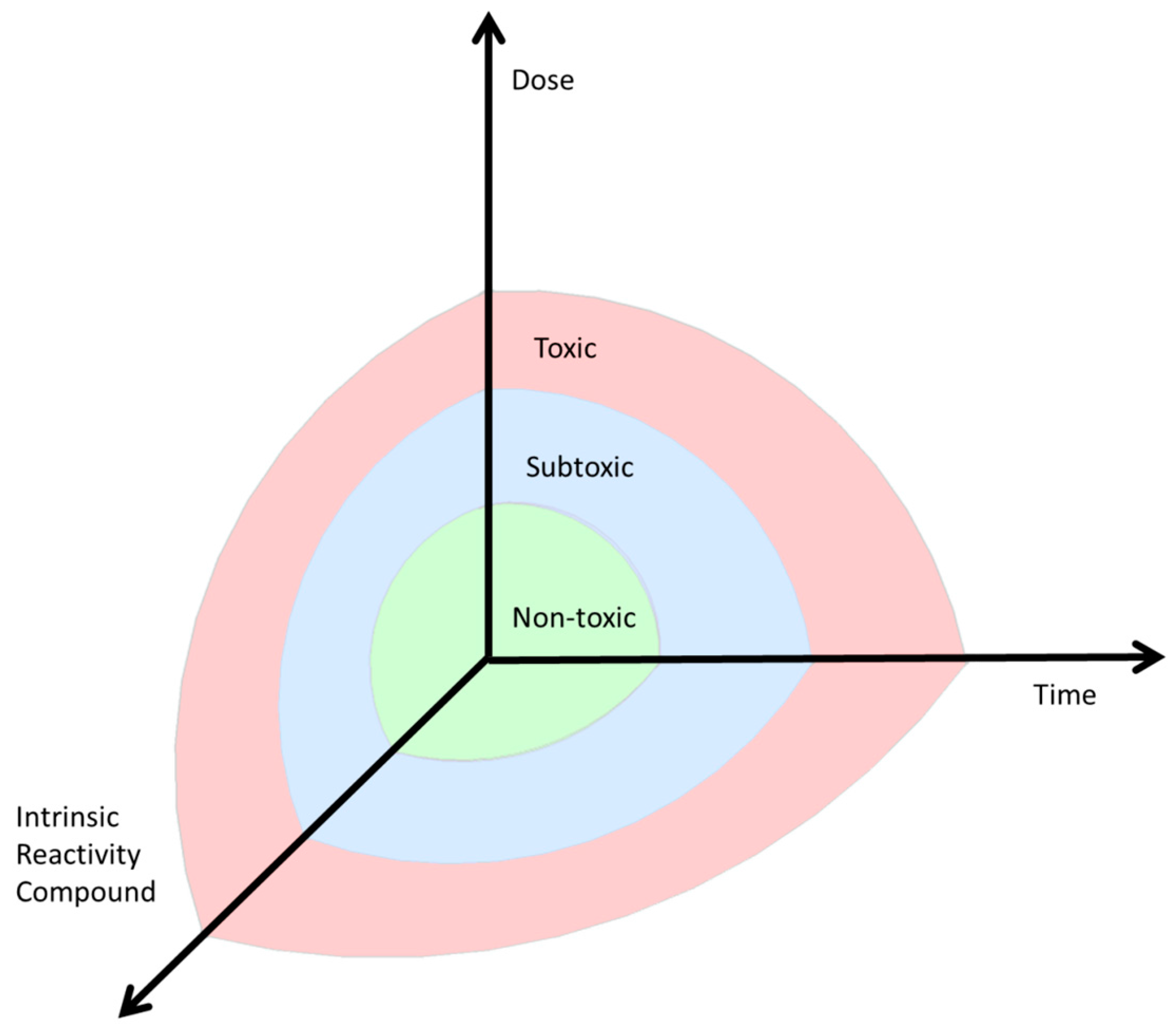
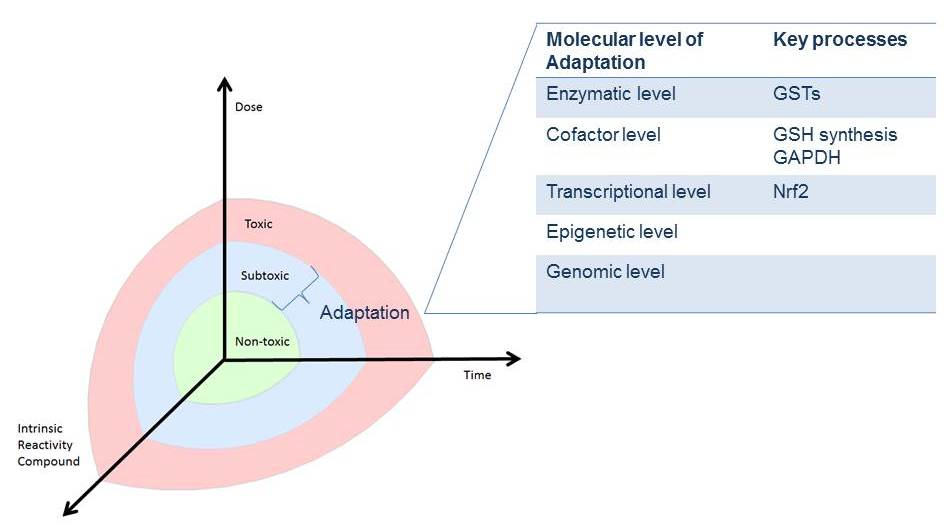
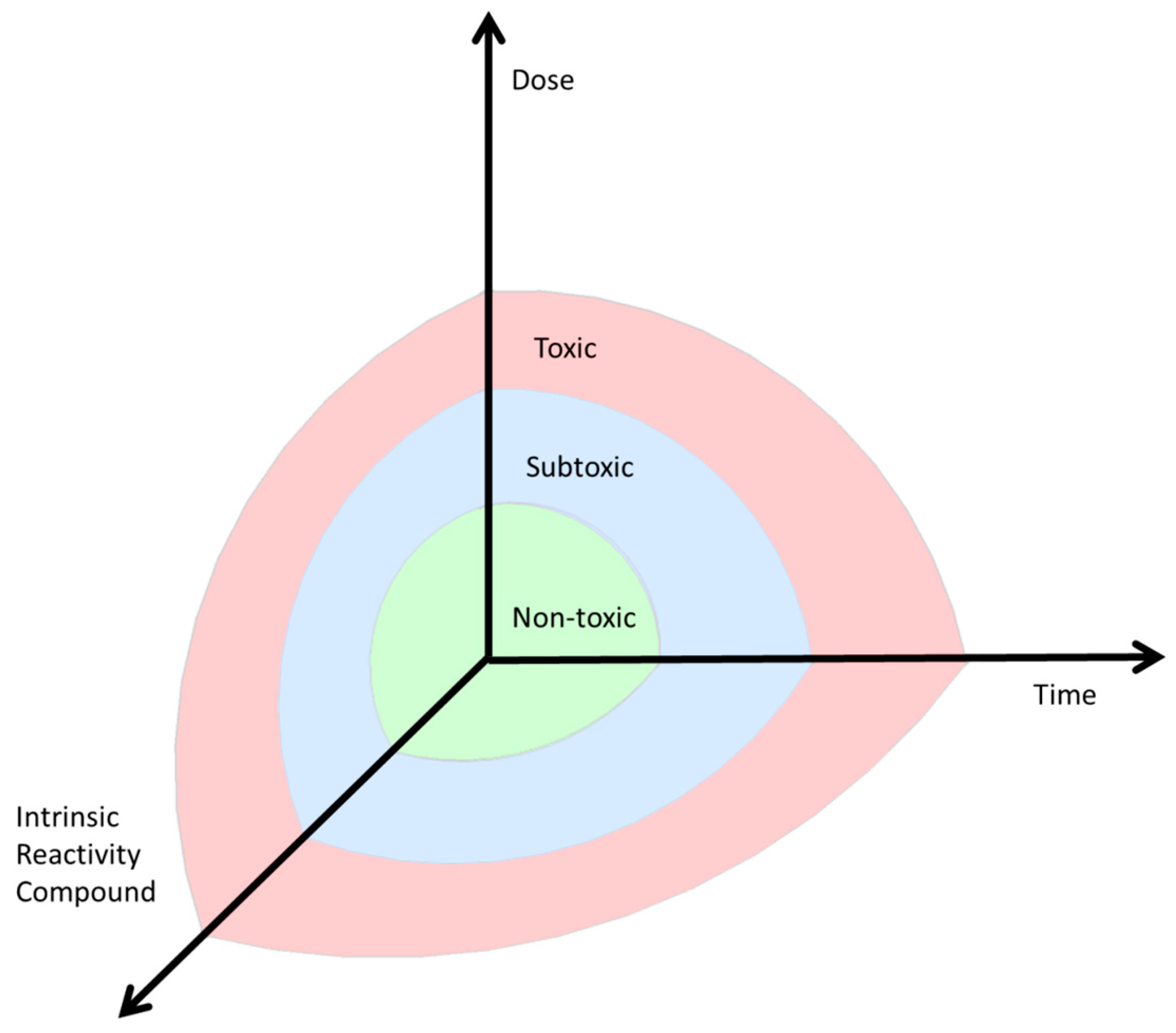
5. Dose and Reactivity of the Compound and Hormesis
6. Adaptation Induced by Acrolein
6.1. Short-Term—Enzymatic Level
6.2. Long-Term Adaptation
6.2.1. Transcriptional Level
6.2.2. Epigenetic Level
6.3. Adaptation Induced by One Compound Protects against Another
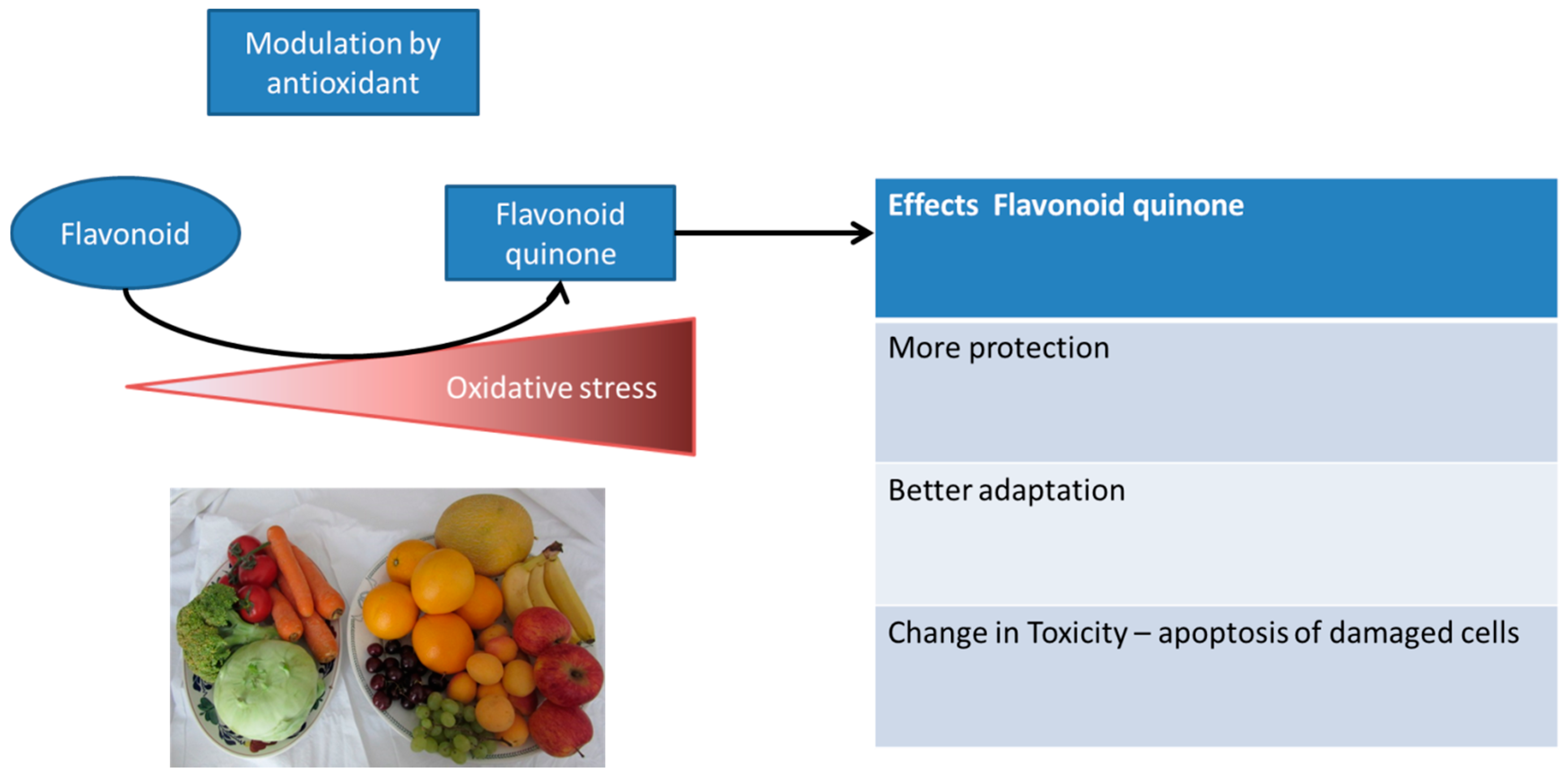
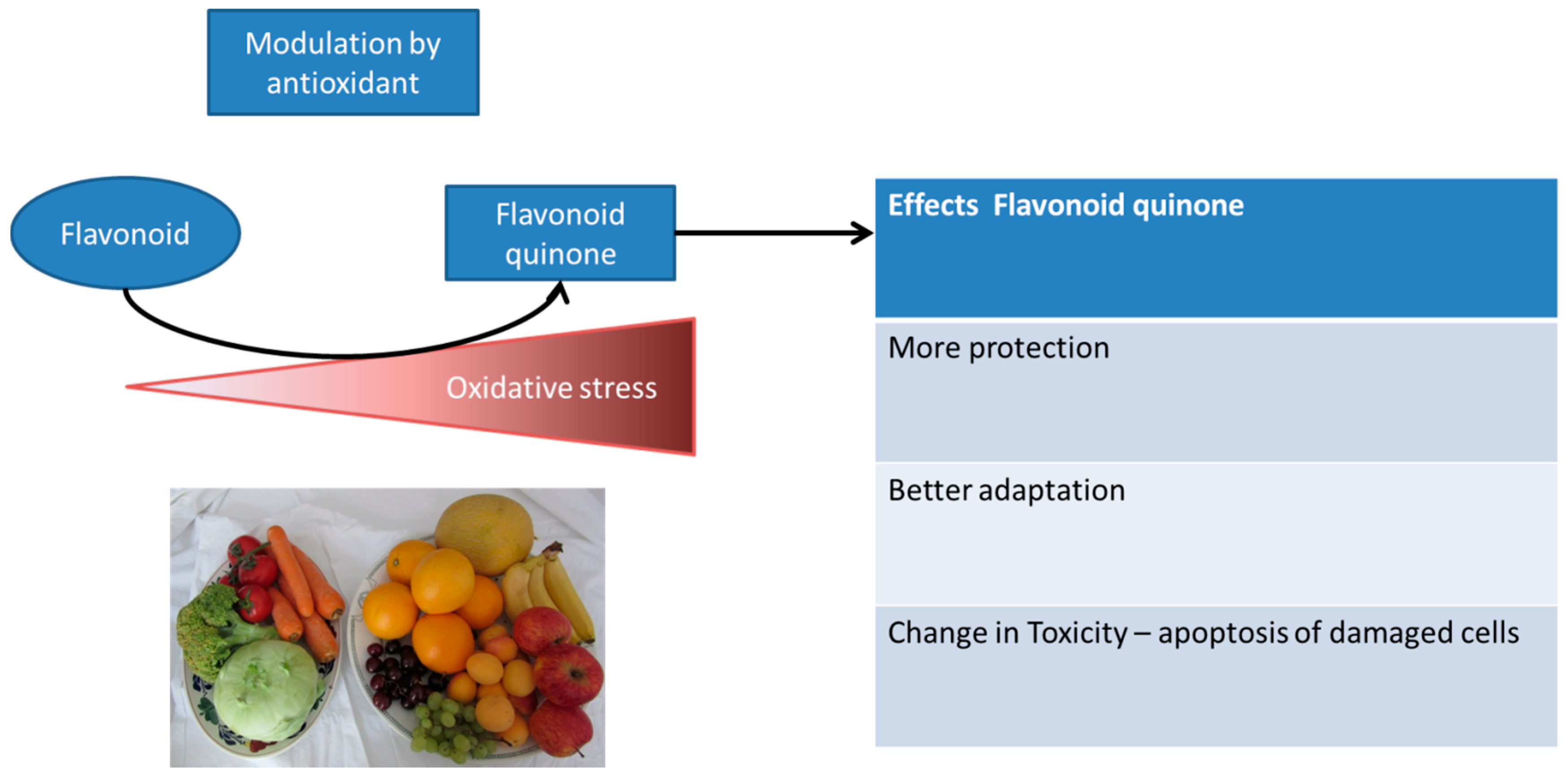
6.4. Adaptation Induced by Dietary Antioxidants
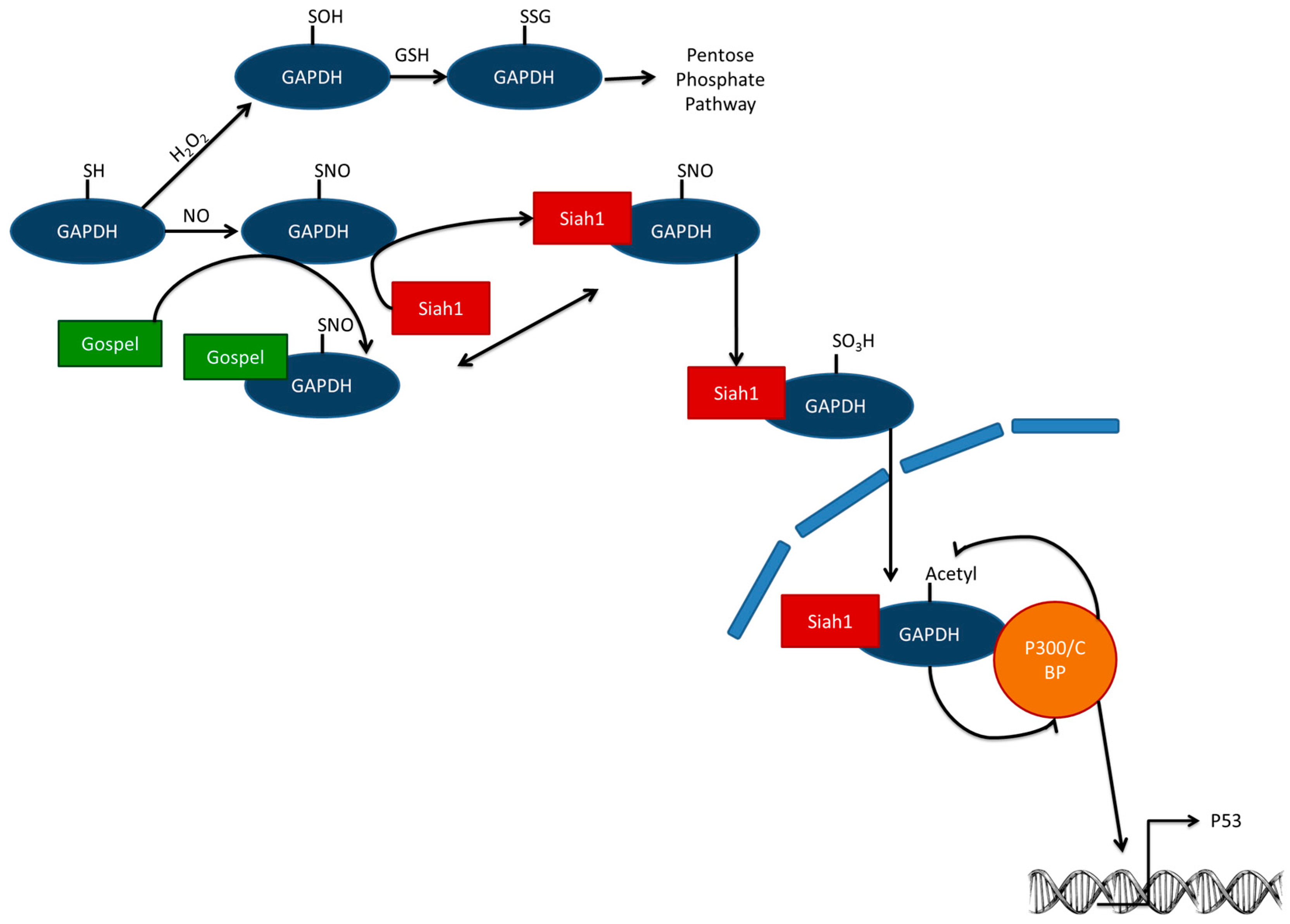
7. Effects of Hormesis—Remodeling of the GSH System
Timing and Hormesis
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lyons, T.W.; Reinhard, C.T.; Planavsky, N.J. The rise of oxygen in earth’s early ocean and atmosphere. Nature 2014, 506, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Oxidative stress: The paradox of aerobic life. Biochem. Soc. Symp. 1995, 61, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Bast, A.; Haenen, G.R.; Doelman, C.J. Oxidants and antioxidants: State of the art. Am. J. Med. 1991, 91, S2–S13. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Adaptive homeostasis. Mol. Asp. Med. 2016, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bernard, C.; Greene, H.C. An introduction to the Study of Experimental Medicine, 3rd ed.; Courier Corporation, Abelard Schuman Inc.: New York, NY, USA, 1957. [Google Scholar]
- Selye, H. Homeostasis and heterostasis. Perspect. Biol. Med. 1973, 16, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Southem, C.M.; Ehrlich, J. Effects of extract of western red-cedar heartwood on certain wood-decaying fungi in culture. Phytopathology 1943, 33, 517–524. [Google Scholar]
- Calabrese, E.J. Hormesis: A revolution in toxicology, risk assessment and medicine. EMBO Rep. 2004, 5, S37–S40. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Pulford, D.J. The glutathione s-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef] [PubMed]
- Haenen, G.R.; Jansen, F.P.; Vermeulen, N.P.; Bast, A. Activation of the microsomal glutathione S-transferase by metabolites of α-methyldopa. Arch. Biochem. Biophys. 1991, 287, 48–52. [Google Scholar] [CrossRef]
- Rahman, I.; MacNee, W. Lung glutathione and oxidative stress: Implications in cigarette smoke-induced airway disease. Am. J. Physiol. 1999, 277, L1067–L1088. [Google Scholar] [PubMed]
- Rahman, I.; Bel, A.; Mulier, B.; Donaldson, K.; MacNee, W. Differential regulation of glutathione by oxidants and dexamethasone in alveolar epithelial cells. Am. J. Physiol. 1998, 275, L80–L86. [Google Scholar] [PubMed]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [PubMed]
- Tian, L.; Shi, M.M.; Forman, H.J. Increased transcription of the regulatory subunit of γ-glutamylcysteine synthetase in rat lung epithelial L2 cells exposed to oxidative stress or glutathione depletion. Arch. Biochem. Biophys. 1997, 342, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Doka, E.; van Laer, K.; Nagy, P.; Grater, F.; Dick, T.P. A proton relay enhances H2O2 sensitivity of gapdh to facilitate metabolic adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of gapdh: Views from different subcellular compartments. Cell Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute activation of oxidative pentose phosphate pathway as first-line response to oxidative stress in human skin cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.Y.; Wang, W.; Schulz, R. Glutathione protects against myocardial ischemia-reperfusion injury by detoxifying peroxynitrite. J. Mol. Cell. Cardiol. 2000, 32, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Thengchaisri, N.; Hein, T.W.; Wang, W.; Xu, X.; Li, Z.; Fossum, T.W.; Kuo, L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.P.; Prasad, A.; Quyyumi, A.A. N-acetylcysteine improves coronary and peripheral vascular function. J. Am. Coll. Cardiol. 2001, 37, 117–123. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [PubMed]
- Touyz, R.M.; Briones, A.M. Reactive oxygen species and vascular biology: Implications in human hypertension. Hypertens. Res. 2011, 34, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015, 5, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.J.; Liu, M.Y.; Nabel, C.S.; Cao, X.J.; Garcia, B.A.; Kohli, R.M. Tet2 catalyzes stepwise 5-methylcytosine oxidation by an iterative and de novo mechanism. J. Am. Chem. Soc. 2016, 138, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimenez, J.L.; Ibanez-Cabellos, J.S.; Seco-Cervera, M.; Pallardo, F.V. Glutathione and cellular redox control in epigenetic regulation. Free Radic. Biol. Med. 2014, 75, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and structural basis for acyl-group selectivity and NAD+ dependence in sirtuin-catalyzed deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Zhong, Q.; Kowluru, R.A. Epigenetic modifications of Nrf2-mediated glutamate-cysteine ligase: Implications for the development of diabetic retinopathy and the metabolic memory phenomenon associated with its continued progression. Free Radic. Biol. Med. 2014, 75, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Molognoni, F.; Melo, F.H.; Galdieri, L.C.; Carneiro, C.R.; D’Almeida, V.; Correa, M.; Jasiulionis, M.G. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia 2007, 9, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Heredia, N.; Senut, M.C.; Land, S.; Hollocher, K.; Lu, X.; Dereski, M.O.; Ruden, D.M. Multigenerational epigenetic inheritance in humans: DNA methylation changes associated with maternal exposure to lead can be transmitted to the grandchildren. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Fahey, R.C.; Buschbacher, R.M.; Newton, G.L. The evolution of glutathione metabolism in phototrophic microorganisms. J. Mol. Evol. 1987, 25, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Pemble, S.E.; Taylor, J.B. An evolutionary perspective on glutathione transferases inferred from class-theta glutathione transferase cDNA sequences. Biochem. J. 1992, 287, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bast, A.; Haenen, G.R. Cytochrome P-450 and glutathione: What is the significance of their interrelationship in lipid peroxidation? Trends Biochem. Sci. 1984, 9, 510–513. [Google Scholar] [CrossRef]
- Johansson, K.; Cebula, M.; Rengby, O.; Dreij, K.; Carlstrom, K.E.; Sigmundsson, K.; Piehl, F.; Arner, E.S. Cross talk in HEK293 cells between Nrf2, HIF, and NF-κB activities upon challenges with redox therapeutics characterized with single-cell resolution. Antioxid. Redox Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Siritantikorn, A.; Johansson, K.; Ahlen, K.; Rinaldi, R.; Suthiphongchai, T.; Wilairat, P.; Morgenstern, R. Protection of cells from oxidative stress by microsomal glutathione transferase 1. Biochem. Biophys. Res. Commun. 2007, 355, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Jarvliden, J.; Gogvadze, V.; Morgenstern, R. Multiple roles of microsomal glutathione transferase 1 in cellular protection: A mechanistic study. Free Radic. Biol. Med. 2010, 49, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; Zhang, J.; Johansson, K. Microsomal glutathione transferase 1: Mechanism and functional roles. Drug Metab. Rev. 2011, 43, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Haenen, G.R.; Vermeulen, N.P.; Tai Tin Tsoi, J.N.; Ragetli, H.M.; Timmerman, H.; Blast, A. Activation of the microsomal glutathione-S-transferase and reduction of the glutathione dependent protection against lipid peroxidation by acrolein. Biochem. Pharmacol. 1988, 37, 1933–1938. [Google Scholar] [CrossRef]
- Sthijns, M.M.; Randall, M.J.; Bast, A.; Haenen, G.R. Adaptation to acrolein through upregulating the protection by glutathione in human bronchial epithelial cells: The materialization of the hormesis concept. Biochem. Biophys. Res. Commun. 2014, 446, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.J.; Tsai, T.; Peng, P.C.; Li, P.T.; Chen, C.T. Histone acetyltransferase p300 is induced by p38MAPK after photodynamic therapy: The therapeutic response is increased by the p300HAT inhibitor anacardic acid. Free Radic. Biol. Med. 2015, 86, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Pi, J.; Woods, C.G.; Andersen, M.E. Phase I to II cross-induction of xenobiotic metabolizing enzymes: A feedforward control mechanism for potential hormetic responses. Toxicol. Appl. Pharmacol. 2009, 237, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Semchyshyn, H.M. Hormetic concentrations of hydrogen peroxide but not ethanol induce cross-adaptation to different stresses in budding yeast. Int. J. Microbiol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, K.J.A.; Sthijns, M.M.J.P.E.; van der Vijgh, W.J.F.; Bast, A.; Haenen, G.R.M.M. The antioxidant flavonoid monoher provides efficient protection and induces the innate Nrf2 mediated adaptation in endothelial cells subjected to oxidative stress. PharmaNutrition 2014, 2, 69–74. [Google Scholar] [CrossRef]
- Boots, A.W.; Li, H.; Schins, R.P.; Duffin, R.; Heemskerk, J.W.; Bast, A.; Haenen, G.R. The quercetin paradox. Toxicol. Appl. Pharmacol. 2007, 222, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.; den Hartog, G.J.; Bast, A. Oxidative damage shifts from lipid peroxidation to thiol arylation by catechol-containing antioxidants. Biochim. Biophys. Acta 2002, 1583, 279–284. [Google Scholar] [CrossRef]
- Cheng, L.; Liu, J.; Li, B.; Liu, S.; Li, X.; Tu, H. Cigarette smoke-induced hypermethylation of the GCLC gene is associated with COPD. Chest 2016, 149, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Carlsohn, A.; Rohn, S.; Bittmann, F.; Raila, J.; Mayer, F.; Schweigert, F.J. Exercise increases the plasma antioxidant capacity of adolescent athletes. Ann. Nutr. Metab. 2008, 53, 96–103. [Google Scholar] [CrossRef] [PubMed]
- El Abed, K.; Rebai, H.; Bloomer, R.J.; Trabelsi, K.; Masmoudi, L.; Zbidi, A.; Sahnoun, Z.; Hakim, A.; Tabka, Z. Antioxidant status and oxidative stress at rest and in response to acute exercise in judokas and sedentary men. J. Strength Cond. Res. 2011, 25, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Falone, S.; Mirabilio, A.; Passerini, A.; Izzicupo, P.; Cacchio, M.; Gallina, S.; Baldassarre, A.D.; Amicarelli, F. Aerobic performance and antioxidant protection in runners. Int. J. Sports Med. 2009, 30, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kwak, Y.S. Impact of aerobic and anaerobic exercise training on oxidative stress and antioxidant defense in athletes. J. Exerc. Rehabil. 2016, 12, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Manley, A.F. Physical Activity and Health: A Report of the Surgeon General; U.S. Department of Health and Human Services: Atlanta, GA, USA, 1996.
- Falone, S.; D’Alessandro, A.; Mirabilio, A.; Petruccelli, G.; Cacchio, M.; di Ilio, C.; di Loreto, S.; Amicarelli, F. Long term running biphasically improves methylglyoxal-related metabolism, redox homeostasis and neurotrophic support within adult mouse brain cortex. PLoS ONE 2012, 7, e31401. [Google Scholar] [CrossRef] [PubMed]
- Falone, S.; D’Alessandro, A.; Mirabilio, A.; Cacchio, M.; di Ilio, C.; di Loreto, S.; Amicarelli, F. Late-onset running biphasically improves redox balance, energy- and methylglyoxal-related status, as well as sirt1 expression in mouse hippocampus. PLoS ONE 2012, 7, e48334. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Radak, Z.; Ji, L.L. Exercise-induced oxidative stress: Past, present and future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, M.; Mladenovic, D.; Ninkovic, M.; Vucevic, D.; Tomasevic, T.; Radosavljevic, T. Effects of caloric restriction on oxidative stress parameters. Gen. Physiol. Biophys. 2013, 32, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M. Radiation hormesis: Historical perspective and implications for low-dose cancer risk assessment. Dose Response 2010, 8, 172–191. [Google Scholar] [CrossRef] [PubMed]
- Feinendegen, L.E. Quantification of adaptive protection following low-dose irradiation. Health Phys. 2016, 110, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Suresh, I.S.R.; Éric, L.B. Hormesis in Health and Disease; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2014; pp. 107–152. [Google Scholar]
- Galvan, I.; Bonisoli-Alquati, A.; Jenkinson, S.; Ghanem, G.; Wakamatsu, K.; Mousseau, T.A.; Møller, A.P. Chronic exposure to low-dose radiation at chernobyl favours adaptation to oxidative stress in birds. Funct. Ecol. 2014, 28, 1387–1403. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short Term GSH-Related Adaptation to O2 | Target |
|---|---|
| Cofactor | GSH |
| Enzyme | γGCS |
| Regeneration | GR- > NAPDH |
| Exposure | Time | Frequency | Adaptation | Example | Effect | Fading Effect | Outcome | Change | |
|---|---|---|---|---|---|---|---|---|---|
| Single exposure of relative low dose | Direct/acute | Occasionally | Enzymatic activity | GSTs | Fast | Fast | Hormetic Response | Protein | Reversible |
| More cofactors | GSH synthesis GAPDH | ||||||||
| Often | More transcription | Nrf2 | Altered Phenotype | ||||||
| Remodeling | Slow | Not | |||||||
| Chronic exposure of relative high dose | Long | Continuously | Epigenetic level | Altered Genotype | |||||
| Genomic level | Gene | Irreversible | |||||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sthijns, M.M.J.P.E.; Weseler, A.R.; Bast, A.; Haenen, G.R.M.M. Time in Redox Adaptation Processes: From Evolution to Hormesis. Int. J. Mol. Sci. 2016, 17, 1649. https://doi.org/10.3390/ijms17101649
Sthijns MMJPE, Weseler AR, Bast A, Haenen GRMM. Time in Redox Adaptation Processes: From Evolution to Hormesis. International Journal of Molecular Sciences. 2016; 17(10):1649. https://doi.org/10.3390/ijms17101649
Chicago/Turabian StyleSthijns, Mireille M. J. P. E., Antje R. Weseler, Aalt Bast, and Guido R. M. M. Haenen. 2016. "Time in Redox Adaptation Processes: From Evolution to Hormesis" International Journal of Molecular Sciences 17, no. 10: 1649. https://doi.org/10.3390/ijms17101649
APA StyleSthijns, M. M. J. P. E., Weseler, A. R., Bast, A., & Haenen, G. R. M. M. (2016). Time in Redox Adaptation Processes: From Evolution to Hormesis. International Journal of Molecular Sciences, 17(10), 1649. https://doi.org/10.3390/ijms17101649