
Homozygous ALOXE3 Nonsense Variant Identified in a Patient with Non-Bullous Congenital Ichthyosiform Erythroderma Complicated by Superimposed Bullous Majocchi’s Granuloma: The Consequences of Skin Barrier Dysfunction

Abstract
:1. Introduction
2. Results
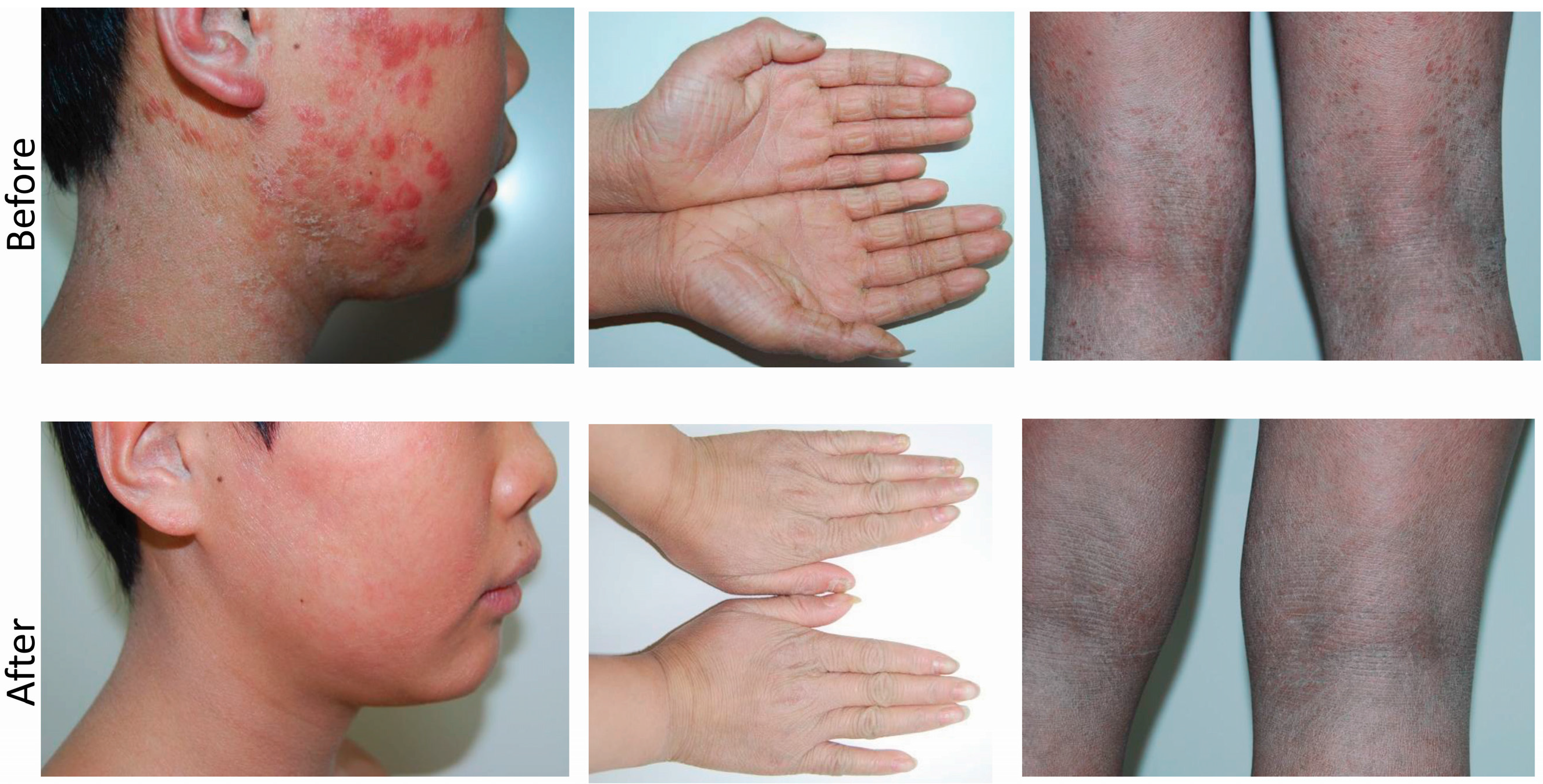
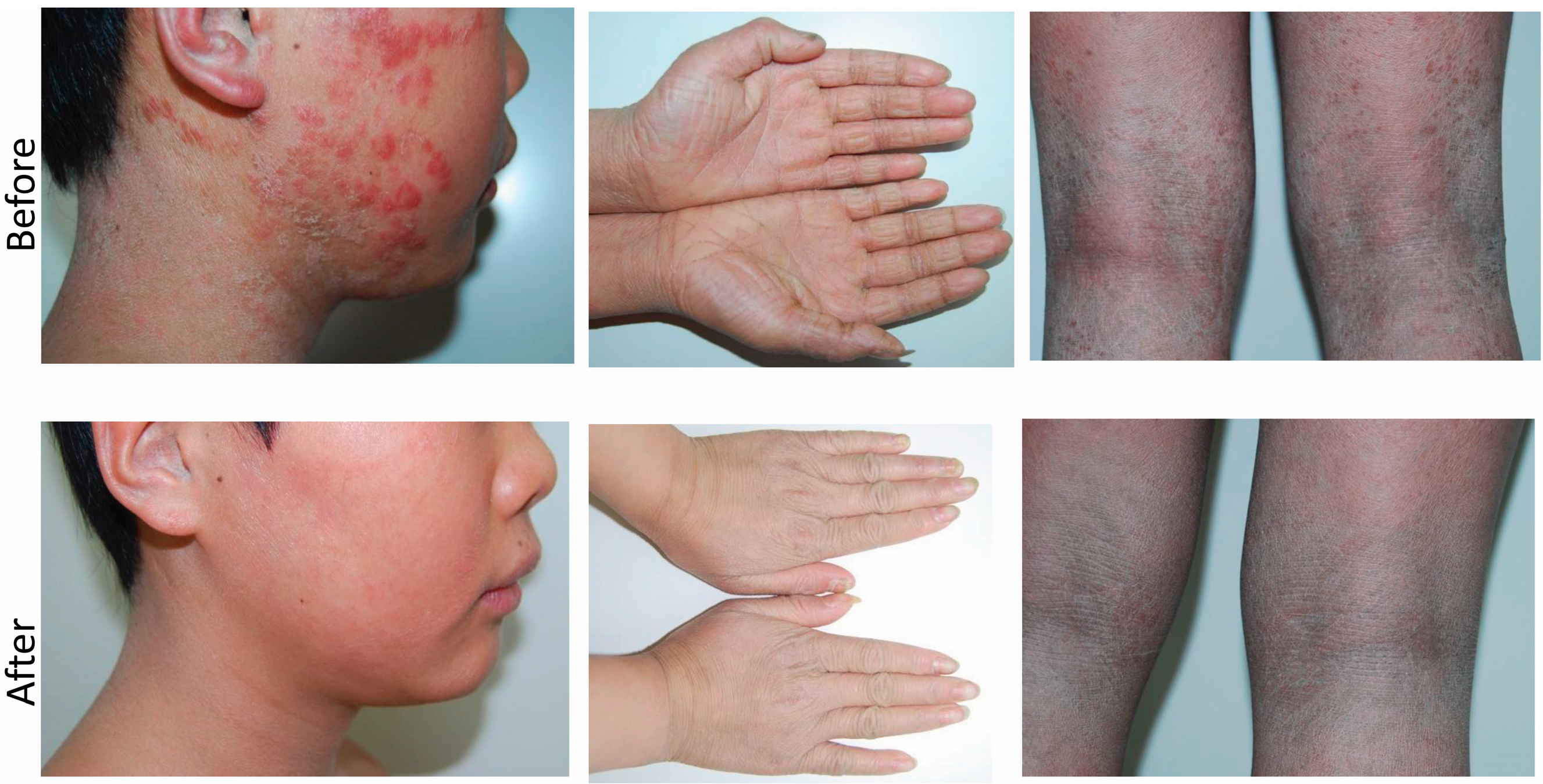
2.1. Clinical Presentation and Family History
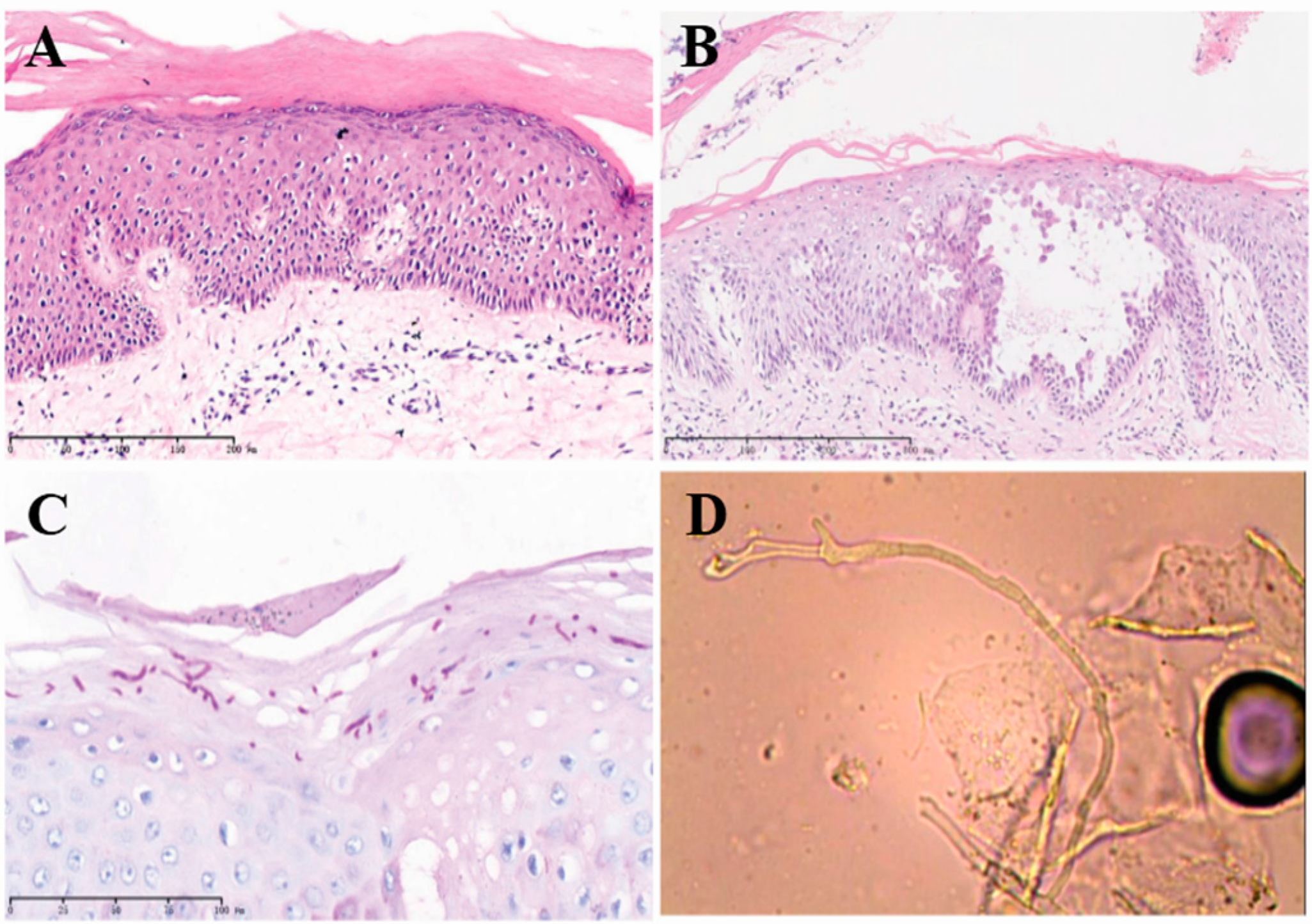
2.2. Histopathological Examination Confirmed the NBCIE Diagnosis
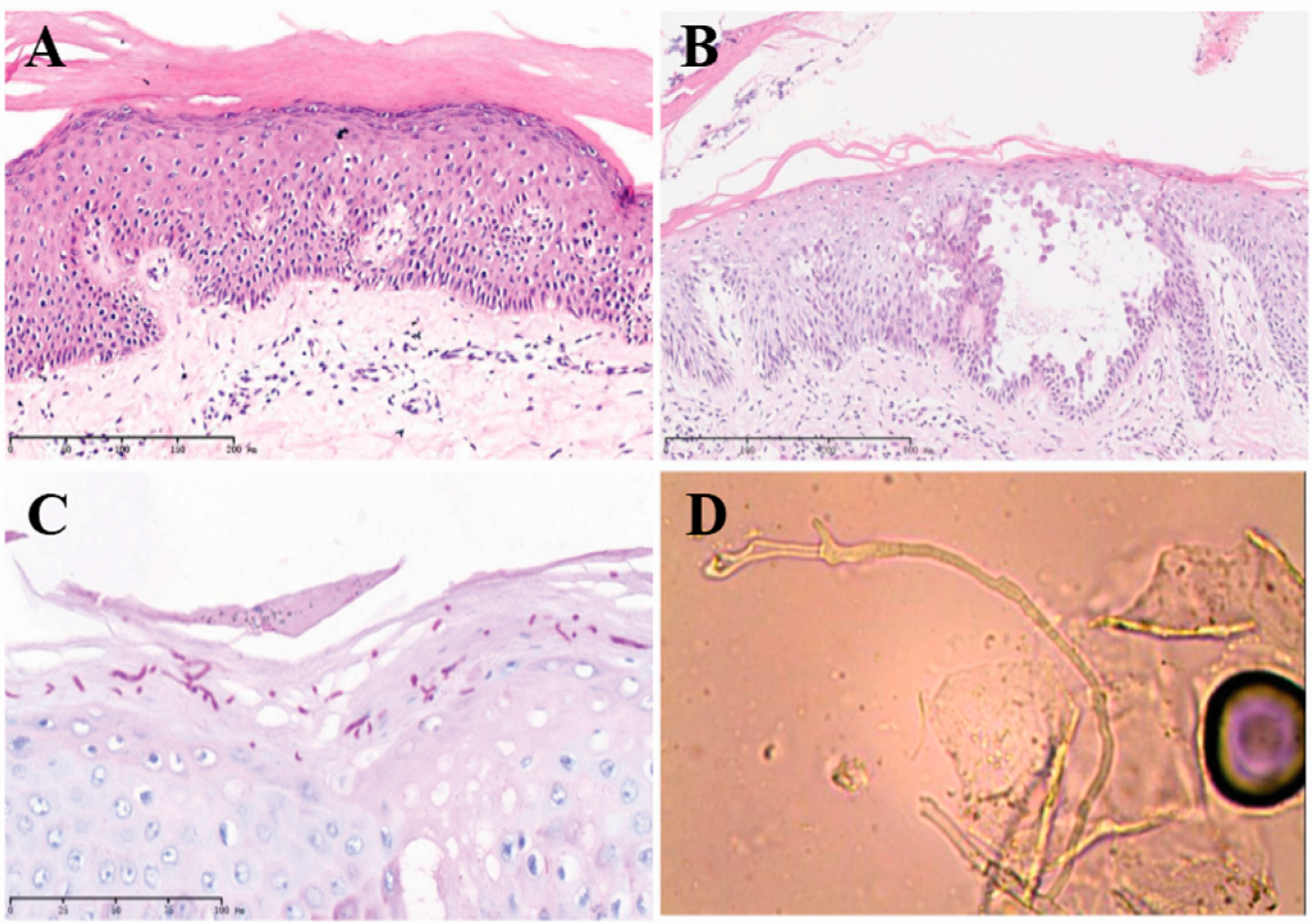
2.3. Superimposed Bullous Majocchi’s Granuloma
2.4. A Homozygous Nonsense Variant in ALOXE3 Was Identified by Genomic Analyses
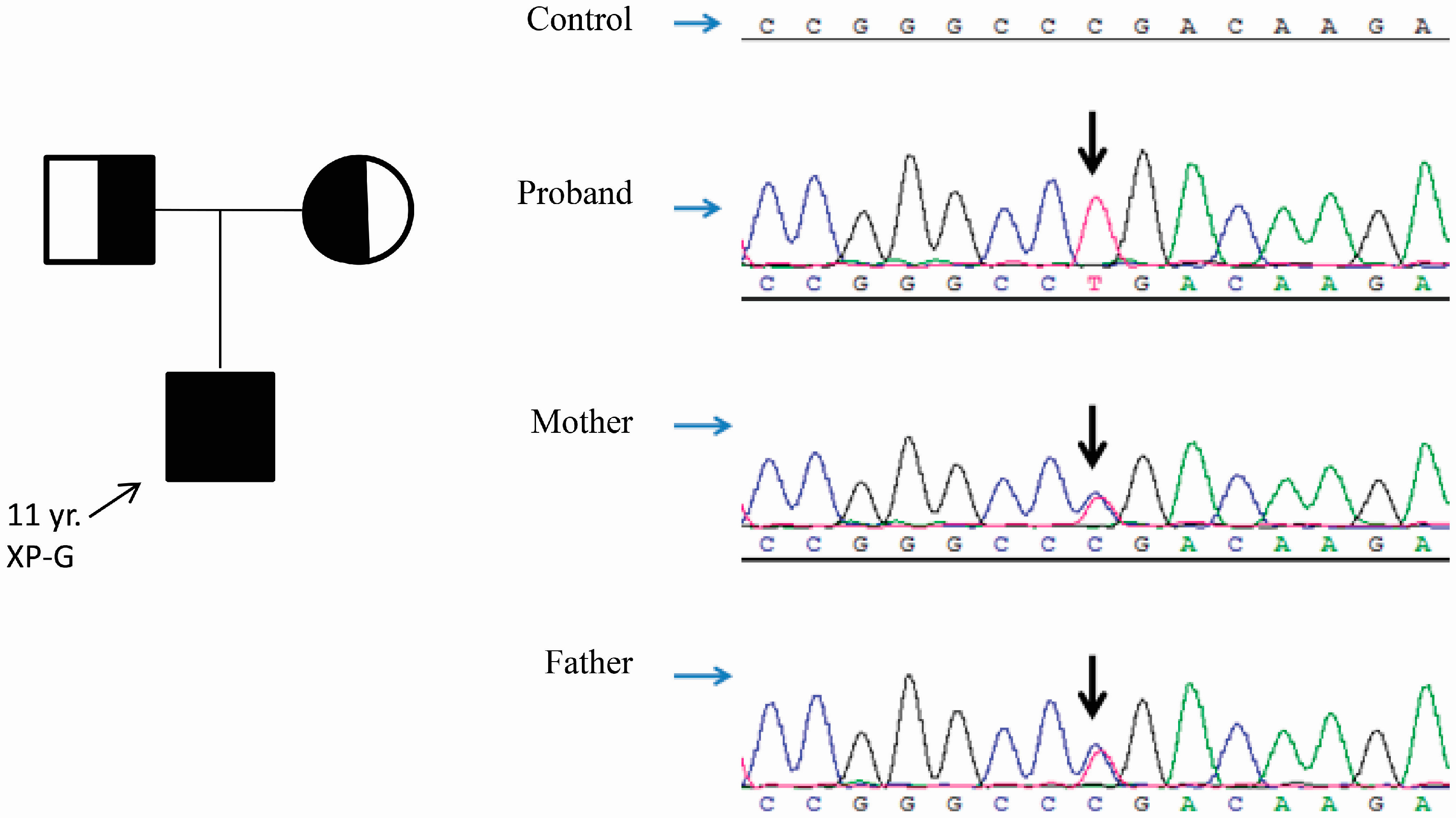

{kind=link}
{kind=link}
{kind=link}
| gDNA | cDNA | Protein | Exon | # of Reported Alleles | Allele Frequency in ExAC |
|---|---|---|---|---|---|
| g.8020119G>T | c.723C>A [9] | p.(Cys241 *) | 3 | 1 | 0 |
| g.8018941G>A | c.814C>T | p.(Arg272 *) | 4 | 2 | 1/121354 (rs370031870) |
| g.8018925C>T | c.830G>A [10] | p.(Arg277fs *12) | 4 | 2 | 3/121250 |
| g.8015495G>A | c.1096C>T [9,10,11,12,13] | p.(Arg366 *) | 7 | 15 | 14/121390 (rs121434233) |
| g.8015476delT | c.1115delA [10] | p.(Lys372fs*40) | 7 | 1 | 0 |
| g.8014792C>A | c.1238G>T [11] | p.(Gly413Val) | 7 | 1 | 0 |
| g.8013765_8013773del | c.1427_1435del9 [10] | p.(Gln476_Ala479delinsPro) | 9 | 2 | 0 |
| g.8013529G>T | c.1582C>A [12] | p.(Arg528Ser) | 10 | 2 | 0 |
| g.8013435A>G | c.1676T>C [9] | p.(Leu559Pro) | 10 | 2 | 0 |
| g.8012556C>A | c.1894G>T [12] | p.(Val632Phe) | 11 | 6 | 0 |
| g.8011840G>A | c.2026C>T [11] | p.(Gln676*) | 13 | 2 | 6/121282 |
| g.8006708G>A | c.2285C>T [9,10,11,12] | p.(Pro762Leu) | 15 | 18 | 114/121404 (rs147149459) |
| g.8013409C>A | c.1701+1G>T [13] | Intron 10 | 1 | 0 | |
| g.8013408A>T | c.1701+2A>T [13] | Intron 10 | 1 | 0 |
2.5. Treatment and Response
3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Histopathological Examination and Special Stains
4.3. Serological Studies
4.4. Genomic DNA Extraction
4.5. Targeted Next Generation Sequencing (NGS) of NBCIE-Associated Genes
4.6. Verification of Variants
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Akiyama, M.; Sawamura, D.; Shimizu, H. The clinical spectrum of nonbullous congenital ichthyosiform erythroderma and lamellar ichthyosis. Clin. Exp. Dermatol. 2003, 28, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Elias, P.M. Heterogeneity in autosomal recessive ichthyosis. Clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch. Dermatol. 1985, 121, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Oji, V.; Tadini, G.; Akiyama, M.; Bardon, C.B.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the first ichthyosis consensus conference in soreze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Ichthyoses, R.F. Erythrokeratodermas and Related Disorders. In Dermatology; Bolognia, J.L., Jorizzo, J.L., Schaffer, J.V., Eds.; Dermatology: Philadelphia, PA, USA, 2012; pp. 840–867. [Google Scholar]
- Elias, P.M.; Williams, M.L.; Holleran, W.M.; Jiang, Y.J.; Schmuth, M. Pathogenesis of permeability barrier abnormalities in the ichthyoses: Inherited disorders of lipid metabolism. J. Lipid Res. 2008, 49, 697–714. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.J.; Doyle, S.Z. The genetics of ichthyosis: A primer for epidemiologists. J. Investig. Dermatol. 1994, 102, 49S–50S. [Google Scholar] [CrossRef] [PubMed]
- Oji, V.; Hautier, J.M.; Ahvazi, B.; Hausser, I.; Aufenvenne, K.; Walker, T.; Seller, N.; Steijlen, P.M.; Küster, W.; Hovnanian, A.; et al. Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: Evidence for a temperature-sensitive phenotype. Hum. Mol. Genet. 2006, 15, 3083–3097. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Ju, X.; Yi, X.; Zhu, Q.; Qu, N.; Liu, T.; Chen, Y.; Jiang, H.; Yang, G.; Zhen, R.; et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS ONE 2011, 6, e29500. [Google Scholar] [CrossRef] [PubMed]
- Vahlquist, A.; Bygum, A.; Gånemo, A.; Virtanen, M.; Hellström-Pigg, M.; Strauss, G.; Brandrup, F.; Fischer, J. Genotypic and clinical spectrum of self-improving collodion ichthyosis: ALOX12B, ALOXE3, and TGM1 mutations in Scandinavian patients. J. Investig. Dermatol. 2010, 130, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Eckl, K.M.; de Juanes, S.; Kurtenbach, J.; Nätebus, M.; Lugassy, J.; Oji, V.; Traupe, H.; Preil, M.-L.; Martínez, F.; Smolle, J.; et al. Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: Evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J. Investig. Dermatol. 2009, 129, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Eckl, K.M.; Krieg, P.; Küster, W.; Traupe, H.; André, F.; Wittstruck, N.; Fürstenberger, G.; Hennies, H.C. Mutation spectrum and functional analysis of epidermis-type lipoxygenases in patients with autosomal recessive congenital ichthyosis. Hum. Mutat. 2005, 26, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Jobard, F.; Lefèvre, C.; Karaduman, A.; Blanchet-Bardon, C.; Emre, S.; Weissenbach, J.; Özgüc, M.; Lathrop, M.; Prud’homme, J.-F.; Fischer, J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum. Mol. Genet. 2002, 11, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Loriè, E.P.; Fischer, J.; Vahlquist, A.; Törmä, H. The expression of epidermal lipoxygenases and transglutaminase-1 is perturbed by NIPAL4 mutations: Indications of a common metabolic pathway essential for skin barrier homeostasis. J. Investig. Dermatol. 2012, 132, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pazos, L.; Bale, S.J. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013, 104, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Krieg, P.; Rosenberger, S.; de Juanes, S.; Latzko, S.; Hou, J.; Dick, A.; Kloz, U.; van der Hoeven, F.; Hausser, I.; Esposito, I.; et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J. Investig. Dermatol. 2013, 133, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Krieg, P.; Furstenberger, G. The role of lipoxygenases in epidermis. Biochim. Biophys. Acta 2014, 1841, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Rabionet, M.; Gorgas, K.; Sandhoff, R. Ceramide synthesis in the epidermis. Biochim. Biophys. Acta 2014, 1841, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Schneider, C.; Boeglin, W.E.; Marnett, L.J.; Brash, A.R. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc. Natl. Acad. Sci. USA 2003, 100, 9162–9167. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J. Autosomal recessive congenital ichthyosis. J. Investig. Dermatol. 2009, 129, 1319–1321. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, F.; Bouadja, B.; Lefèvre, C.; Jobard, F.; Audebert, S.; Lakhdar, H.; Martin, L.; Tadini, G.; Karaduman, A.; Emre, S.; et al. Novel mutations in ALOX12B in patients with autosomal recessive congenital ichthyosis and evidence for genetic heterogeneity on chromosome 17p13. J. Investig. Dermatol. 2007, 127, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Fang, X.; Yang, M.; Wang, J.; Kristiansen, K.; Wang, J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009, 19, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Xu, C.; Zhou, X.; Li, C.; Zhang, H.; Lian, B.Q.; Lee, J.J.; Shen, J.; Liu, Y.; Lian, C.G. Homozygous ALOXE3 Nonsense Variant Identified in a Patient with Non-Bullous Congenital Ichthyosiform Erythroderma Complicated by Superimposed Bullous Majocchi’s Granuloma: The Consequences of Skin Barrier Dysfunction. Int. J. Mol. Sci. 2015, 16, 21791-21801. https://doi.org/10.3390/ijms160921791
Wang T, Xu C, Zhou X, Li C, Zhang H, Lian BQ, Lee JJ, Shen J, Liu Y, Lian CG. Homozygous ALOXE3 Nonsense Variant Identified in a Patient with Non-Bullous Congenital Ichthyosiform Erythroderma Complicated by Superimposed Bullous Majocchi’s Granuloma: The Consequences of Skin Barrier Dysfunction. International Journal of Molecular Sciences. 2015; 16(9):21791-21801. https://doi.org/10.3390/ijms160921791
Chicago/Turabian StyleWang, Tao, Chenchen Xu, Xiping Zhou, Chunjia Li, Hongbing Zhang, Bill Q. Lian, Jonathan J. Lee, Jun Shen, Yuehua Liu, and Christine Guo Lian. 2015. "Homozygous ALOXE3 Nonsense Variant Identified in a Patient with Non-Bullous Congenital Ichthyosiform Erythroderma Complicated by Superimposed Bullous Majocchi’s Granuloma: The Consequences of Skin Barrier Dysfunction" International Journal of Molecular Sciences 16, no. 9: 21791-21801. https://doi.org/10.3390/ijms160921791
APA StyleWang, T., Xu, C., Zhou, X., Li, C., Zhang, H., Lian, B. Q., Lee, J. J., Shen, J., Liu, Y., & Lian, C. G. (2015). Homozygous ALOXE3 Nonsense Variant Identified in a Patient with Non-Bullous Congenital Ichthyosiform Erythroderma Complicated by Superimposed Bullous Majocchi’s Granuloma: The Consequences of Skin Barrier Dysfunction. International Journal of Molecular Sciences, 16(9), 21791-21801. https://doi.org/10.3390/ijms160921791