
Rational Protein Engineering Guided by Deep Mutational Scanning


Abstract
:1. Protein Engineering in the Ultrahigh-Throughput Sequencing (uHTS) Era
2. Deep Mutational Scanning
2.1. Overview
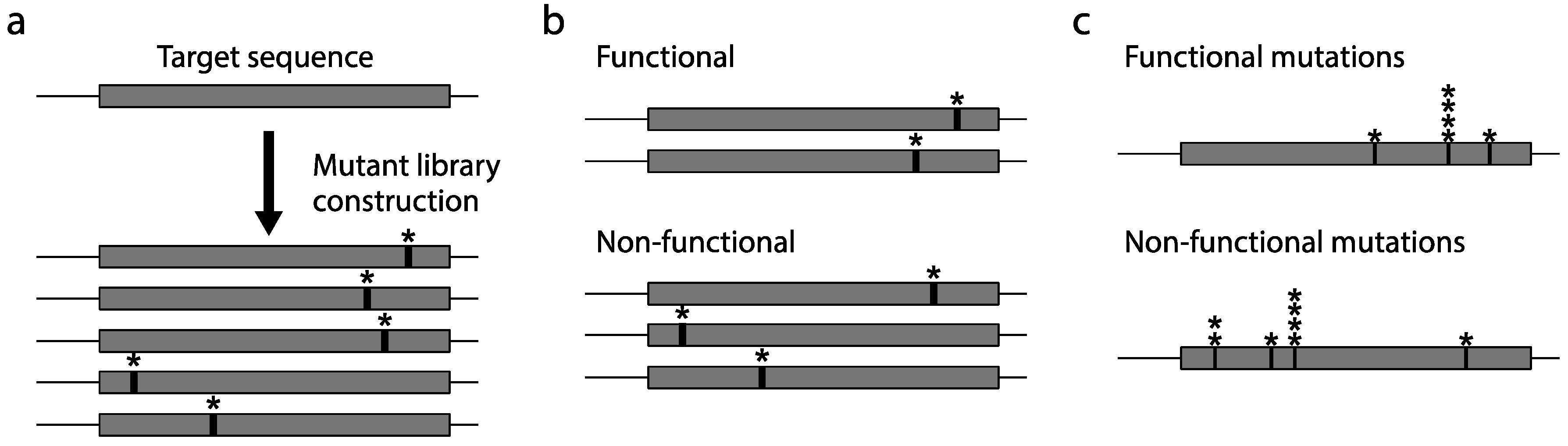
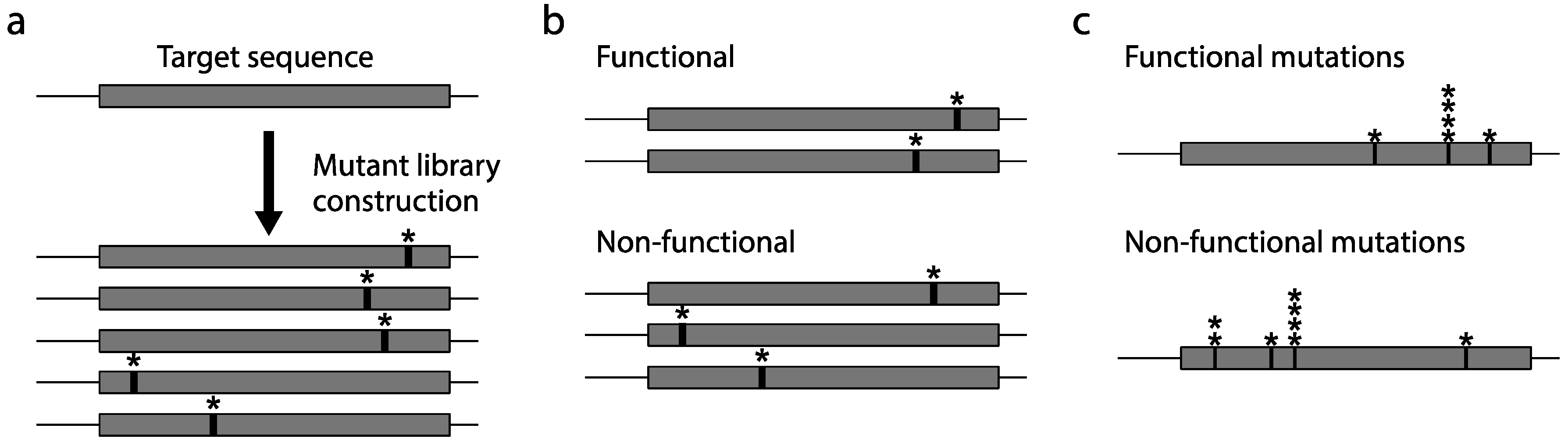
2.2. Mutagenesis
2.3. Construction of a Protein Variant Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Generation Method 1 | Variant Library | Sequencing Method 2 | Target Protein 3 | Reference |
|---|---|---|---|---|
| ORM | Phage display | Solexa/PE | PSD95pdz3 | [10] |
| ORM | Bacterial two-hybrid | Illumina/PE | hYAP65 | [22] |
| ORM | Yeast two-hybrid | Illumina/SE | BRCA1 | [27] |
| PRM | Plasmid | Illumina/SE | EcFbFP | [21] |
| SM | Yeast display | Illumina/PE | HB80.3 | [24] |
| ORM | Plasmid | Illumina/PE | APH(3′)II | [28] |
| SM | Plasmid | Illumina/PE | Bgl3 | [23] |
| SM | Plasmid | 454 | CcdB | [26] |
| ORM | Plasmid | Illumina/PE | Pab1 | [29] |
| ORM | Mammalian display vectors | 454 | IgG | [30] |
| ORM | Ribosome display | 454 | CDR loops of Fab | [47] |
| ORM | Phage display | Illumina/PE | hYAP65 | [48] |
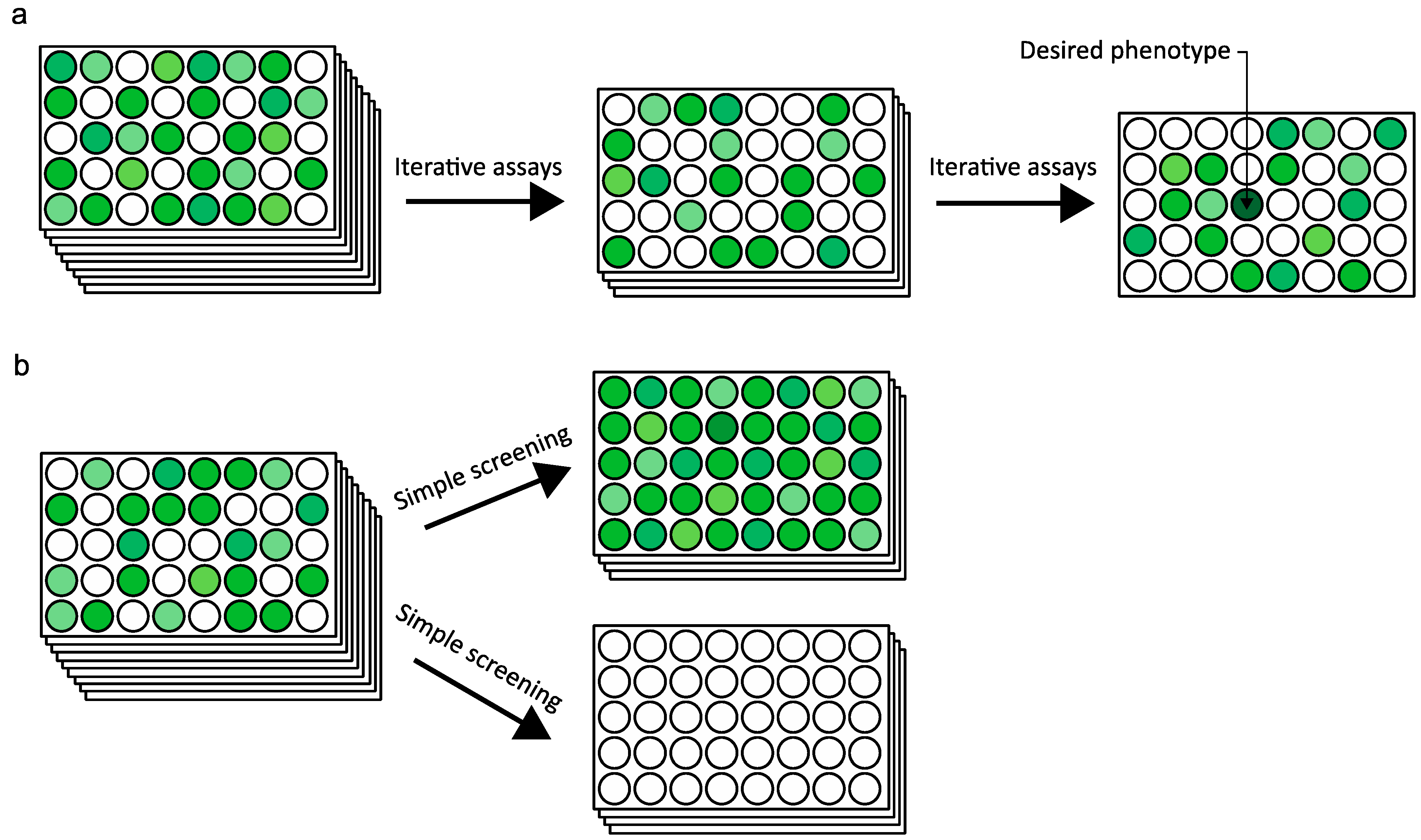
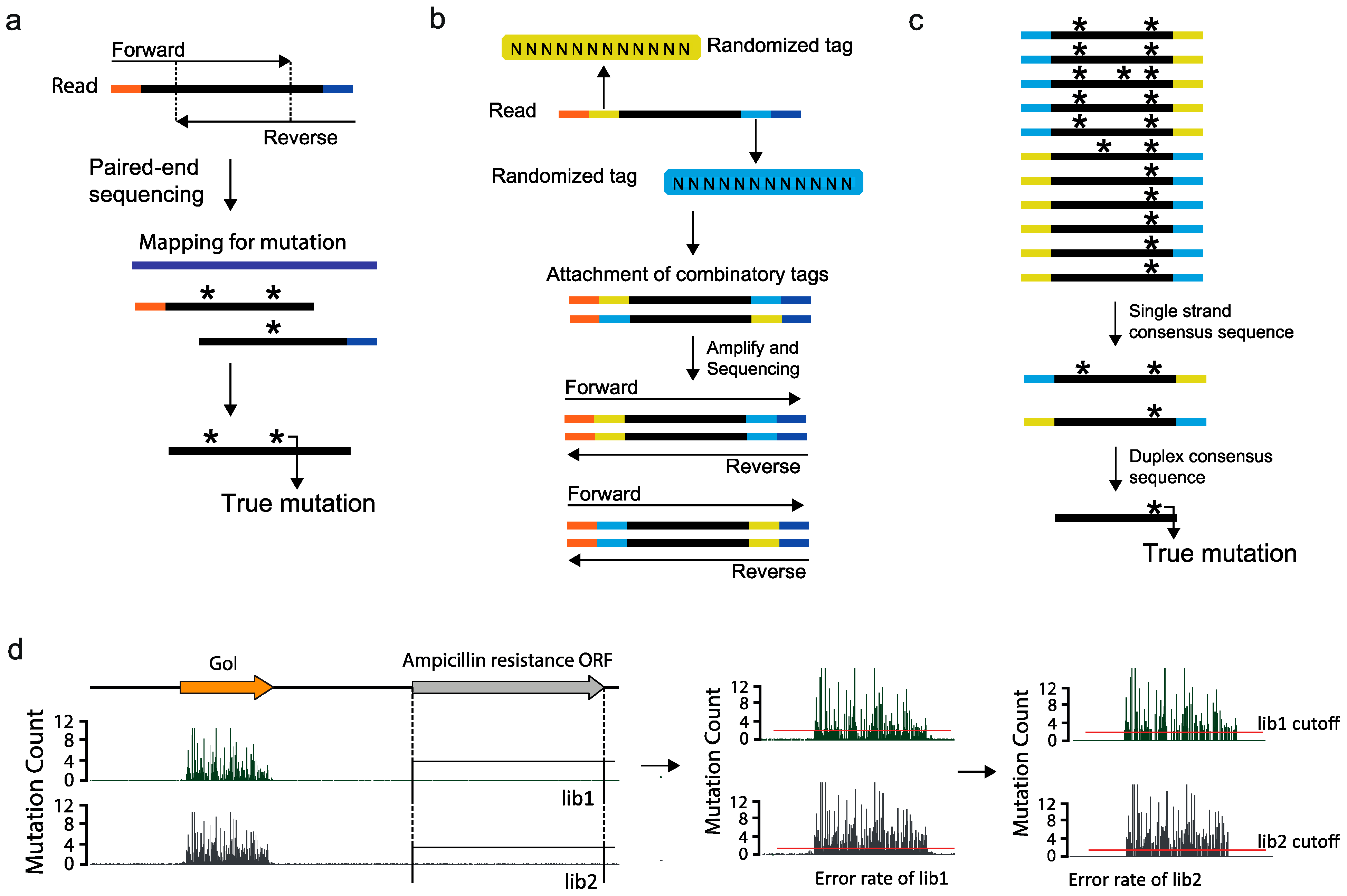
2.4. Ultra High-Throughput Sequencing (uHTS)
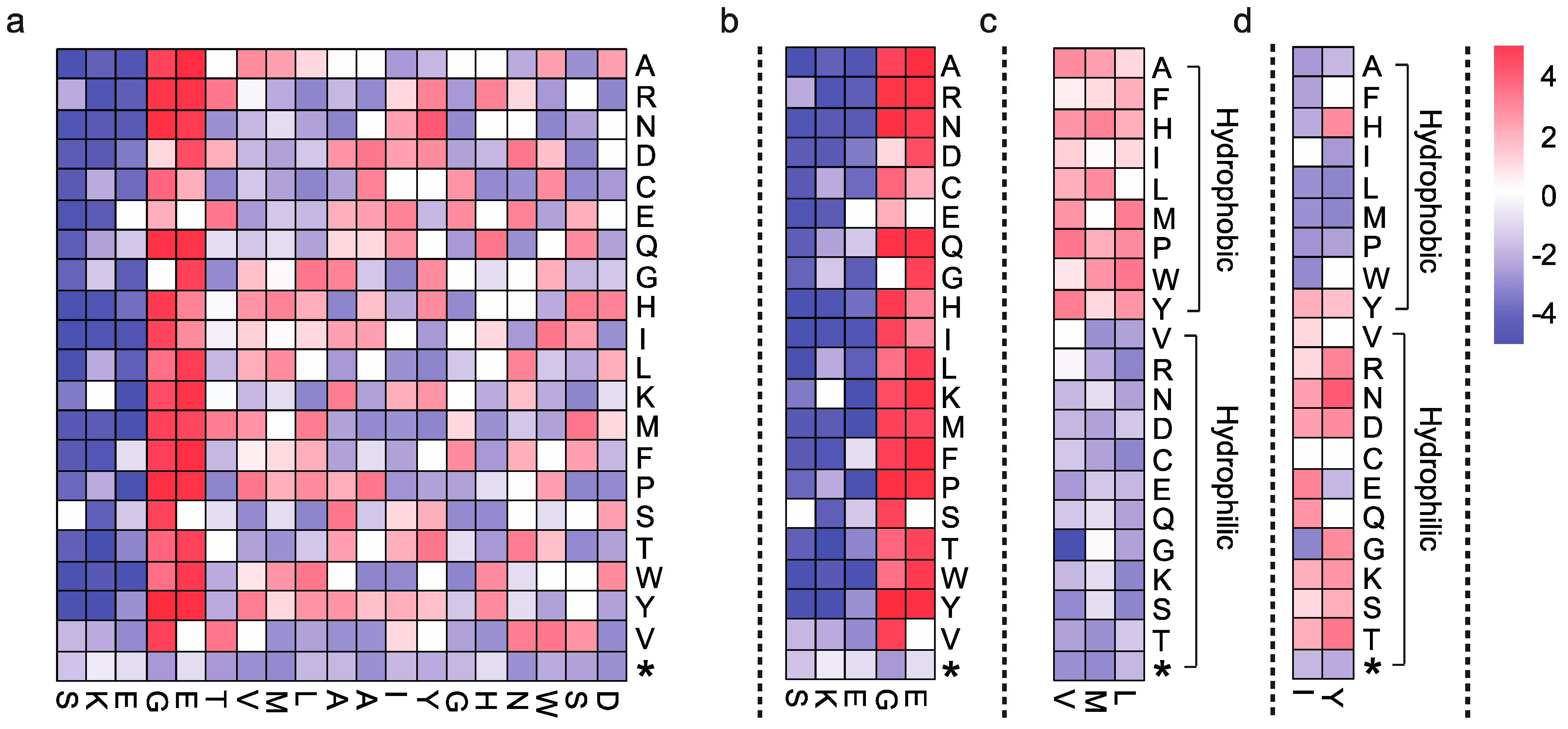
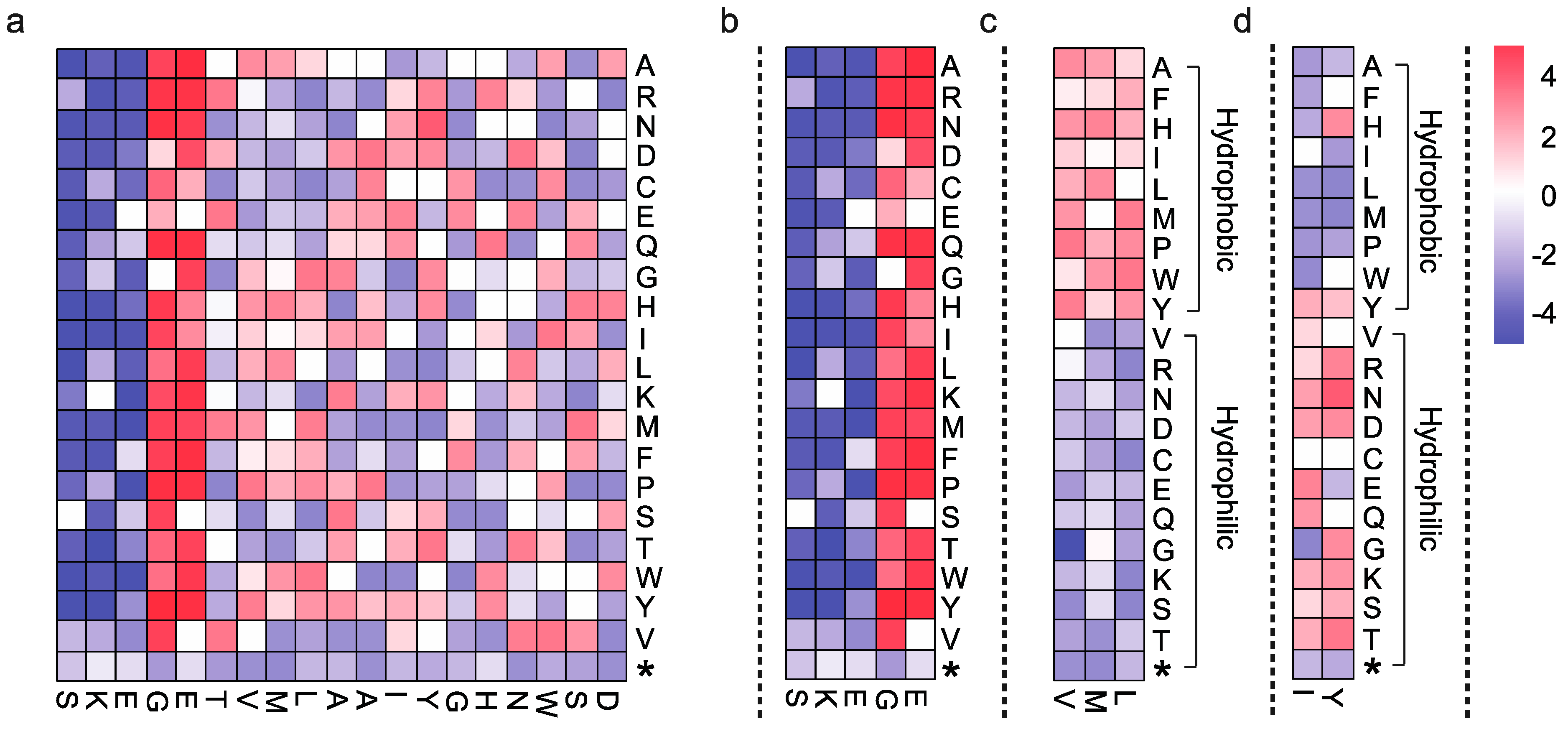
2.5. Data Interpretation
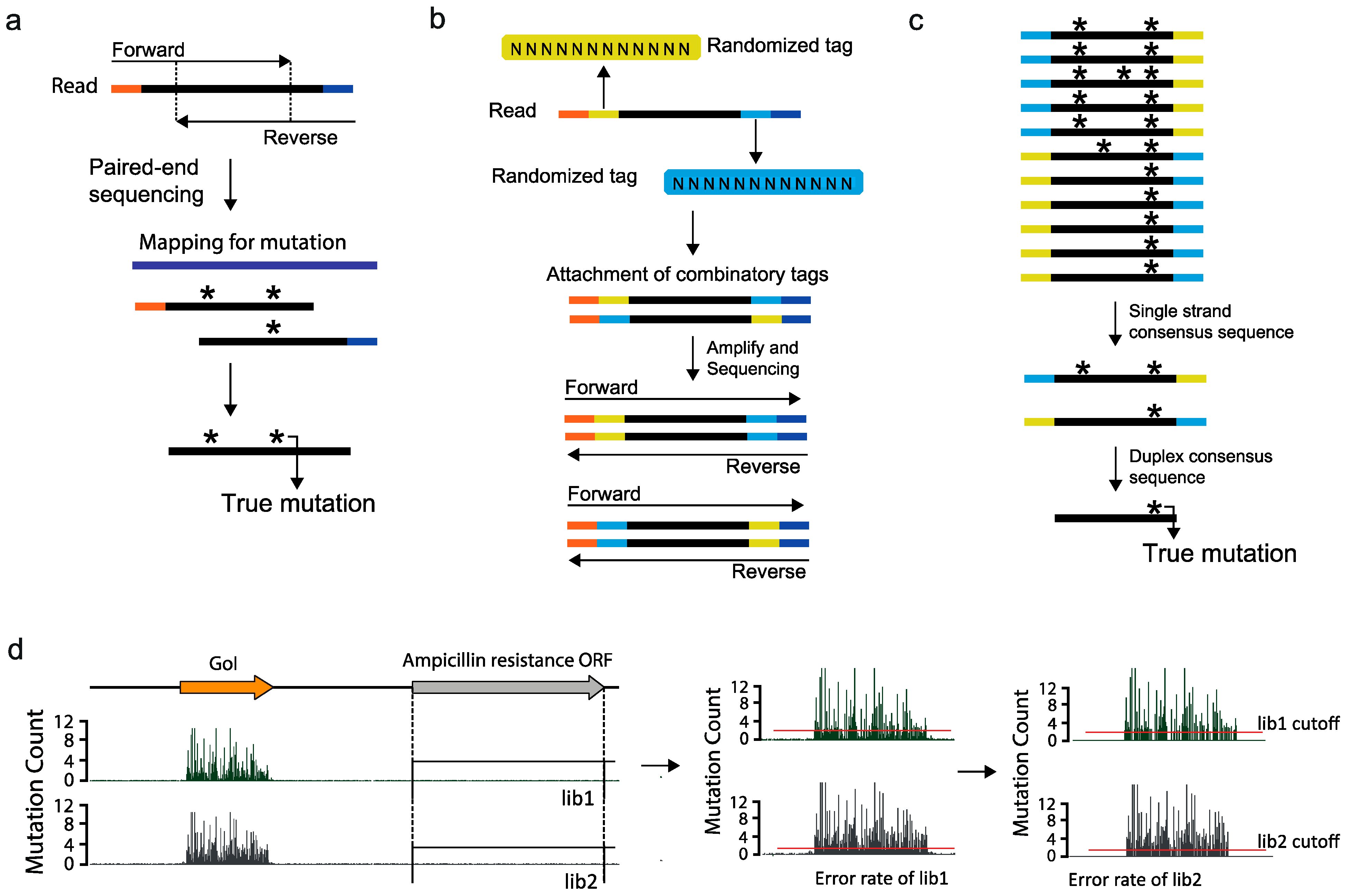
2.6. Limitations and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dalbadie-McFarland, G.; Cohen, L.W.; Riggs, A.D.; Morin, C.; Itakura, K.; Richards, J.H. Oligonucleotide-directed mutagenesis as a general and powerful method for studies of protein function. Proc. Natl. Acad. Sci. USA 1982, 79, 6409–6413. [Google Scholar] [CrossRef] [PubMed]
- Blundell, T.L. Problems and solutions in protein engineering—Towards rational design. Trends Biotechnol. 1994, 12, 145–148. [Google Scholar] [CrossRef]
- McManus, S.; Riechmann, L. Use of 2D NMR, protein engineering, and molecular modeling to study the hapten-binding site of an antibody Fv fragment against 2-phenyloxazolone. Biochemistry 1991, 30, 5851–5857. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T.; Toda, S.; Sugio, S.; Tomita, K.; Nishikawa, S.; Morioka, H.; Fuchimura, K.; Kimura, T.; Uesugi, S.; Ohtsuka, E.; et al. Conformational properties of the guanine-binding site of ribonuclease T1 inferred from the X-ray structure and protein engineering. Protein Eng. 1988, 2, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein structure prediction using rosetta. Methods Enzymol. 2004, 383, 66–93. [Google Scholar] [PubMed]
- Liu, Y.; Kuhlman, B. Rosettadesign server for protein design. Nucleic Acids Res. 2006, 34, W235–W238. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S. Beyond directed evolution—Semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Gombault, A.; Godin, F.; Sy, D.; Legrand, B.; Chautard, H.; Vallee, B.; Vovelle, F.; Benedetti, H. Molecular basis of the Tfs1/Ira2 interaction: A combined protein engineering and molecular modelling study. J. Mol. Biol. 2007, 374, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry 1971, 8, 871–874. [Google Scholar] [CrossRef]
- McLaughlin, R.N., Jr.; Poelwijk, F.J.; Raman, A.; Gosal, W.S.; Ranganathan, R. The spatial architecture of protein function and adaptation. Nature 2012, 491, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.L.; Fowler, D.M. Deep mutational scanning: Assessing protein function on a massive scale. Trends Biotechnol. 2011, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Natural selection and the concept of a protein space. Nature 1970, 225, 563–564. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.M.; Mole, B.M.; Silversmith, R.E.; Bourret, R.B. Action at a distance: Amino acid substitutions that affect binding of the phosphorylated chey response regulator and catalysis of dephosphorylation can be far from the chez phosphatase active site. J. Bacteriol. 2011, 193, 4709–4718. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef]
- O’Neil, K.T.; Hoess, R.H. Phage display: Protein engineering by directed evolution. Curr. Opin. Struct. Biol. 1995, 5, 443–449. [Google Scholar] [CrossRef]
- Fernandez-Gacio, A.; Uguen, M.; Fastrez, J. Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol. 2003, 21, 408–414. [Google Scholar] [CrossRef]
- Chen, Y.; Wiesmann, C.; Fuh, G.; Li, B.; Christinger, H.W.; McKay, P.; de Vos, A.M.; Lowman, H.B. Selection and analysis of an optimized anti-VEGF antibody: Crystal structure of an affinity-matured fab in complex with antigen. J. Mol. Biol. 1999, 293, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Hibbert, E.G.; Dalby, P.A. Directed evolution strategies for improved enzymatic performance. Microb. Cell Factories 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.R.; Jayaram, B. Proteins: Sequence to structure and function—Current status. Curr. Protein Pept. Sci. 2010, 11, 498–514. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.I.; Jones, D.T. The sequence-structure relationship and protein function prediction. Curr. Opin. Struct. Biol. 2009, 19, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Cho, Y.; Choe, D.H.; Jeong, Y.; Cho, S.; Kim, S.C.; Cho, B.K. Exploring the functional residues in a flavin-binding fluorescent protein using deep mutational scanning. PLoS ONE 2014, 9, e97817. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Araya, C.L.; Fleishman, S.J.; Kellogg, E.H.; Stephany, J.J.; Baker, D.; Fields, S. High-resolution mapping of protein sequence-function relationships. Nat. Methods 2010, 7, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, T.A.; Chevalier, A.; Song, Y.; Dreyfus, C.; Fleishman, S.J.; de Mattos, C.; Myers, C.A.; Kamisetty, H.; Blair, P.; Wilson, I.A.; et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012, 30, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Baumann, U.; Reymond, J.L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004, 32. [Google Scholar] [CrossRef] [PubMed]
- Adkar, B.V.; Tripathi, A.; Sahoo, A.; Bajaj, K.; Goswami, D.; Chakrabarti, P.; Swarnkar, M.K.; Gokhale, R.S.; Varadarajan, R. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure 2012, 20, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Starita, L.M.; Young, D.L.; Islam, M.; Kitzman, J.O.; Gullingsrud, J.; Hause, R.J.; Fowler, D.M.; Parvin, J.D.; Shendure, J.; Fields, S. Massively parallel functional analysis of brca1 ring domain variants. Genetics 2015, 200, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, A.; Rogov, P.; Wang, L.; Gnirke, A.; Mikkelsen, T.S. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Melamed, D.; Young, D.L.; Gamble, C.E.; Miller, C.R.; Fields, S. Deep mutational scanning of an RRM domain of the Saccharomyces cerevisiae poly(A)-binding protein. RNA 2013, 19, 1537–1551. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.M.; Juan, V.; Akamatsu, Y.; DuBridge, R.B.; Doan, M.; Ivanov, A.V.; Ma, Z.; Polakoff, D.; Razo, J.; Wilson, K.; et al. Deep mutational scanning of an antibody against epidermal growth factor receptor using mammalian cell display and massively parallel pyrosequencing. MAbs 2013, 5, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol. Biol. Evol. 2014, 31, 1956–1978. [Google Scholar] [CrossRef] [PubMed]
- Firnberg, E.; Labonte, J.W.; Gray, J.J.; Ostermeier, M. A comprehensive, high-resolution map of a gene’s fitness landscape. Mol. Biol. Evol. 2014, 31, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.A.; Wu, N.C.; Sun, R. A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain. Curr. Biol. 2014, 24, 2643–2651. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Olson, C.A.; Wu, N.C.; Ke, R.; Loverdo, C.; Chu, V.; Truong, S.; Remenyi, R.; Chen, Z.; Du, Y.; et al. A quantitative high-resolution genetic profile rapidly identifies sequence determinants of hepatitis C viral fitness and drug sensitivity. PLoS Pathog. 2014, 10, e1004064. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Bloom, J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Stiffler, M.A.; Hekstra, D.R.; Ranganathan, R. Evolvability as a function of purifying selection in TEM-1 β-lactamase. Cell 2015, 160, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Stephany, J.J.; Fields, S. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 2014, 9, 2267–2284. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawsawi, L.Q.; Wu, N.C.; Olson, C.A.; Shi, V.C.; Qi, H.; Zheng, X.; Wu, T.T.; Sun, R. High-throughput profiling of point mutations across the HIV-1 genome. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Young, A.P.; Al-Mawsawi, L.Q.; Olson, C.A.; Feng, J.; Qi, H.; Chen, S.H.; Lu, I.H.; Lin, C.Y.; Chin, R.G.; et al. High-throughput profiling of influenza a virus hemagglutinin gene at single-nucleotide resolution. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Young, A.P.; Al-Mawsawi, L.Q.; Olson, C.A.; Feng, J.; Qi, H.; Luan, H.H.; Li, X.; Wu, T.T.; Sun, R. High-throughput identification of loss-of-function mutations for anti-interferon activity in the influenza a virus ns segment. J. Virol. 2014, 88, 10157–10164. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Olson, C.A.; Du, Y.; Le, S.; Tran, K.; Remenyi, R.; Gong, D.; Al-Mawsawi, L.Q.; Qi, H.; Wu, T.T.; et al. Functional constraint profiling of a viral protein reveals discordance of evolutionary conservation and functionality. PLoS Genet. 2015, 11, e1005310. [Google Scholar] [CrossRef] [PubMed]
- McCullum, E.O.; Williams, B.A.; Zhang, J.; Chaput, J.C. Random mutagenesis by error-prone PCR. Methods Mol. Biol. 2010, 634, 103–109. [Google Scholar] [PubMed]
- Currin, A.; Swainston, N.; Day, P.J.; Kell, D.B. Synthetic biology for the directed evolution of protein biocatalysts: Navigating sequence space intelligently. Chem. Soc. Rev. 2015, 44, 1172–1239. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Temirov, J.; Pesavento, E.; Kiss, C.; Velappan, N.; Pavlik, P.; Werner, J.H.; Bradbury, A.R. Using T7 phage display to select GFP-based binders. Protein Eng. Des. Sel. 2008, 21, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Fujino, Y.; Fujita, R.; Wada, K.; Fujishige, K.; Kanamori, T.; Hunt, L.; Shimizu, Y.; Ueda, T. Robust in vitro affinity maturation strategy based on interface-focused high-throughput mutational scanning. Biochem. Biophys. Res. Commun. 2012, 428, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Araya, C.L.; Fowler, D.M.; Chen, W.; Muniez, I.; Kelly, J.W.; Fields, S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. USA 2012, 109, 16858–16863. [Google Scholar] [CrossRef] [PubMed]
- Pellis, M.; Muyldermans, S.; Vincke, C. Bacterial two hybrid: A versatile one-step intracellular selection method. Methods Mol. Biol. 2012, 911, 135–150. [Google Scholar] [PubMed]
- Evangelista, C.; Lockshon, D.; Fields, S. The yeast two-hybrid system: Prospects for protein linkage maps. Trends Cell Biol. 1996, 6, 196–199. [Google Scholar] [CrossRef]
- Monti-Bragadin, C.; Babudri, N.; Samer, L. Expression of the plasmid pKM101—Determined DNA repair system in recA- and lex- strains of Escherichia coli. Mol. Gen. Genet. 1976, 145, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ren, L.; Meng, Q.; Li, Y.; Yu, Y.; Yu, J. The next-generation sequencing technology and application. Protein Cell 2010, 1, 520–536. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihara, K. Clinical application of next-generation sequencing technologies to achieve cancer precision medicine. Gan To Kagaku Ryoho Cancer Chemother. 2014, 41, 1–6. [Google Scholar]
- Shin, H.; Hong, S.J.; Kim, H.; Yoo, C.; Lee, H.; Choi, H.K.; Lee, C.G.; Cho, B.K. Elucidation of the growth delimitation of Dunaliella tertiolecta under nitrogen stress by integrating transcriptome and peptidome analysis. Bioresour. Technol. 2015, 194, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Kozarewa, I.; Smith, F.; Scally, A.; Stephens, P.J.; Durbin, R.; Swerdlow, H.; Turner, D.J. A large genome center’s improvements to the illumina sequencing system. Nat. Methods 2008, 5, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Gout, J.F.; Thomas, W.K.; Smith, Z.; Okamoto, K.; Lynch, M. Large-scale detection of in vivo transcription errors. Proc. Natl. Acad. Sci. USA 2013, 110, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.C.; Risques, R.A.; et al. Detecting ultralow-frequency mutations by duplex sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Skrzypczak, M.; Ginalski, K.; Rowicka, M. Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using illumina platform. PLoS ONE 2015, 10, e0120520. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Morrison, H.G.; Benjamino, J.; Grim, S.L.; Graf, J. Analysis, optimization and verification of illumina-generated 16S rRNA gene amplicon surveys. PLoS ONE 2014, 9, e94249. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Araya, C.L.; Gerard, W.; Fields, S. Enrich: Software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 2011, 27, 3430–3431. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D. Software for the analysis and visualization of deep mutational scanning data. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, S.; Kuramitsu, S. Thermodynamics and molecular simulation analysis of hydrophobic substrate recognition by aminotransferases. J. Biol. Chem. 1998, 273, 18353–18364. [Google Scholar] [CrossRef] [PubMed]
- Bank, C.; Hietpas, R.T.; Jensen, J.D.; Bolon, D.N. A systematic survey of an intragenic epistatic landscape. Mol. Biol. Evol. 2015, 32, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.B.; Patwardhan, R.P.; Turner, E.H.; Lee, C.; Shendure, J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat. Methods 2010, 7, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.Z.; Hong, S.; Wong, H.T.; Aw, P.P.; Cheng, Y.; Wilm, A.; de Sessions, P.F.; Lim, S.G.; Nagarajan, N.; Hibberd, M.L.; et al. BAsE-Seq: A method for obtaining long viral haplotypes from short sequence reads. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Wu, N.C.; de La Cruz, J.; Al-Mawsawi, L.Q.; Olson, C.A.; Qi, H.; Luan, H.H.; Nguyen, N.; Du, Y.; Le, S.; Wu, T.T.; et al. HIV-1 quasispecies delineation by tag linkage deep sequencing. PLoS ONE 2014, 9, e97505. [Google Scholar] [CrossRef] [PubMed]
- Borgstrom, E.; Redin, D.; Lundin, S.; Berglund, E.; Andersson, A.F.; Ahmadian, A. Phasing of single DNA molecules by massively parallel barcoding. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.; Cho, B.-K. Rational Protein Engineering Guided by Deep Mutational Scanning. Int. J. Mol. Sci. 2015, 16, 23094-23110. https://doi.org/10.3390/ijms160923094
Shin H, Cho B-K. Rational Protein Engineering Guided by Deep Mutational Scanning. International Journal of Molecular Sciences. 2015; 16(9):23094-23110. https://doi.org/10.3390/ijms160923094
Chicago/Turabian StyleShin, HyeonSeok, and Byung-Kwan Cho. 2015. "Rational Protein Engineering Guided by Deep Mutational Scanning" International Journal of Molecular Sciences 16, no. 9: 23094-23110. https://doi.org/10.3390/ijms160923094
APA StyleShin, H., & Cho, B.-K. (2015). Rational Protein Engineering Guided by Deep Mutational Scanning. International Journal of Molecular Sciences, 16(9), 23094-23110. https://doi.org/10.3390/ijms160923094