
Identification of Ramie Genes in Response to Pratylenchus coffeae Infection Challenge by Digital Gene Expression Analysis




Abstract
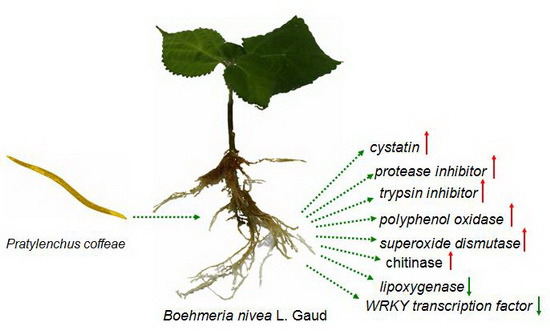
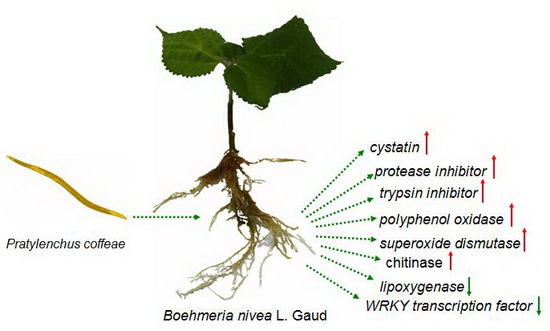
:
1. Introduction
2. Results
2.1. De Novo Transcriptome Assembly

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragments Length | Total Number | ||
|---|---|---|---|
| Contigs | Transcripts | Unigenes | |
| 0–300 | 1,908,616 | 15,757 | 12,877 |
| 300–500 | 12,017 | 14,229 | 9821 |
| 500–1000 | 7806 | 15,142 | 6865 |
| 1000–2000 | 7608 | 18,196 | 7302 |
| 2000+ | 4359 | 10,207 | 3961 |
| Total number | 1,940,406 | 73,531 | 40,826 |
| Total length | 140,000,159 | 76,345,971 | 33,878,532 |
| N50 | 99 | 1665 | 1491 |
| Mean length | 72 | 1038 | 830 |
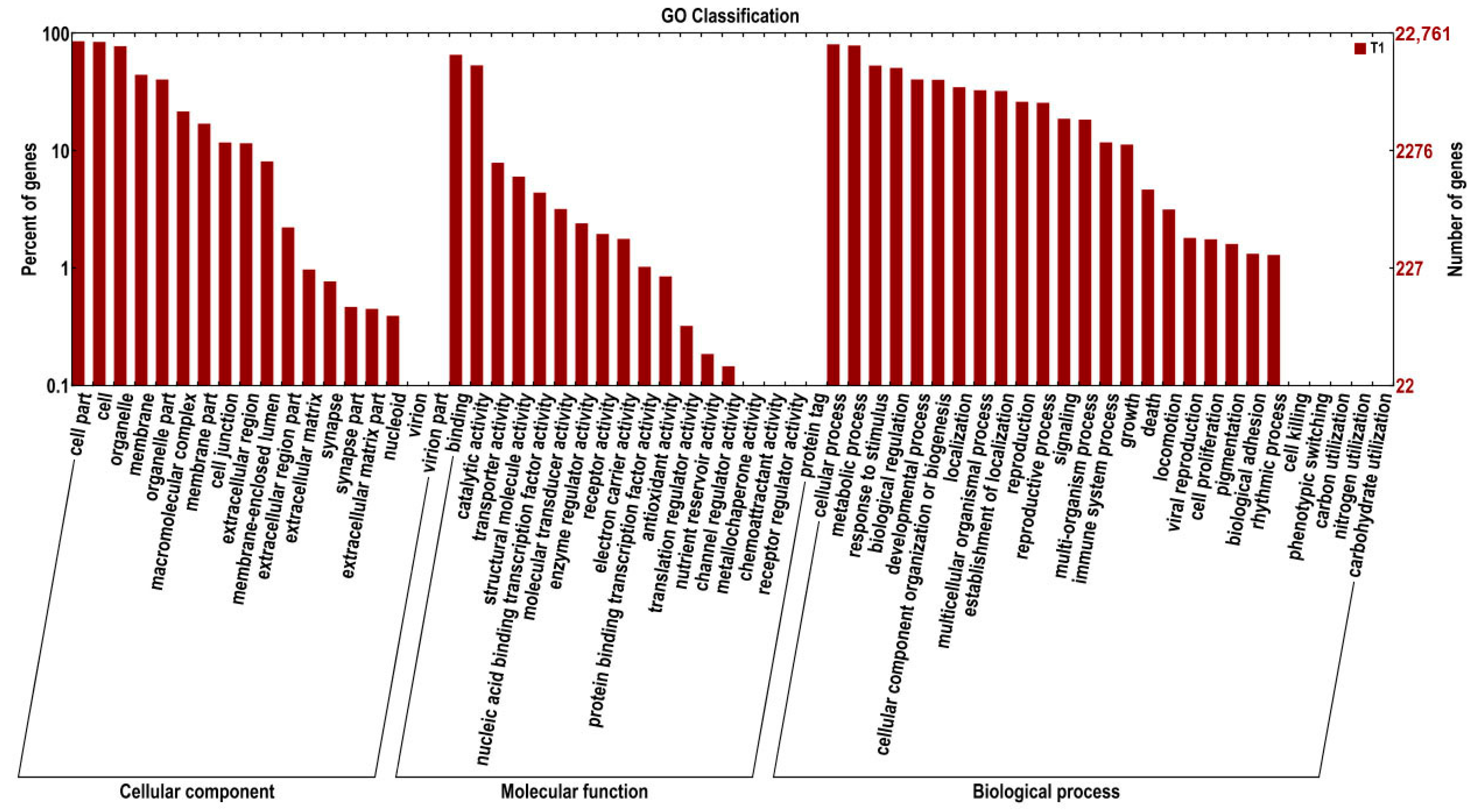
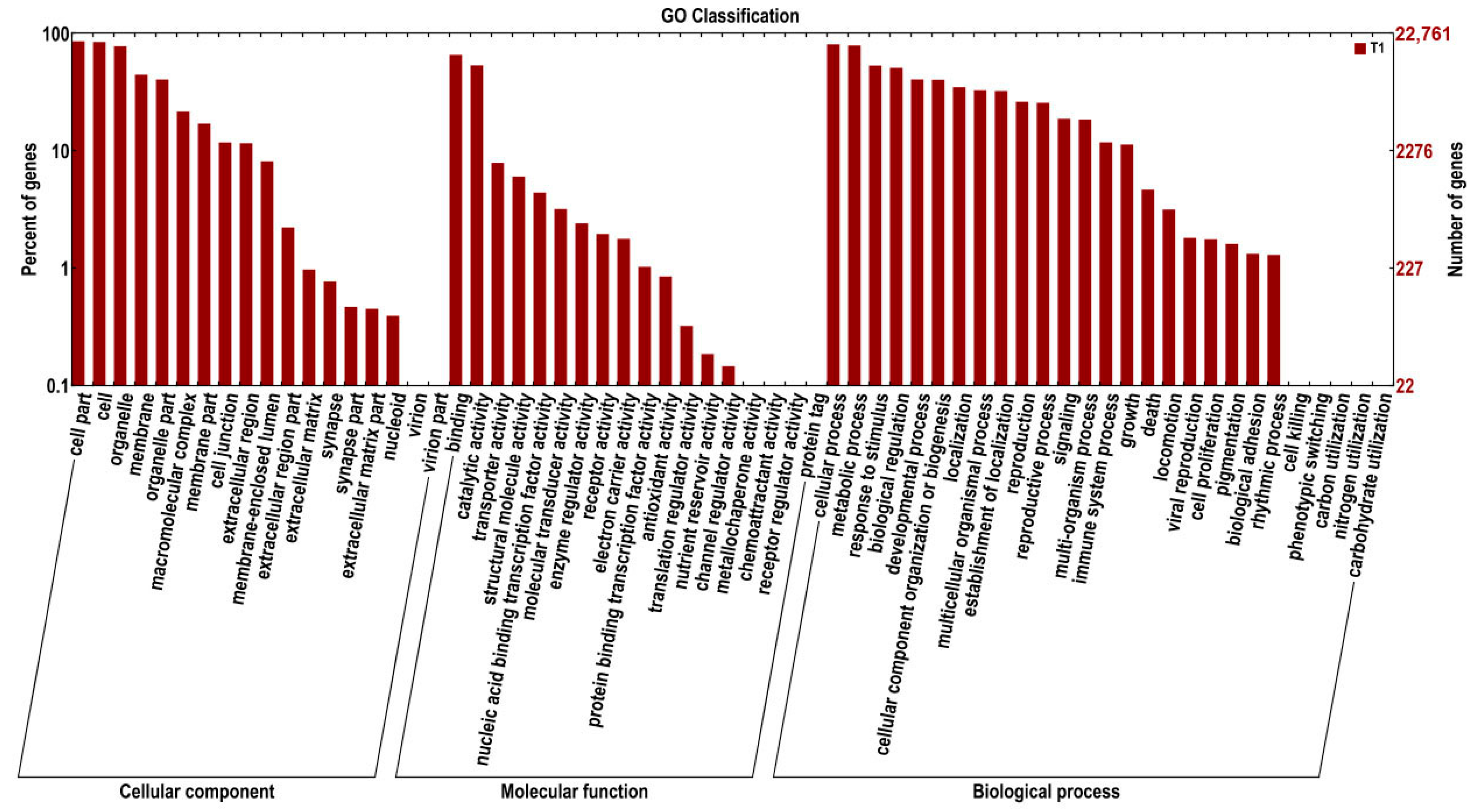
2.2. Functional Annotation and Classification
| Annotated Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| Nr | 26,685 | 10,427 | 11,092 |
| SwissProt | 18,851 | 7019 | 8632 |
| COG | 9657 | 2991 | 5132 |
| KEGG | 6243 | 2092 | 2843 |
| Total annotated | 26,851 | 10,510 | 11,105 |
2.3. Digital Gene Expression (DGE) Library Sequencing and Mapping to the Reference Transcriptome
2.4. Identification of Differentially Expressed Genes (DEGs)
| Gene ID | CH | CK | log2(CH/CK) | Regulated | Gene Annotation |
|---|---|---|---|---|---|
| Protease Inhibitor | |||||
| Unigene11292 | 271 | 134 | 1.02 | Up | Cysteine protease inhibitor |
| Unigene2589 | 41 | 7 | 2.55 | Up | Protease inhibitor |
| Unigene9323 | 38 | 3 | 3.66 | Up | Protease inhibitor |
| Unigene19135 | 1589 | 504 | 1.66 | Up | Trypsin inhibitor |
| Unigene13433 | 468 | 217 | 1.11 | Up | Endogenous α-amylase/subtilisin inhibitor |
| Unigene16750 | 73 | 22 | 1.73 | Up | Xyloglucanase inhibitor 3 |
| Heat Shock Protein | |||||
| Unigene2183 | 25 | 2 | 3.64 | Up | 17.9 kDa class II heat shock protein |
| Unigene11009 | 61 | 14 | 2.12 | Up | 23.6 kDa heat shock protein |
| Unigene3206 | 29 | 3 | 3.27 | Up | Heat shock protein 70 |
| Unigene3204 | 23 | 2 | 3.52 | Up | Heat shock70 kDa protein |
| Transcription Factor | |||||
| Unigene17588 | 88 | 26 | 1.76 | Up | Ethylene-responsive transcription factor |
| Unigene9042 | 148 | 12 | 3.62 | Up | Ethylene-responsive transcription factor |
| Unigene9043 | 131 | 10 | 3.71 | Up | Ethylene-responsive transcription factor |
| Unigene12273 | 116 | 258 | −1.15 | Down | WRKY transcription factor |
| Antioxidant Enzyme | |||||
| Unigene13732 | 48 | 7 | 2.78 | Up | Polyphenol oxidase |
| Unigene7762 | 359 | 168 | 1.10 | Up | Superoxide dismutase |
| Cell Wall Reinforcement | |||||
| Unigene24997 | 19 | 1 | 4.25 | Up | Arabinogalactan protein 23 |
| Unigene24306 | 36 | 3 | 3.58 | Up | Cell wall-associated hydrolase |
| Unigene9085 | 258 | 104 | 1.31 | Up | Proline-rich cell wall protein |
| Unigene5928 | 24 | 2 | 3.58 | Up | Cytochrome P450 |
| Unigene14594 | −60 | 127 | −1.08 | Down | Cellulose synthase-like protein |
| Pathogenesis-Related Protein | |||||
| Unigene24094 | 4572 | 1738 | 1.40 | Up | Chitinase |
| Unigene10467 | 157 | 50 | 1.65 | Up | Endochitinase |
| Unigene13343 | −53 | 111 | −1.07 | Down | Non-specific lipid-transfer protein-like protein |
| Others | |||||
| Unigene47 | 21 | 1 | 4.39 | Up | E3 ubiquitin protein ligase |
| Unigene20652 | 81 | 29 | 1.48 | Up | E3 ubiquitin-protein ligase |
| Unigene15546 | 84 | 37 | 1.18 | Up | F-box/kelch-repeat protein |
| Unigene23894 | 240 | 109 | 1.14 | Up | Nitrate reductase |
| Unigene23899 | 90 | 42 | 1.10 | Up | Nitrate reductase |
| Unigene8357 | 1008 | 369 | 1.45 | Up | Non-symbiotic hemoglobin |
| Unigene10901 | 253 | 60 | 2.08 | Up | Universal stress protein A-like protein |
| Unigene22463 | −55 | 131 | −1.25 | Down | Lipoxygenase |
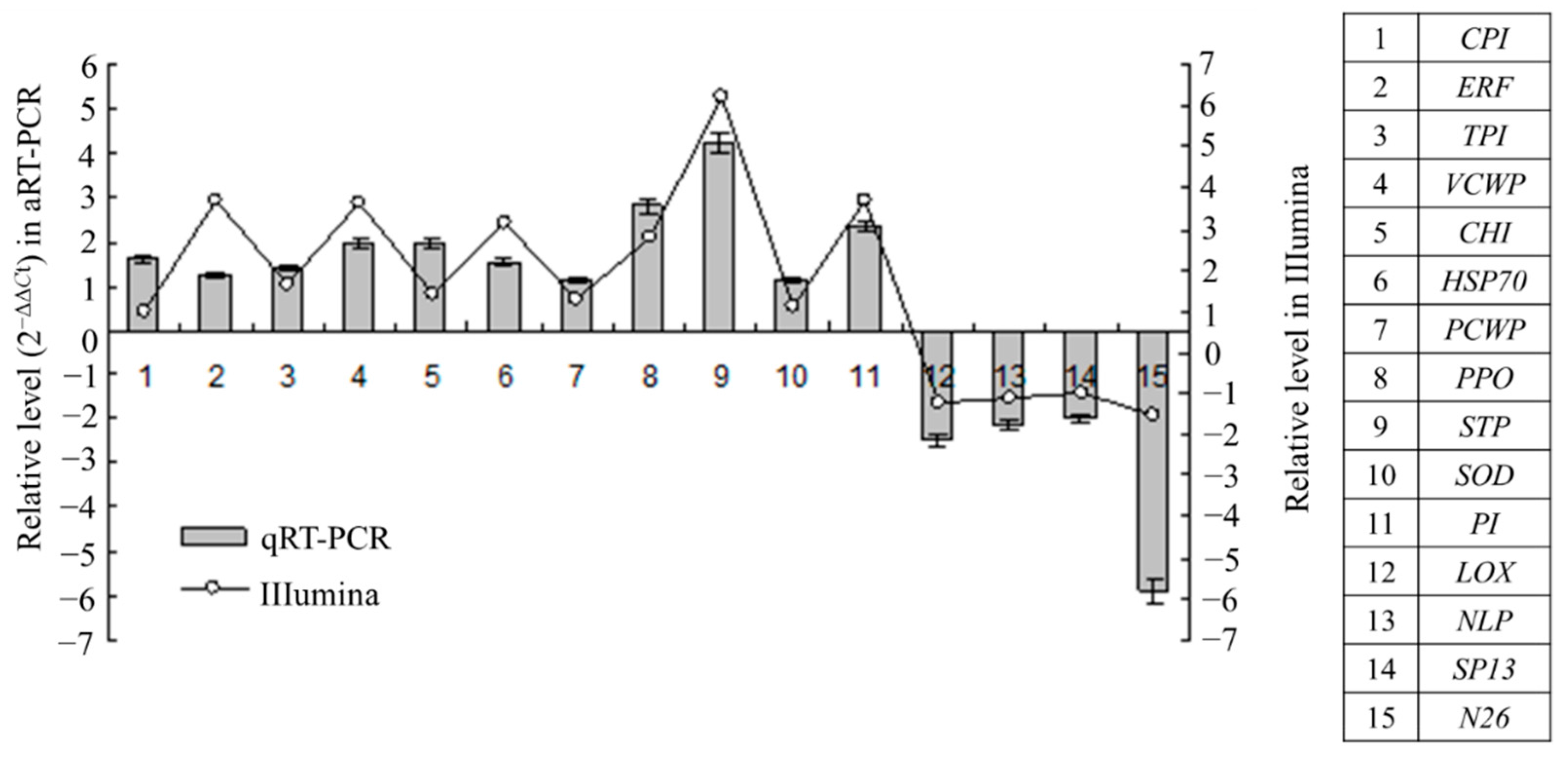
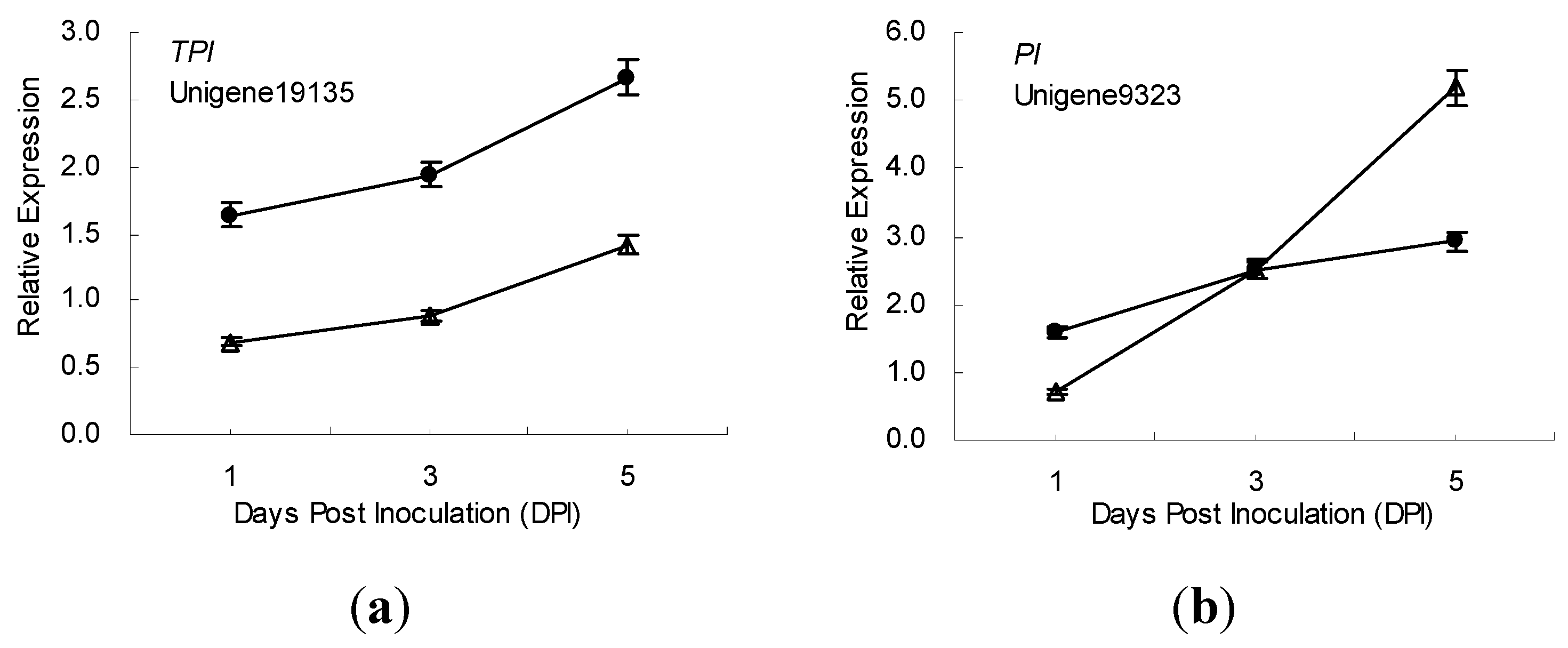
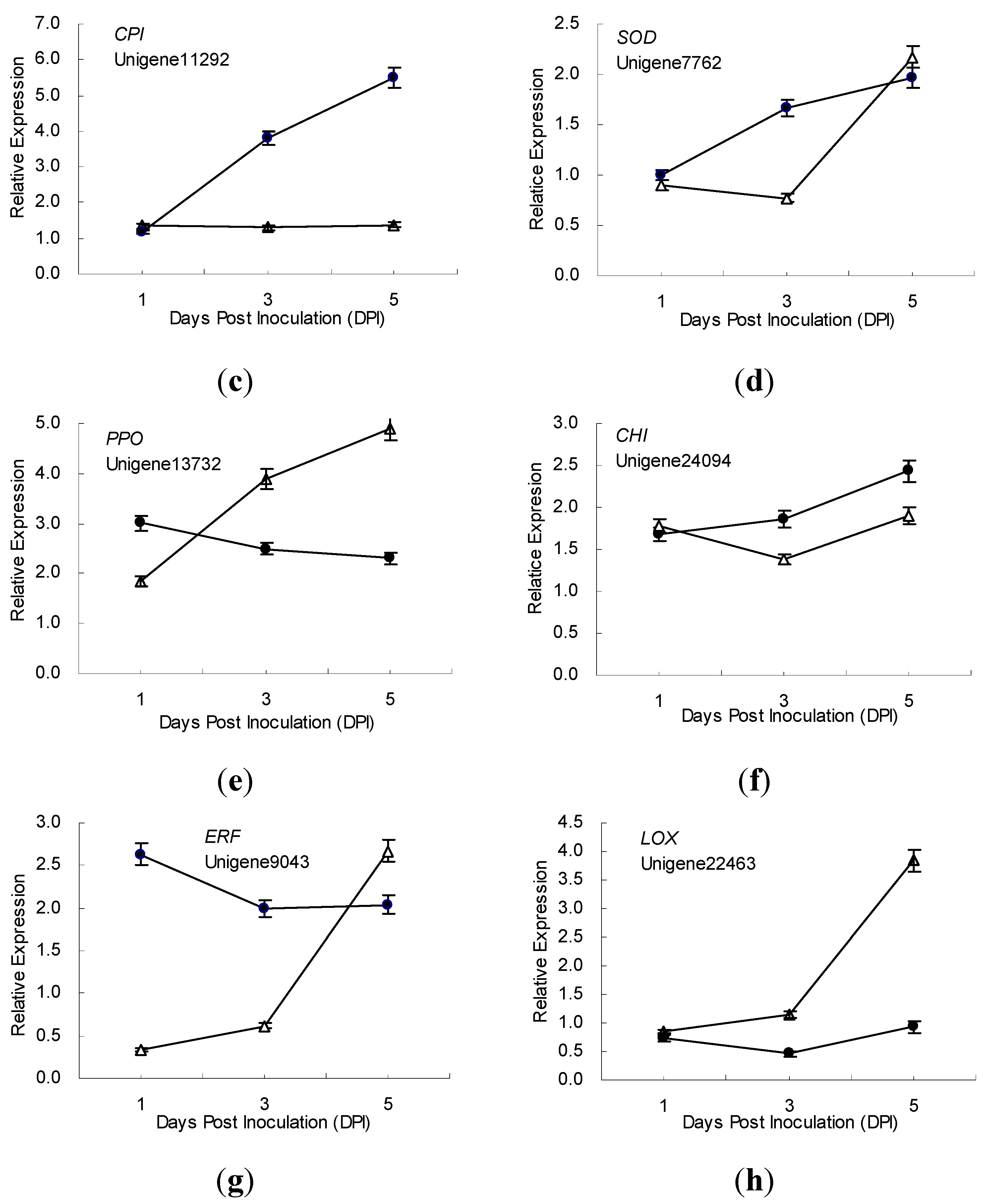
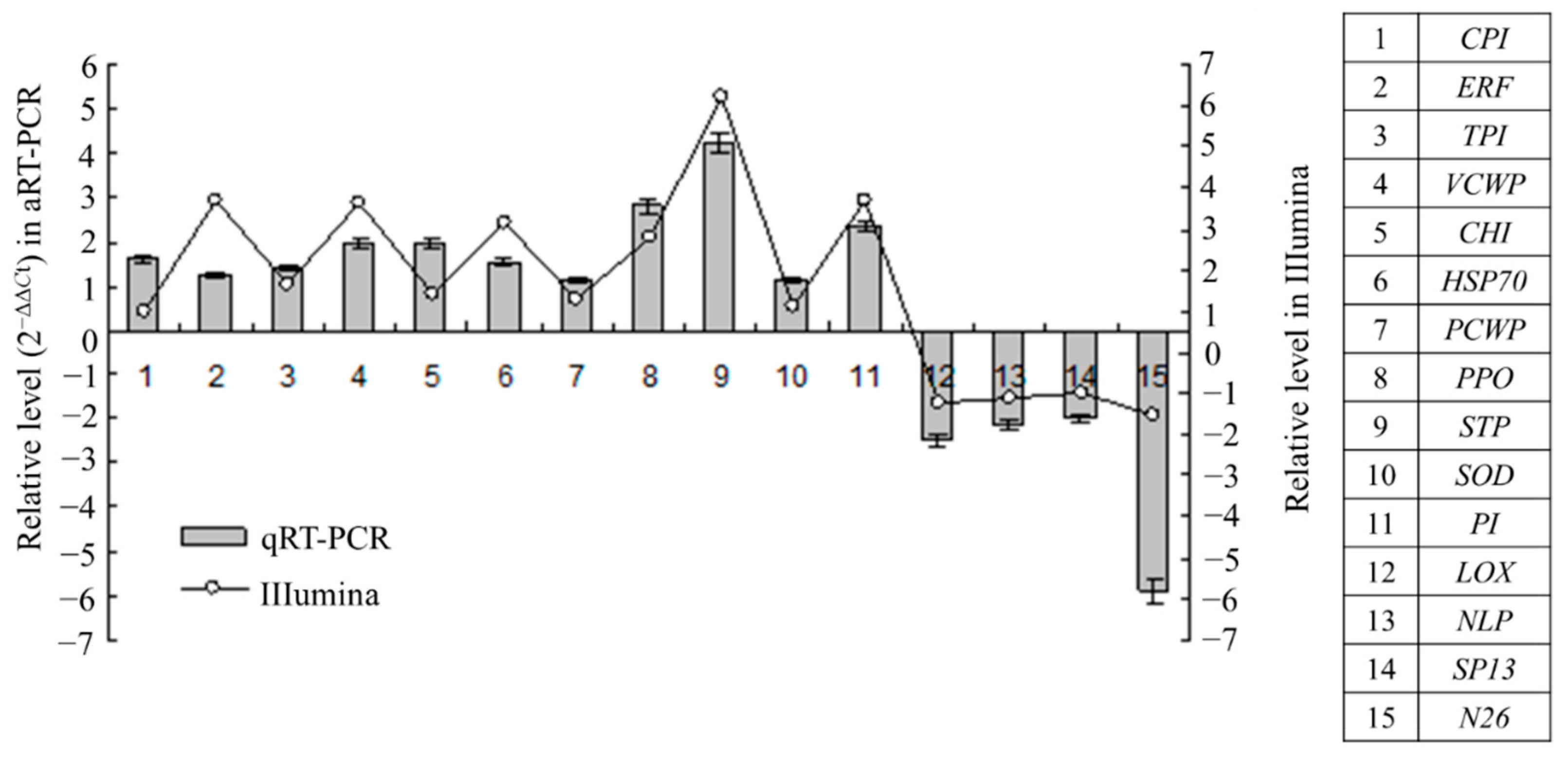
2.5. Gene Expression Patterns Analysis Using qRT-PCR
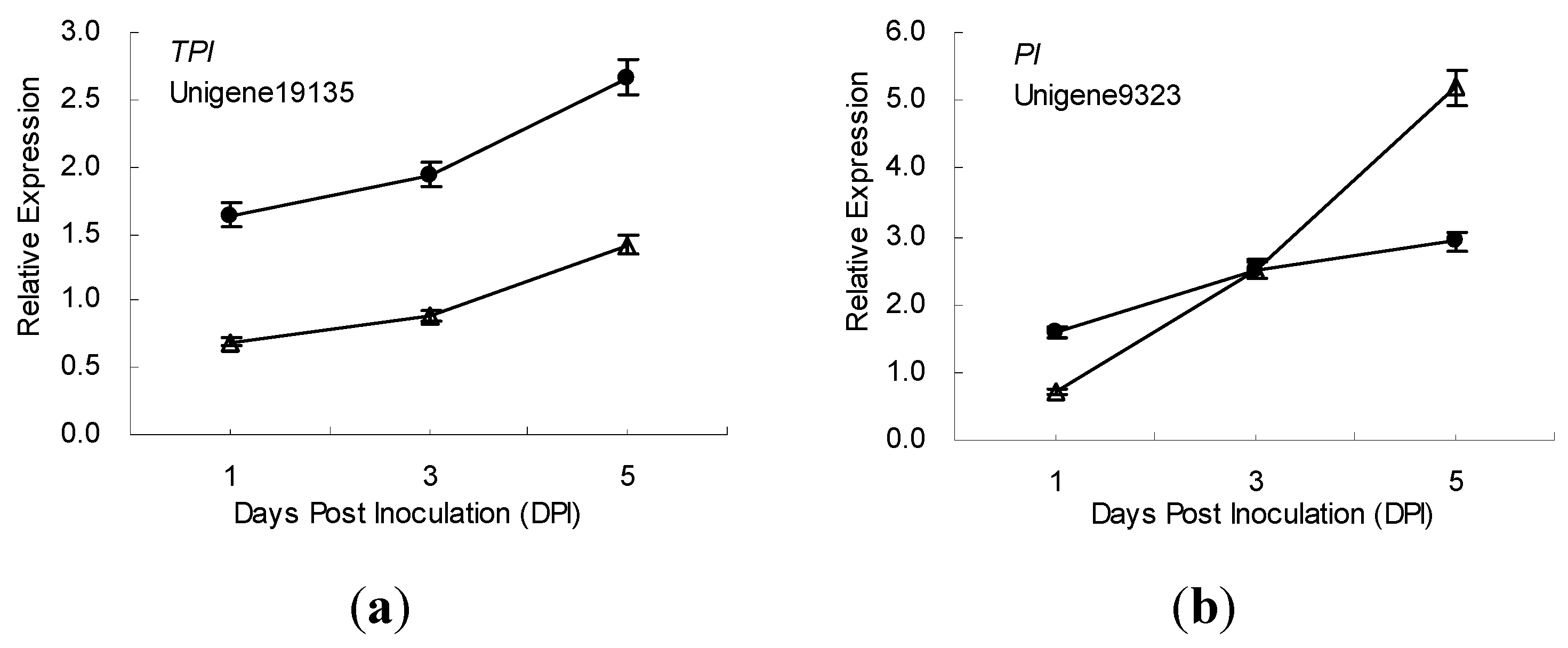
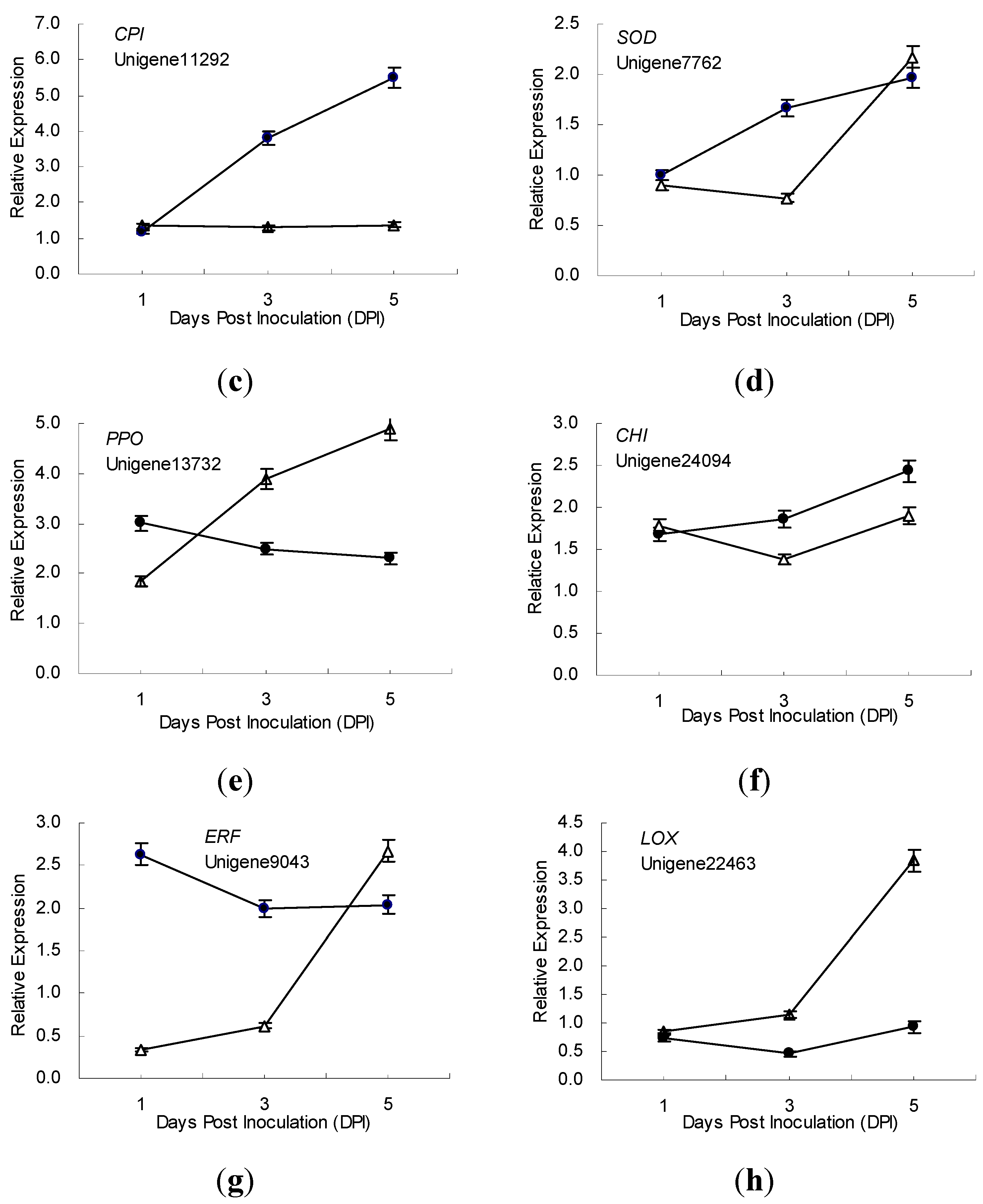
3. Discussion
4. Experimental Section
4.1. Plants and Nematode Inoculation
4.2. RNA Preparation
4.3. Transcriptome Library Preparation and Sequencing
4.4. De Novo Assembly and Functional Annotation
4.5. Digital Gene Expression (DGE) Library Preparation and Sequencing
4.6. Identification of Differentially Expressed Genes (DEGs)
4.7. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, S.; Xiao, Y.; Zhao, J.; Wang, F.; Zheng, Y. Digital gene expression analysis of early root infection resistance to Sporisorium. reilianum f. sp. zeae in maize. Mol. Genet. Genom. 2013, 288, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Zhu, S.Y.; Tang, Q.M.; Chen, P.; Yu, Y.T.; Tang, S.W. De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud). BMC Genom. 2013, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Kravchik, M.; Bernstein, N. Effects of salinity on the transcriptome of growing maize leaf cells point at cell-age specificity in the involvement of the antioxidative response in cell growth restriction. BMC Genom. 2013, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.F.; Fang, H.M.; Tian, Q.N.; Bao, Z.X.; Lu, P.; Zhao, J.M.; Mai, J.; Zhu, Z.Y.; Shu, L.L.; Zhao, L.; et al. Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica. Genomics 2011, 97, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.N.; Chen, S.C. Ramie—An effective plant for soil and water conservation in the southern slopled farmlands. Glob. Seabuckthorn Res. Dev. 2007, 5, 45–48. (In Chinese) [Google Scholar]
- Yu, Y.T.; Liu, H.L.; Zhu, A.G.; Zhang, G.; Zeng, L.B.; Xue, Z.D. A review of root lesion nematode: Identification and plant resistance. Adv. Microbiol. 2012, 2, 411–416. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Tang, S.W.; Tang, Q.M.; Liu, T.M. Genome-wide transcriptional changes of ramie (Boehmeria. nivea L. Gaud) in response to root-lesion nematode infection. Gene 2014, 552, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Taylor, P.; Bogacki, P.; Pallotta, M.; Bariana, S.; Wallwork, H. Mapping of the root lesion nematode (Pratylenchus neglectus) resistance gene Rlnn1 in wheat. Theor. Appl. Genet. 2002, 104, 874–879. [Google Scholar] [PubMed]
- Nagy, E.D.; Chu, Y.; Guo, Y.F.; Khanal, S.; Tang, S.X.; Li, Y.; Dong, W.B.; Timper, P.; Taylor, C.; Ozias-Akins, P.; et al. Recombination is suppressed in an alien introgression in peanut harboring Rma, a dominant root-knot nematode resistance gene. Mol. Breed. 2010, 26, 357–370. [Google Scholar] [CrossRef]
- Montes, M.J.; Andres, M.F.; Sin, E.; Lopez-Brana, I.; Martin-Sanchez, J.A.; Romero, M.D.; Delibes, A. Cereal cyst nematode resistance conferred by the Cre7 gene from Aegilops triuncialis and its relationship with Cre genes from Australian wheat cultivars. Genome 2008, 51, 315–319. [Google Scholar] [PubMed]
- Fuller, V.L.; Lilley, C.J.; Urwin, P.E. Nematode resistance. New Phytol. 2008, 180, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, H.; Zhang, L.; Zhang, J.; Xiao, J.; Ye, Z. CaMi, a root-knot nematode resistance gene from hot pepper (Capsium annuum L.) confers nematode resistance in tomato. Plant Cell. Rep. 2007, 26, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.S.; Wang, W.H.; Wang, L.X.; Ma, F.M.; Wang, P.W.; Chang, R.Z.; Qiu, L.J. Genetic diversity of soybean and the establishment of a core collection focused on resistance to soybean cyst nematode. J. Integr. Plant Biol. 2006, 48, 722–731. [Google Scholar] [CrossRef]
- Bird, D.M.; Wilson, M.A. DNA sequence and expression analysis of root-knot nematode-elicited giant cell transcripts. Mol. Plant Microbe Interact. 1994, 7, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Hermsmeier, D.; Mazarei, M.; Baum, T.J. Differential display analysis of the early compatible interaction between soybean and the soybean cyst nematode. Mol. Plant Microbe Interact. 1998, 11, 1258–1263. [Google Scholar] [CrossRef]
- Schaff, J.E.; Nielsen, D.M.; Smith, C.P.; Scholl, E.H.; Bird, D.M. Comprehensive transcriptome profiling in tomato reveals a role for glycosyltransferase in Mi-mediated nematode resistance. Plant Physiol. 2007, 144, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; Joseph, S.; Gheysen, G. Analysis of the transcriptome of the root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology. Mol. Biochem. Parasitol. 2011, 178, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Nicol, P.; Gill, R.; Fosu-Nyarko, J.; Jones, M.G. De novo analysis and functional classification of the transcriptome of the root lesion nematode, Pratylenchus thornei, after 454 GS FLX sequencing. Int. J. Parasitol. 2012, 42, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Backiyarani, S.; Uma, S.; Arunkumar, G.; Saraswathi, M.S.; Sundararaju, P. Differentially expressed genes in incompatible interactions of Pratylenchus coffeae with Musa using suppression subtractive hybridization. Physiol. Mol. Plant Pathol. 2014, 86, 11–18. [Google Scholar] [CrossRef]
- Castillo, P.; Vovlas, N.; Jiménez-Díaz, R.M. Pathogenicity and histopathology of Pratylenchus thornei populations on selected chickpea genotypes. Plant Pathol. 1998, 47, 370–376. [Google Scholar] [CrossRef]
- Acosta, N.; Malek, R.B. Symptomatology and histopathology of soybean roots infected by Pratylenchus scribneri and P. alleni. J. Nematol. 1981, 13, 6–12. [Google Scholar] [PubMed]
- Radewald, J.D.; O'Bannon, J.H.; Tomerlin, A.T. Anatomical studies of Citrus jambhiri Lush. roots infected by Pratylenchus coffeae. J. Nematol. 1971, 3, 409–416. [Google Scholar] [PubMed]
- Urwin, P.E.; Atkinson, H.J.; Waller, D.A.; McPherson, M.J. Engineered oryzacystatin-I expressed in transgenic hairy roots confers resistance to Globodera pallida. Plant J. 1995, 8, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Urwin, P.E.; Levesley, A.; McPherson, M.J.; Atkinson, H.J. Transgenic resistance to the nematode Rotylenchulus reniformis conferred by Arabidopsis thaliana plants expressing proteinase inhibitors. Mol. Breed. 2000, 6, 257–264. [Google Scholar] [CrossRef]
- Urwin, P.E.; Lilley, C.J.; McPherson, M.J.; Atkinson, H.J. Resistance to both cyst and root-knot nematodes conferred by transgenic Arabidopsis expressing a modified plant cystatin. Plant J. 1997, 12, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Samac, D.A.; Smigocki, A.C. Expression of oryzacystatin I and II in alfalfa increases resistance to the root-lesion nematode. Phytopathology 2003, 93, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.J.; Grimwood, S.; Johnston, K.; Green, J. Prototype demonstration of transgenic resistance to the nematode Radopholus similis conferred on banana by a cystatin. Transgenic Res. 2004, 13, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yu, B.; Zhai, H.; He, S.Z.; Liu, Q.C. Enhanced stem nematode resistance of transgenic sweetpotato plants expressing oryzacystatin-I gene. Agric. Sci. China 2011, 10, 519–525. [Google Scholar] [CrossRef]
- Vieira, P.; Wantoch, S.; Lilley, C.J.; Chitwood, D.J.; Atkinson, H.J.; Kamo, K. Expression of a cystatin transgene can confer resistance to root lesion nematodes in Lilium. longiflorum cv. “Nellie White”. Transgenic Res. 2015, 24, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.; Tripathi, L.; Babirye, A.; Wang, D.; Tripathi, J.; Urwin, P.E.; Atkinson, H.J. Generation of transgenic plantain (Musa spp.) with resistance to plant pathogenic nematodes. Mol. Plant Pathol. 2012, 13, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Yang, A.H.; Chen, J.T.; Yeh, K.W.; Chan, M.T. Heterologous expression of taro cystatin protects transgenic tomato against Meloidogyne incognita infection by means of interfering sex determination and suppressing gall formation. Plant Cell. Rep. 2010, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Gaur, V.S.; Taj, G.; Kumar, A. Differential induction of two different cystatin genes during pathogenesis of Karnal bunt (Tilletia indica) in wheat under the influence of jasmonic acid. Gene 2012, 506, 253–560. [Google Scholar] [CrossRef] [PubMed]
- Farmer, E.E.; Ryan, C.A. Octadecanoid precursors of jasmonic acid activate the synthesis of wound-inducible proteinase inhibitors. Plant Cell 1992, 4, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Tomitaka, Y.; Abe, H.; Tsuda, S.; Futai, K.; Mizukubo, T. Expression profile of jasmonic acid-induced genes and the induced resistance against the root-knot nematode (Meloidogyne incognita) in tomato plants (Solanum lycopersicum) after foliar treatment with methyl jasmonate. J. Plant Physiol. 2011, 168, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Howe, G.A.; Jander, G. Plant immunity to insect herbivores. Annu. Rev. Plant Biol. 2008, 59, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Starr, J.; Gobel, C.; Engelberth, J.; Feussner, I.; Tumlinson, J.; Kolomiets, M. Maize 9-lipoxygenase ZmLOX3 controls development, root-specific expression of defense genes, and resistance to root-knot nematodes. Mol. Plant Microbe Interact. 2008, 21, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Shim, W.B.; Gobel, C.; Kunze, S.; Feussner, I.; Meeley, R.; Balint-Kurti, P.; Kolomiets, M. Disruption of a maize 9-lipoxygenase results in increased resistance to fungal pathogens and reduced levels of contamination with mycotoxin fumonisin. Mol. Plant Microbe Interact. 2007, 20, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Brodhagen, M.; Isakeit, T.; Brown, S.H.; Gobel, C.; Betran, J.; Feussner, I.; Keller, N.P.; Kolomiets, M.V. Inactivation of the lipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp. Mol. Plant Microbe Interact. 2009, 22, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Ozalvo, R.; Cabrera, J.; Escobar, C.; Christensen, S.A.; Borrego, E.J.; Kolomiets, M.V.; Castresana, C.; Iberkleid, I.; Brown Horowitz, S. Two closely related members of Arabidopsis 13-lipoxygenases (13-LOXs), LOX3 and LOX4, reveal distinct functions in response to plant-parasitic nematode infection. Mol. Plant Pathol. 2014, 15, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rashotte, A.M.; Singh, N.K.; Weaver, D.B.; Lawrence, K.S.; Locy, R.D. Integrated signaling networks in plant responses to sedentary endoparasitic nematodes: A perspective. Plant Cell. Rep. 2015, 34, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.; Turner, J.G. Regulation of jasmonate-mediated plant responses in Arabidopsis. Ann. Bot. 2003, 92, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Wenig, M.; Langen, G.; Sharma, S.; Kugler, K.G.; Knappe, C.; Hause, B.; Bichlmeier, M.; Babaeizad, V.; Imani, J.; et al. Bacteria-triggered systemic immunity in barley is associated with WRKY and ETHYLENE RESPONSIVE FACTORs but not with salicylic acid. Plant Physiol. 2014, 166, 2133–2151. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ju, H.; Zhou, G.; Zhu, C.; Erb, M.; Wang, X.; Wang, P.; Lou, Y. An EAR-motif-containing ERF transcription factor affects herbivore-induced signaling, defense and resistance in rice. Plant J. 2011, 68, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Skibbe, M.; Qu, N.; Galis, I.; Baldwin, I.T. Induced plant defenses in the natural environment: Nicotiana attenuata WRKY3 and WRKY6 coordinate responses to herbivory. Plant Cell. 2008, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Duan, M.; Wei, C.; Li, Y. WRKY62 transcription factor acts downstream of cytosolic NPR1 and negatively regulates jasmonate-responsive gene expression. Plant Cell. Physiol. 2007, 48, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Xiao, J.; Ding, X.; Xiong, M.; Cai, M.; Cao, Y.; Li, X.; Xu, C.; Wang, S. OsWRKY13 mediates rice disease resistance by regulating defense-related genes in salicylate- and jasmonate-dependent signaling. Mol. Plant Microbe Interact. 2007, 20, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Jiang, W.; Zhang, L.; Zhang, F.; Zhang, F.; Shen, Q.; Wang, G.; Tang, K. AaERF1 positively regulates the resistance to Botrytis cinerea in Artemisia annua. PLoS ONE 2013, 8, e57657. [Google Scholar] [CrossRef] [PubMed]
- Moffat, C.S.; Ingle, R.A.; Wathugala, D.L.; Saunders, N.J.; Knight, H.; Knight, M.R. ERF5 and ERF6 play redundant roles as positive regulators of JA/Et-mediated defense against Botrytis cinerea in Arabidopsis. PLoS ONE 2012, 7, e35995. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.C.; Dombrecht, B.; Manners, J.M.; Schenk, P.M.; Edgar, C.I.; Maclean, D.J.; Scheible, W.R.; Udvardi, M.K.; Kazan, K. Repressor- and activator-type ethylene response factors functioning in jasmonate signaling and disease resistance identified via a genome-wide screen of Arabidopsis transcription factor gene expression. Plant Physiol. 2005, 139, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.Q.; Lin, H.R.; Luo, L.Y.; Chen, X.Y.; Lai, Z.J.; Yin, Z.G.; Lu, X.Y. Ramie Cultivar Records in China; Agriculture Press: Beijing, China, 1992. (In Chinese) [Google Scholar]
- Yu, Y.T.; Xue, S.D.; Zeng, L.B.; Zhang, G.; Chen, Q.; Zhu, A.G. Identification of a nematode isolate from rot root of ramie. J. Northwest. A&F Univ. 2011, 39, 105–109. (In Chinese) [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Zeng, L.; Yan, Z.; Liu, T.; Sun, K.; Zhu, T.; Zhu, A. Identification of Ramie Genes in Response to Pratylenchus coffeae Infection Challenge by Digital Gene Expression Analysis. Int. J. Mol. Sci. 2015, 16, 21989-22007. https://doi.org/10.3390/ijms160921989
Yu Y, Zeng L, Yan Z, Liu T, Sun K, Zhu T, Zhu A. Identification of Ramie Genes in Response to Pratylenchus coffeae Infection Challenge by Digital Gene Expression Analysis. International Journal of Molecular Sciences. 2015; 16(9):21989-22007. https://doi.org/10.3390/ijms160921989
Chicago/Turabian StyleYu, Yongting, Liangbin Zeng, Zhun Yan, Touming Liu, Kai Sun, Taotao Zhu, and Aiguo Zhu. 2015. "Identification of Ramie Genes in Response to Pratylenchus coffeae Infection Challenge by Digital Gene Expression Analysis" International Journal of Molecular Sciences 16, no. 9: 21989-22007. https://doi.org/10.3390/ijms160921989
APA StyleYu, Y., Zeng, L., Yan, Z., Liu, T., Sun, K., Zhu, T., & Zhu, A. (2015). Identification of Ramie Genes in Response to Pratylenchus coffeae Infection Challenge by Digital Gene Expression Analysis. International Journal of Molecular Sciences, 16(9), 21989-22007. https://doi.org/10.3390/ijms160921989