
Advances in the Study of the Antiatherogenic Function and Novel Therapies for HDL

Abstract
: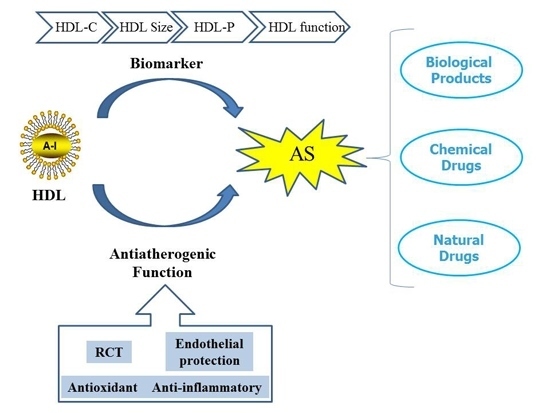
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Classification | Drug | Mechanism | TC | TG | VLDL | LDL | HDL | Advantage | Disadvantage |
|---|---|---|---|---|---|---|---|---|---|
| Stains | Lovastatin | As inhibition of HMG CoA reductase, reduce cholesterol synthesis | ↓ | ↓ | ↓ | ↑ | The advantage of these drugs is a low incidence of adverse reaction, and can be suitable for a variety of hypercholesterolemia except hypertriglyceridemia , is a lipid-lowering drug with rapid development in recent years | Gastrointestinal symptoms and rash, and the residual risk | |
| Atorvastatin | ↓ | ||||||||
| Fibrates | Gemfibrozil | The drug can increase Lp(a) Lipase activity to remove VLDL, TG; | ↓ | ↓ | ↓ | ↑ | These drugs do not cause the increase of diabetic insulin resistance or affect the control of blood sugar, therefore this kind of drugs is the first choice for treating the diabetic patients with hyperlipidemia | Gastrointestinal reactions, allergic reaction, due the drugs increase the concentration of cholesterol in bile, it may cause gallstones, occasional eyesight obstacle and hematological abnormalities | |
| Fenofibrate | Thus reducing VLDL and TG, TC and LDL can also be reduced | ↓ | |||||||
| Nicotinic Acids | Niacin; Inositol Aluminum | The drug can prevent fat decomposition, prevent free fatty acid formation, inhibit synthesis of TG and secretion of VLDL in liver | ↓ | ↓ | ↓ | ↑ | Cheap, and it is the only lipid-lowering drug can also reduce risk and mortality of cardiovascular disease | It is not suitable for diabetes patients, overdose adverse reactions (toxic to the liver, high blood sugar) has a high incidence common adverse reactions are skin flushing, itching, rash | |
| Aluminum Nicotinate | ↓ | ||||||||
| Cholesterol Absorption Inhibitor | Ezetimibe | The drug can combine with bile acid to block the bile acid absorption; | ↓ | ↓ | ↓ | ↑ | This kind of medicine is recognized as TC lowering drugs, when treats together with statins, the risk of accidental heart disease related to decrease the occurrence of 50% or more | The common adverse reactions are mild nausea and abdominal distension, constipation, therefore, it is not suitable for intestinal diseases and intractable constipation patients | |
| Prompte the translation of the cholesterol into bile in the gallbladder, then bile binding to drug is eliminated from the body | ↓ | ||||||||
| Polyene Unsaturated Fatty Acids | Duoxikang | This drug can combine with total cholesterol to be ester; Then promotes the degradation of bile acid excreted along with the bile, decreases plasma total cholesterol concentration | ↓ | ↓ | ↓ | These drugs in combination with statins can reduce the level of TG, and play an effective role in the prevention and treatment of coronary heart disease | This kind of medicaments is easy to be oxidized to atherogenic substance, has inhibitory effect on platelet aggregation, so it needs to be used with caution | ||
| Ecosapeatanolic acid | |||||||||
| ↓ |
2. HDL and AS
2.1. HDL as the Biomarker for AS
2.1.1. HDL-C
2.1.2. HDL Particle Size
2.1.3. HDL Particles Concentration (HDL-P)
2.1.4. Other Biomarkers
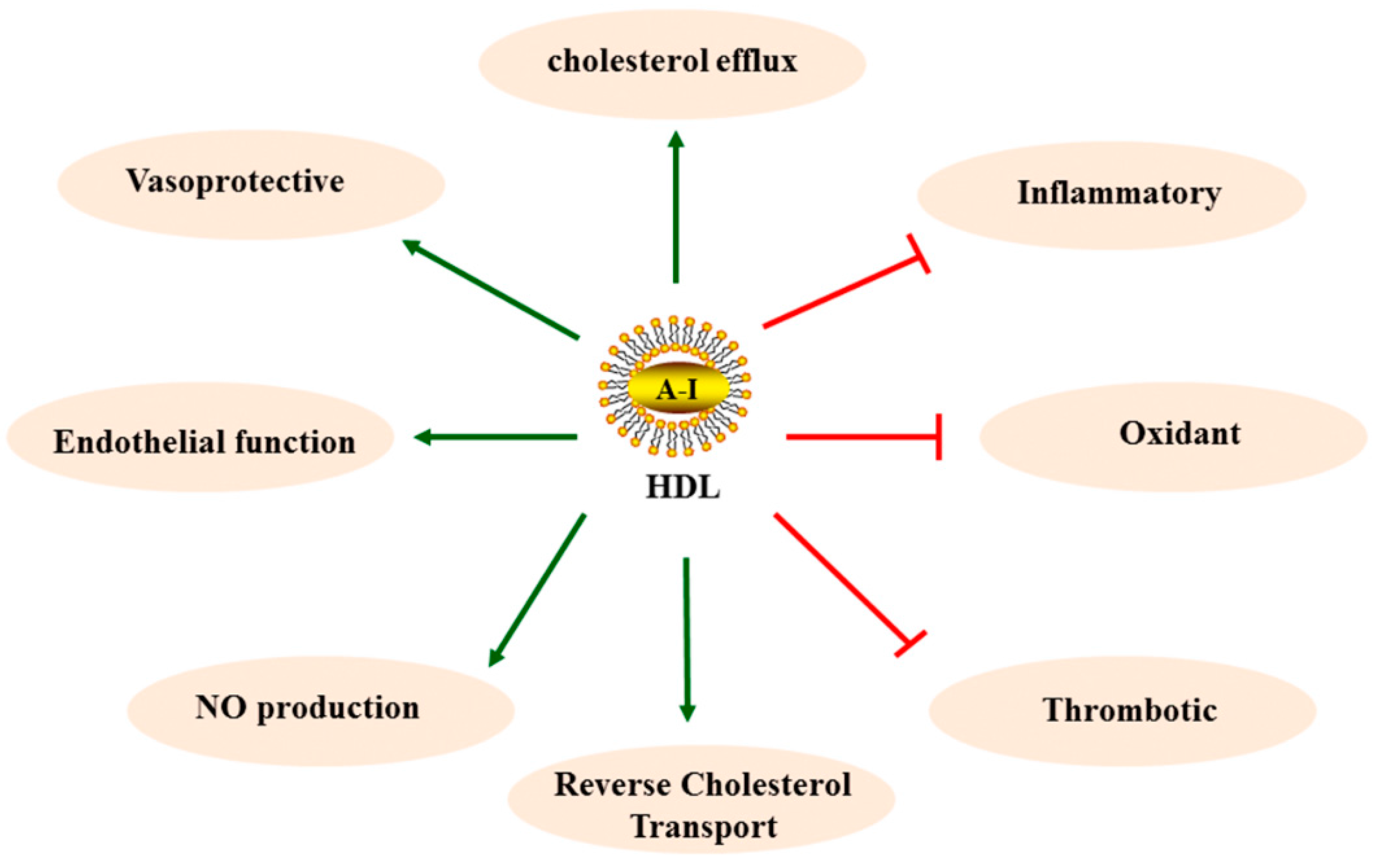
2.2. HDL in Anti-Atherosclerosis
 (promotion),
(promotion),  (inhibition).
(inhibition).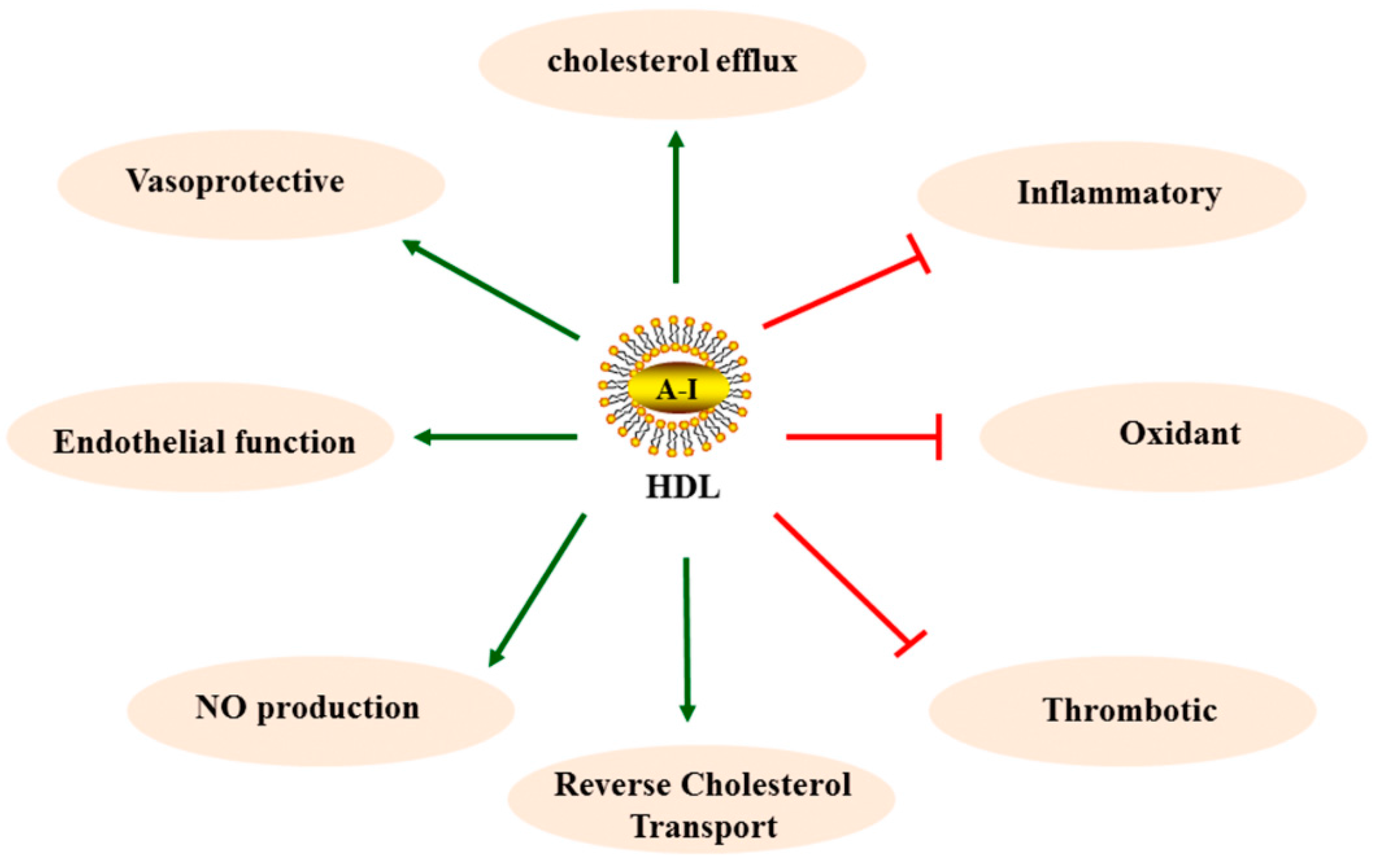
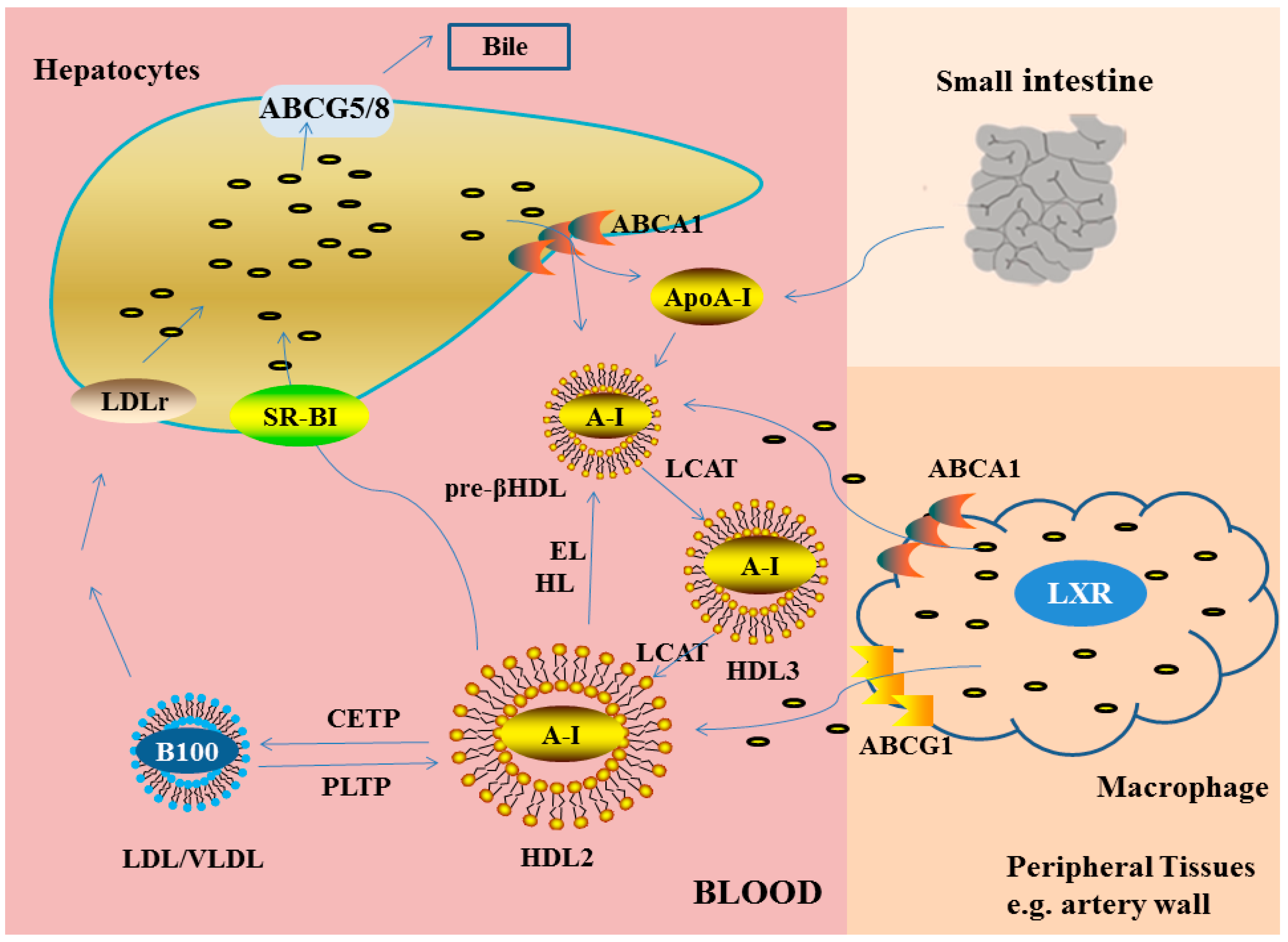
2.2.1. Reverse Cholesterol Transport
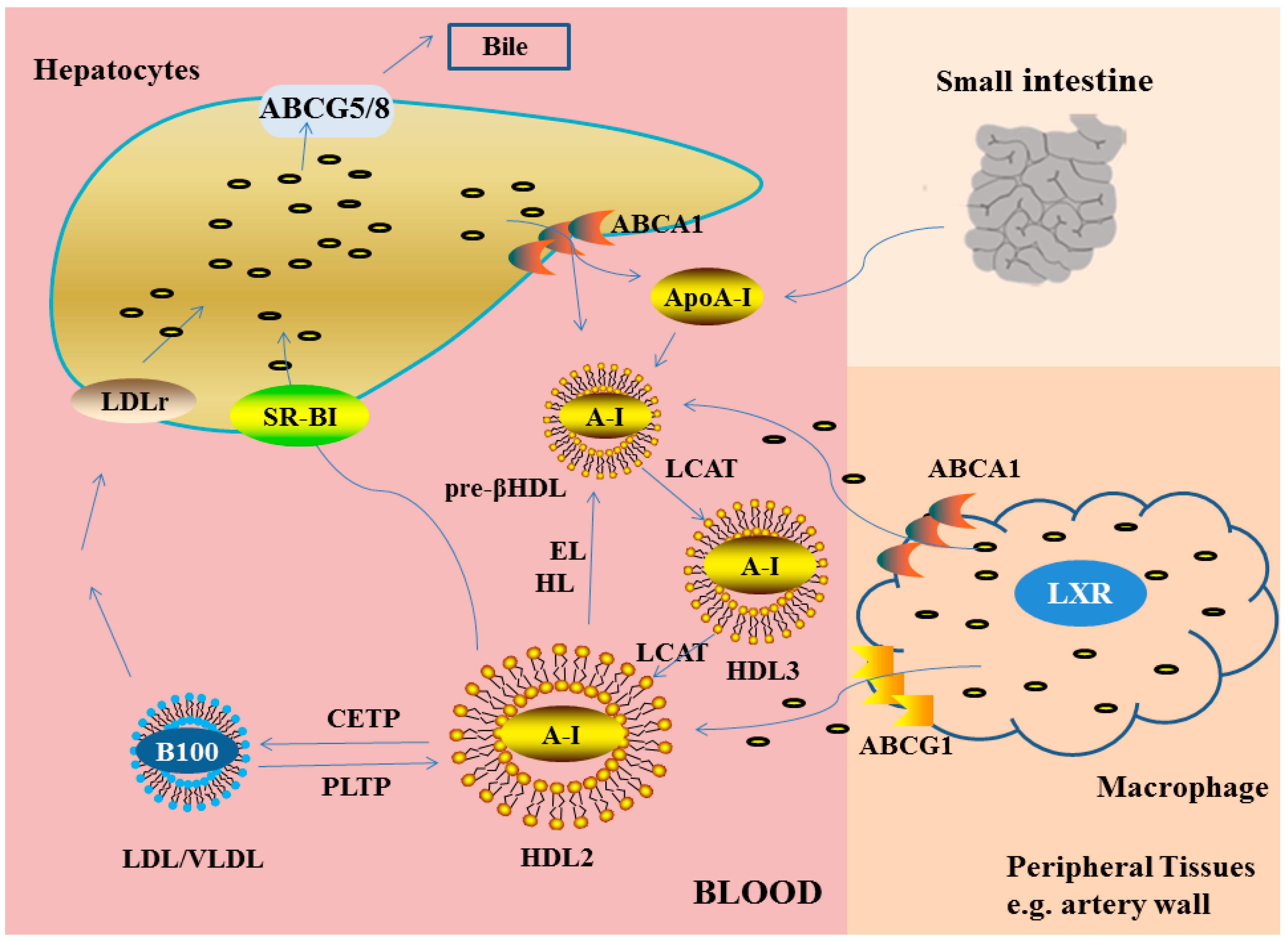
Part One: The Formation of Nascent HDL
Part Two: The Process of Cholesterol Esterified by LCAT
Part Three: The Exchange of CE (Cholesteryl Esters) Mediated by CETP
Part Four: Catabolism of HDL Cholesterol in Biliary Pathway
2.2.2. Antioxidant Properties of High-Density Lipoprotein
2.2.3. HDL in the Endothelium
2.2.4. Anti-inflammatory Properties of HDL
3. Novel Therapies with HDL as the Key Components
3.1. Novel Pharmacotherapeutic Strategies in Increasing HDL
3.1.1. Infusions of Special HDL
3.1.2. Autologous Delipidated HDL
3.2. Novel Pharmacotherapeutic Strategies in Four Steps of RCT
3.2.1. First Part of RCT
ApoA-I Mimetic Peptides
Regulators of ABCA1 and ABCG1
3.2.2. Second Part of RCT
LCAT Agonists
3.2.3. Third Part of RCT
CETP Inhibitor
3.2.4. Fourth Part of RCT
SR-BI Activators
3.3. Natural Drugs Associated with HDL at the Future Stage
3.3.1. Chitosan
3.3.2. Chitosan Oligosaccharides
4. Discussion
| Strategies Increasing HDL | Strategies of RCT in Four Steps | Natural Drugs | |||||
|---|---|---|---|---|---|---|---|
| ● Infusions of special HDL | ♦ rHDL: CSL-111 CER-001 CSL-112 | ● First part of RCT | ● Second part of RCT | ● Polysaccharide: | Chitosan; Chitosan oligosaccharides | ||
| ApoA-I mimetic peptides; ApoA-I upregulator | LCAT Agonists; ♦ rLCAT: ACP-501 | ||||||
| LXR agonists; Mir-33; PPAR modulators | ● Anthocyanins | ||||||
| ● Autologous delipidated HDL | ● Third part of RCT | ● Fourth part of RCT | |||||
| ♦ CETP inhibitor: | Anacetrapib; Evacetrapib; BAY 60-5521; TA-8995; CETP ASO | ♦ SR-BI activators: | Traditional drugs; Gene therapeutics | ● Sesamin | |||
| ● 24(S)-Saringosterol | |||||||
| ● Others | |||||||
Acknowledgments
Conflicts of Interests
References
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Executive summary: Heart disease and stroke statistics-2012 update: A report from the American heart association. Circulation 2012, 125, 188–197. [Google Scholar] [PubMed]
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The framingham study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef]
- Baigent, C.; Blackwell, L.; Emberson, J.; Holland, L.E.; Reith, C.; Bhala, N.; Peto, R.; Barnes, E.H.; Keech, A.; Simes, J.; et al. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [PubMed]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, L.; Goodloe, R.; Bradford, Y.; Farber-Eger, E.; Boston, J.; Crawford, D.C. The effects of electronic medical record phenotyping details on genetic association studies: HDL-C as a case study. Biodata Min. 2015, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Remaley, A.T.; Norata, G.D.; Catapano, A.L. Novel concepts in HDL pharmacology. Cardiovasc. Res. 2014, 103, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Saemann, M.D.; Heinemann, A.; Holzer, M. Inflammation alters HDL composition and function: Implications for HDL-raising therapies. Pharmacol. Ther. 2013, 137, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K. Atherosclerosis: Targeting endogenous apo AI—A new approach for raising HDL. Nat. Rev. Cardiol. 2011, 8, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Holleboom, A.G.; Jakulj, L.; Franssen, R.; Decaris, J.; Vergeer, M.; Koetsveld, J.; Luchoomun, J.; Glass, A.; Hellerstein, M.K.; Kastelein, J.J.; et al. In vivo tissue cholesterol efflux is reduced in carriers of a mutation in APOA1. J. Lipid Res. 2013, 54, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Umemoto, T.; Han, C.Y.; Mitra, P.; Averill, M.M.; Tang, C.; Goodspeed, L.; Omer, M.; Subramanian, S.; Wang, S.; den Hartigh, L.J.; et al. Apolipoprotein AI and high-density lipoprotein have anti-inflammatory effects on adipocytes via cholesterol transporters: ATP-binding cassette A-1, ATP-binding cassette G-1, and scavenger receptor B-1. Circ. Res. 2013, 112, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Reddy, S.T.; van Lenten, B.J.; Fogelman, A.M. HDL and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat. Rev. Cardiol. 2011, 8, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.; Didonato, J.A.; et al. Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.; Frishman, W.H. Inflammation and atherosclerosis: A review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol. Rev. 2014, 22, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Bonow, R.O. The cardiovascular biomarker conundrum: Challenges and solutions. JAMA 2011, 306, 2151–2152. [Google Scholar] [CrossRef] [PubMed]
- The AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol: baseline characteristics of study participants. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: Impact on Global Health outcomes (AIM-HIGH) trial. Am. Heart J. 2011, 161, 538–543. [Google Scholar]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [PubMed]
- Li, C.; Zhang, W.; Zhou, F.; Chen, C.; Zhou, L.; Li, Y.; Liu, L.; Pei, F.; Luo, H.; Hu, Z.; et al. Cholesteryl ester transfer protein inhibitors in the treatment of dyslipidemia: A systematic review and meta-analysis. PLoS ONE 2013, 8, e77049. [Google Scholar] [CrossRef] [PubMed]
- Boekholdt, S.M.; Arsenault, B.J.; Hovingh, G.K.; Mora, S.; Pedersen, T.R.; Larosa, J.C.; Welch, K.M.; Amarenco, P.; Demicco, D.A.; Tonkin, A.M.; et al. Levels and changes of HDL cholesterol and apolipoprotein A-I in relation to risk of cardiovascular events among statin-treated patients: A meta-analysis. Circulation 2013, 128, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Keene, D.; Price, C.; Shun-Shin, M.J.; Francis, D.P. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: Meta-analysis of randomised controlled trials including 117,411 patients. BMJ 2014, 349, g4379. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Saely, C.H.; Vonbank, A.; Drexel, H. HDL cholesterol and residual risk of first cardiovascular events. Lancet 2010, 376, 1738–1739. [Google Scholar] [CrossRef]
- Mora, S.; Glynn, R.J.; Boekholdt, S.M.; Nordestgaard, B.G.; Kastelein, J.J.; Ridker, P.M. On-treatment non-high-density lipoprotein cholesterol, apolipoprotein B, triglycerides, and lipid ratios in relation to residual vascular risk after treatment with potent statin therapy: JUPITER (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). J. Am. Coll. Cardiol. 2012, 59, 1521–1528. [Google Scholar] [PubMed]
- Rye, K.A.; Bursill, C.A.; Lambert, G.; Tabet, F.; Barter, P.J. The metabolism and anti-atherogenic properties of HDL. J. Lipid Res. 2009, 50, S195–S200. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.; Kastelein, J.; Nunn, A.; Hobbs, R. High density lipoproteins (HDLs) and atherosclerosis: The unanswered questions. Atherosclerosis 2003, 168, 195–211. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Lemieux, I.; Despres, J.P.; Gagnon, P.; Wareham, N.J.; Stroes, E.S.; Kastelein, J.J.; Khaw, K.T.; Boekholdt, S.M. HDL particle size and the risk of coronary heart disease in apparently healthy men and women: the EPIC-Norfolk prospective population study. Atherosclerosis 2009, 206, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.H.; Wajngarten, M.; Prete, A.C.; Diament, J.; Maranhao, R.C. Simultaneous transfer of cholesterol, triglycerides, and phospholipids to high-density lipoprotein in aging subjects with or without coronary artery disease. Clinics 2011, 66, 1543–1548. [Google Scholar] [PubMed]
- Pascot, A.; Lemieux, I.; Prud'Homme, D.; Tremblay, A.; Nadeau, A.; Couillard, C.; Bergeron, J.; Lamarche, B.; Despres, J.P. Reduced HDL particle size as an additional feature of the atherogenic dyslipidemia of abdominal obesity. J. Lipid Res. 2001, 42, 2007–2014. [Google Scholar] [PubMed]
- Du, X.M.; Kim, M.J.; Hou, L.; le Goff, W.; Chapman, M.J.; van Eck, M.; Curtiss, L.K.; Burnett, J.R.; Cartland, S.P.; Quinn, C.M.; et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ. Res. 2015, 116, 1133–1142. [Google Scholar] [PubMed]
- Kim, D.S.; Burt, A.A.; Rosenthal, E.A.; Ranchalis, J.E.; Eintracht, J.F.; Hatsukami, T.S.; Furlong, C.E.; Marcovina, S.; Albers, J.J.; Jarvik, G.P. HDL-3 is a superior predictor of carotid artery disease in a case-control cohort of 1725 participants. J. Am. Heart Assoc. 2014, 3, e902. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Genest, J.; Boekholdt, S.M.; Libby, P.; Gotto, A.M.; Nordestgaard, B.G.; Mora, S.; MacFadyen, J.G.; Glynn, R.J.; Kastelein, J.J. HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: An analysis from the JUPITER trial. Lancet 2010, 376, 333–339. [Google Scholar] [CrossRef]
- Mora, S.; Glynn, R.J.; Ridker, P.M. High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation 2013, 128, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- El, H.K.; Arsenault, B.J.; Franssen, R.; Despres, J.P.; Hovingh, G.K.; Stroes, E.S.; Otvos, J.D.; Wareham, N.J.; Kastelein, J.J.; Khaw, K.T.; et al. High-density lipoprotein particle size and concentration and coronary risk. Ann. Intern. Med. 2009, 150, 84–93. [Google Scholar]
- Mora, S.; Otvos, J.D.; Rifai, N.; Rosenson, R.S.; Buring, J.E.; Ridker, P.M. Lipoprotein particle profiles by nuclear magnetic resonance compared with standard lipids and apolipoproteins in predicting incident cardiovascular disease in women. Circulation 2009, 119, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Ala-Korpela, M.; Soininen, P.; Savolainen, M.J. Letter by Ala-Korpela et al regarding article, “Lipoprotein particle profiles by nuclear magnetic resonance compared with standard lipids and apolipoproteins in predicting incident cardiovascular disease in women”. Circulation 2009, 120, e149–e150. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, P.M.; Ronsein, G.E.; Monette, J.S.; Pamir, N.; Wimberger, J.; He, Y.; Anantharamaiah, G.M.; Kim, D.S.; Ranchalis, J.E.; Jarvik, G.P.; et al. Quantification of HDL particle concentration by calibrated ion mobility analysis. Clin. Chem. 2014, 60, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B.; Nielsen, L.B. Apolipoprotein M—A novel player in high-density lipoprotein metabolism and atherosclerosis. Curr. Opin. Lipidol. 2006, 17, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Elsoe, S.; Christoffersen, C.; Luchoomun, J.; Turner, S.; Nielsen, L.B. Apolipoprotein M promotes mobilization of cellular cholesterol in vivo. Biochim. Biophys. Acta 2013, 1831, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Poy, M.N.; Stoffel, M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat. Med. 2005, 11, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Jiao, G.; Yang, C.; Ye, Y. Evaluation of apolipoprotein M as a biomarker of coronary artery disease. Clin. Biochem. 2009, 42, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Borup, A.; Christensen, P.M.; Nielsen, L.B.; Christoffersen, C. Apolipoprotein M in lipid metabolism and cardiometabolic diseases. Curr. Opin. Lipidol. 2015, 26, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Brea, D.; Sobrino, T.; Blanco, M.; Fraga, M.; Agulla, J.; Rodriguez-Yanez, M.; Rodriguez-Gonzalez, R.; Perez, D.L.O.N.; Leira, R.; Forteza, J.; et al. Usefulness of haptoglobin and serum amyloid A proteins as biomarkers for atherothrombotic ischemic stroke diagnosis confirmation. Atherosclerosis 2009, 205, 561–567. [Google Scholar] [CrossRef] [PubMed]
- King, V.L.; Thompson, J.; Tannock, L.R. Serum amyloid A in atherosclerosis. Curr. Opin. Lipidol. 2011, 22, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Delanghe, J.R.; Langlois, M.R.; de Bacquer, D.; Mak, R.; Capel, P.; van Renterghem, L.; de Backer, G. Discriminative value of serum amyloid A and other acute-phase proteins for coronary heart disease. Atherosclerosis 2002, 160, 471–476. [Google Scholar] [CrossRef]
- Lepedda, A.J.; Nieddu, G.; Zinellu, E.; de Muro, P.; Piredda, F.; Guarino, A.; Spirito, R.; Carta, F.; Turrini, F.; Formato, M. Proteomic analysis of plasma-purified VLDL, LDL, and HDL fractions from atherosclerotic patients undergoing carotid endarterectomy: Identification of serum amyloid A as a potential marker. Oxid. Med. Cell. Longev. 2013, 2013, 385214. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Grossman, J.; FitzGerald, J.; Dahlin-Lee, E.; Wallace, D.J.; Thong, B.Y.; Badsha, H.; Kalunian, K.; Charles, C.; Navab, M.; et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.T.; Thulasiraman, V.; Weinberger, S.R.; Dalmasso, E.A. Protein biochips for differential profiling. Curr. Opin. Biotechnol. 2001, 12, 65–69. [Google Scholar] [CrossRef]
- Issaq, H.J.; Veenstra, T.D.; Conrads, T.P.; Felschow, D. The SELDI-TOF MS approach to proteomics: Protein profiling and biomarker identification. Biochem. Biophys. Res. Commun. 2002, 292, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Chou, K.J.; Liao, J.C.; Miao, Y.; Meng, H.H.; Ge, H.; Grijalva, V.; Hama, S.; Kozak, K.; Buga, G.; et al. Differential association of hemoglobin with proinflammatory high density lipoproteins in atherogenic/hyperlipidemic mice. A novel biomarker of atherosclerosis. J. Biol. Chem. 2007, 282, 23698–23707. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Al-Sarraf, A. Cholesterol efflux capacity and atherosclerosis. N. Engl. J. Med. 2011, 364, 1474–1475. [Google Scholar] [PubMed]
- Uto-Kondo, H.; Ayaori, M.; Ogura, M.; Nakaya, K.; Ito, M.; Suzuki, A.; Takiguchi, S.; Yakushiji, E.; Terao, Y.; Ozasa, H.; et al. Coffee consumption enhances high-density lipoprotein-mediated cholesterol efflux in macrophages. Circ. Res. 2010, 106, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Lawn, R.M.; Garvin, M.R.; Wade, D.P. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J. Biol. Chem. 2000, 275, 34508–34511. [Google Scholar] [CrossRef] [PubMed]
- Santamarina-Fojo, S.; Peterson, K.; Knapper, C.; Qiu, Y.; Freeman, L.; Cheng, J.F.; Osorio, J.; Remaley, A.; Yang, X.P.; Haudenschild, C.; et al. Complete genomic sequence of the human ABCA1 gene: analysis of the human and mouse ATP-binding cassette A promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kennedy, M.A.; Baldan, A.; Bojanic, D.D.; Lyons, K.; Edwards, P.A. Expression and regulation of multiple murine ATP-binding cassette transporter G1 mRNAs/isoforms that stimulate cellular cholesterol efflux to high density lipoprotein. J. Biol. Chem. 2004, 279, 45980–45989. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.G.; Bortnick, A.E.; Kellner-Weibel, G.; de la Llera-Moya, M.; Phillips, M.C.; Rothblat, G.H. Importance of different pathways of cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Dikkers, A.; Freak, D.B.J.; Annema, W.; Groen, A.K.; Tietge, U.J. Scavenger receptor BI and ABCG5/G8 differentially impact biliary sterol secretion and reverse cholesterol transport in mice. Hepatology 2013, 58, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Von Eckardstein, A.; Nofer, J.R.; Assmann, G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.J.; Davidson, W.S.; Fayad, Z.A.; Fuster, V.; Goldstein, J.; Hellerstein, M.; Jiang, X.C.; Phillips, M.C.; Rader, D.J.; et al. Cholesterol efflux and atheroprotection: Advancing the concept of reverse cholesterol transport. Circulation 2012, 125, 1905–1919. [Google Scholar] [CrossRef] [PubMed]
- Joy, T.; Hegele, R.A. The end of the road for CETP inhibitors after torcetrapib? Curr. Opin. Cardiol. 2009, 24, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tall, A.R. Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.K.; Valenta, D.T.; Hime, N.J.; Rye, K.A. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.R.; Jin, X.; Anzinger, J.J.; Xu, Q.; Purushothaman, S.; Fessler, M.B.; Addadi, L.; Kruth, H.S. ABCG1-mediated generation of extracellular cholesterol microdomains. J. Lipid Res. 2014, 55, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Song, G.J.; Kim, S.M.; Park, K.H.; Kim, J.; Choi, I.; Cho, K.H. SR-BI mediates high density lipoprotein (HDL)-induced anti-inflammatory effect in macrophages. Biochem. Biophys. Res. Commun. 2015, 457, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Collins, H.L.; Ranalletta, M.; Fuki, I.V.; Billheimer, J.T.; Rothblat, G.H.; Tall, A.R.; Rader, D.J. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2007, 117, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Daniil, G.; Zannis, V.I.; Chroni, A. Effect of apoA-I Mutations in the capacity of reconstituted HDL to promote ABCG1-mediated cholesterol efflux. PLoS ONE 2013, 8, e67993. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Bultel, S.; Helin, L.; Clavey, V.; Chinetti-Gbaguidi, G.; Rigamonti, E.; Colin, M.; Fruchart, J.C.; Staels, B.; Lestavel, S. Liver X receptor activation induces the uptake of cholesteryl esters from high density lipoproteins in primary human macrophages. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Hanf, R.; Millatt, L.J.; Cariou, B.; Noel, B.; Rigou, G.; Delataille, P.; Daix, V.; Hum, D.W.; Staels, B. The dual peroxisome proliferator-activated receptor α/δ agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma-associated adverse cardiac effects. Diabetes Vasc. Dis. Res. 2014, 11, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Briand, O.; Touche, V.; Wouters, K.; Baron, M.; Pattou, F.; Hanf, R.; Tailleux, A.; Chinetti, G.; Staels, B.; et al. Activation of intestinal peroxisome proliferator-activated receptor-α increases high-density lipoprotein production. Eur. Heart J. 2013, 34, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Rousset, X.; Shamburek, R.; Vaisman, B.; Amar, M.; Remaley, A.T. Lecithin cholesterol acyltransferase: An anti- or pro-atherogenic factor? Curr. Atheroscler. Rep. 2011, 13, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Soran, H.; Hama, S.; Yadav, R.; Durrington, P.N. HDL functionality. Curr. Opin. Lipidol. 2012, 23, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, G.; Olivecrona, T. Triglyceride lipases and atherosclerosis. Curr. Opin. Lipidol. 2010, 21, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Sparks, D.L. Hepatic lipase, high density lipoproteins, and hypertriglyceridemia. Am. J. Pathol. 2011, 178, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Ishida, T.; Rader, D.J. Update on the role of endothelial lipase in high-density lipoprotein metabolism, reverse cholesterol transport, and atherosclerosis. Circ. J. 2010, 74, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Annema, W.; Tietge, U.J. Role of hepatic lipase and endothelial lipase in high-density lipoprotein-mediated reverse cholesterol transport. Curr. Atheroscler. Rep. 2011, 13, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Choi, S.; Kundu, R.K.; Hirata, K.; Rubin, E.M.; Cooper, A.D.; Quertermous, T. Endothelial lipase is a major determinant of HDL level. J. Clin. Investig. 2003, 111, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ruel, I.L.; Couture, P.; Cohn, J.S.; Bensadoun, A.; Marcil, M.; Lamarche, B. Evidence that hepatic lipase deficiency in humans is not associated with proatherogenic changes in HDL composition and metabolism. J. Lipid Res. 2004, 45, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.; Amar, M.J.; Martin, P.; Fruchart-Najib, J.; Foger, B.; Shamburek, R.D.; Brewer, H.J.; Santamarina-Fojo, S. Hepatic lipase deficiency decreases the selective uptake of HDL-cholesteryl esters in vivo. J. Lipid Res. 2000, 41, 667–672. [Google Scholar] [PubMed]
- Jaye, M.; Lynch, K.J.; Krawiec, J.; Marchadier, D.; Maugeais, C.; Doan, K.; South, V.; Amin, D.; Perrone, M.; Rader, D.J. A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 1999, 21, 424–428. [Google Scholar] [PubMed]
- Escola-Gil, J.C.; Chen, X.; Julve, J.; Quesada, H.; Santos, D.; Metso, J.; Tous, M.; Jauhiainen, M.; Blanco-Vaca, F. Hepatic lipase- and endothelial lipase-deficiency in mice promotes macrophage-to-feces RCT and HDL antioxidant properties. Biochim. Biophys. Acta 2013, 1831, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, F.; Zhang, S.; Lei, D.; Charles, M.A.; Cavigiolio, G.; Oda, M.; Krauss, R.M.; Weisgraber, K.H.; Rye, K.A.; et al. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 2012, 8, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; le Goff, W.; Guerin, M.; Kontush, A. Cholesteryl ester transfer protein: At the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur. Heart J. 2010, 31, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Albers, J.J.; Wolfbauer, G.; Pownall, H.J. Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry 1997, 36, 3645–3653. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, S.; Feng, Y.; Feng, L.; Cui, Y.; Song, G.; Luo, T.; Zhang, K.; Wang, Y.; Jiang, X.C.; et al. Phospholipid transfer protein deficiency decreases the content of S1P in HDL via the loss of its transfer capability. Lipids 2014, 49, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.J.; Vuletic, S.; Cheung, M.C. Role of plasma phospholipid transfer protein in lipid and lipoprotein metabolism. Biochim. Biophys. Acta 2012, 1821, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Bruce, C.; Mar, J.; Lin, M.; Ji, Y.; Francone, O.L.; Tall, A.R. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high-density lipoprotein levels. J. Clin. Investig. 1999, 103, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Yazdanyar, A.; Quan, W.; Jin, W.; Jiang, X.C. Liver-specific phospholipid transfer protein deficiency reduces high-density lipoprotein and non-high-density lipoprotein production in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Qin, S.; Qiao, C.; Kawano, K.; Lin, M.; Skold, A.; Xiao, X.; Tall, A.R. Apolipoprotein B secretion and atherosclerosis are decreased in mice with phospholipid-transfer protein deficiency. Nat. Med. 2001, 7, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Shelly, L.; Sand, T.; Reidich, B.; Chang, G.; Macdougall, M.; Peakman, M.C.; Jiang, X.C. Pharmacologic inhibition of phospholipid transfer protein activity reduces apolipoprotein-B secretion from hepatocytes. J. Pharmacol. Exp. Ther. 2010, 332, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Rinninger, F.; Heine, M.; Singaraja, R.; Hayden, M.; Brundert, M.; Ramakrishnan, R.; Heeren, J. High density lipoprotein metabolism in low density lipoprotein receptor-deficient mice. J. Lipid Res. 2014, 55, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, A.E.; Vrins, C.L.; van den Oever, K.; Kunne, C.; Oude, E.R.; Kuipers, F.; Groen, A.K. Direct intestinal cholesterol secretion contributes significantly to total fecal neutral sterol excretion in mice. Gastroenterology 2007, 133, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Van der Velde, A.E.; Brufau, G.; Groen, A.K. Transintestinal cholesterol efflux. Curr. Opin. Lipidol. 2010, 21, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Moreau, F.; Cariou, B.; Le May, C. Trans-intestinal cholesterol excretion (TICE): A new route for cholesterol excretion. Med. Sci. 2014, 30, 896–901. [Google Scholar]
- Vrins, C.L.; van der Velde, A.E.; van den Oever, K.; Levels, J.H.; Huet, S.; Oude, E.R.; Kuipers, F.; Groen, A.K. Peroxisome proliferator-activated receptor delta activation leads to increased transintestinal cholesterol efflux. J. Lipid Res. 2009, 50, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Vrins, C.L.; Ottenhoff, R.; van den Oever, K.; de Waart, D.R.; Kruyt, J.K.; Zhao, Y.; van Berkel, T.J.; Havekes, L.M.; Aerts, J.M.; van Eck, M.; et al. Trans-intestinal cholesterol efflux is not mediated through high density lipoprotein. J. Lipid Res. 2012, 53, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chantepie, S.; Chapman, M.J. Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Chapman, M.J. Antiatherogenic function of HDL particle subpopulations: Focus on antioxidative activities. Curr. Opin. Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Mackness, B.; Mackness, M. The antioxidant properties of high-density lipoproteins in atherosclerosis. Panminerva Med. 2012, 54, 83–90. [Google Scholar] [PubMed]
- Vohl, M.C.; Neville, T.A.; Kumarathasan, R.; Braschi, S.; Sparks, D.L. A novel lecithin-cholesterol acyltransferase antioxidant activity prevents the formation of oxidized lipids during lipoprotein oxidation. Biochemistry 1999, 38, 5976–5981. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Anantharamaiah, G.M.; Hassan, K.; Hough, G.P.; Watson, A.D.; Reddy, S.T.; Sevanian, A.; Fonarow, G.C.; Fogelman, A.M. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: Steps 2 and 3. J. Lipid Res. 2000, 41, 1495–1508. [Google Scholar] [PubMed]
- Turunen, P.; Jalkanen, J.; Heikura, T.; Puhakka, H.; Karppi, J.; Nyyssonen, K.; Yla-Herttuala, S. Adenovirus-mediated gene transfer of Lp-PLA2 reduces LDL degradation and foam cell formation in vitro. J. Lipid Res. 2004, 45, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Liu, Y.; Greiner, C.D.; Holtzman, J.L. Physiologic concentrations of homocysteine inhibit the human plasma GSH peroxidase that reduces organic hydroperoxides. J. Lab. Clin. Med. 2000, 136, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horvath, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.M.; Parker, T.S.; Donnelly, T.M.; Walsh, A.; Rubin, A.L. In vivo protection against endotoxin by plasma high density lipoprotein. Proc. Natl. Acad. Sci. USA 1993, 90, 12040–12044. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; Giannarelli, C.; Cimmino, G.; Santos-Gallego, C.G.; Alique, M.; Pinero, A.; Vilahur, G.; Fuster, V.; Badimon, L.; Badimon, J.J. Recombinant HDL(Milano) exerts greater anti-inflammatory and plaque stabilizing properties than HDL(wild-type). Atherosclerosis 2012, 220, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Goffinet, M.; Boubekeur, N.; Ackermann, R.; Sy, G.; Bluteau, A.; Cholez, G.; Keyserling, C.; Lalwani, N.; Paolini, J.F.; et al. CER-001, a HDL-mimetic, stimulates the reverse lipid transport and atherosclerosis regression in high cholesterol diet-fed LDL-receptor deficient mice. Atherosclerosis 2014, 232, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Ballantyne, C.M.; Barter, P.; Dasseux, J.L.; Fayad, Z.A.; Guertin, M.C.; Kastelein, J.J.; Keyserling, C.; Klepp, H.; Koenig, W.; et al. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: A randomized trial. Eur. Heart J. 2014, 35, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Funt, S.; Gorman, D.; Tall, A.R.; Wang, N. Pegylation of high-density lipoprotein decreases plasma clearance and enhances antiatherogenic activity. Circ. Res. 2013, 113, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- Waksman, R.; Torguson, R.; Kent, K.M.; Pichard, A.D.; Suddath, W.O.; Satler, L.F.; Martin, B.D.; Perlman, T.J.; Maltais, J.A.; Weissman, N.J.; et al. A first-in-man, randomized, placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J. Am. Coll. Cardiol. 2010, 55, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Rudel, L.L.; Conner, A.; Akeefe, H.; Kostner, G.; Baki, T.; Rothblat, G.; de la Llera-Moya, M.; Asztalos, B.; Perlman, T.; et al. Selective delipidation of plasma HDL enhances reverse cholesterol transport in vivo. J. Lipid Res. 2009, 50, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Van Capelleveen, J.C.; Brewer, H.B.; Kastelein, J.J.; Hovingh, G.K. Novel therapies focused on the high-density lipoprotein particle. Circ. Res. 2014, 114, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Imura, T.; Leman, L.J.; Curtiss, L.K.; Maryanoff, B.E.; Ghadiri, M.R. Mimicry of high-density lipoprotein: Functional peptide-lipid nanoparticles based on multivalent peptide constructs. J. Am. Chem. Soc. 2013, 135, 13414–13424. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.G.; Murzilli, S.; Salvatore, L.; D’Errico, I.; Petruzzelli, M.; Conca, P.; Jiang, Z.Y.; Calabresi, L.; Parini, P.; Moschetta, A. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010, 12, 187–193. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Feng, J.; He, G. miR-613 regulates cholesterol efflux by targeting LXRα and ABCA1 in PPARβ activated THP-1 macrophages. Biochem. Biophys. Res. Commun. 2014, 448, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Simonelli, S.; Tinti, C.; Salvini, L.; Tinti, L.; Ossoli, A.; Vitali, C.; Sousa, V.; Orsini, G.; Nolli, M.L.; Franceschini, G.; et al. Recombinant human LCAT normalizes plasma lipoprotein profile in LCAT deficiency. Biologicals 2013, 41, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Espinosa, S.L.; Mendoza-Espinosa, P.; Delgado-Coello, B.; Mas-Oliva, J. Lecithin cholesterol acyltransferase (LCAT) activity in the presence of Apo-AI-derived peptides exposed to disorder-order conformational transitions. Biochem. Biophys. Res. Commun. 2013, 441, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Barylski, M.; Toth, P.P.; Nikolic, D.; Banach, M.; Rizzo, M.; Montalto, G. Emerging therapies for raising high-density lipoprotein cholesterol (HDL-C) and augmenting HDL particle functionality. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Rye, K.A. Cholesteryl ester transfer protein inhibition as a strategy to reduce cardiovascular risk. J. Lipid Res. 2012, 53, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, A.H.; Akhlaghi, F. Future of cholesteryl ester transfer protein (CETP) inhibitors: A pharmacological perspective. Clin. Pharmacokinet. 2013, 52, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Beyer, T.P.; Zhang, Y.; Schmidt, R.J.; Chen, Y.Q.; Cockerham, S.L.; Zimmerman, K.M.; Karathanasis, S.K.; Cannady, E.A.; Fields, T.; et al. Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure. J. Lipid Res. 2011, 52, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Lawson, M.; Fowler, D.; Maruyama, N.; Mito, S.; Tomiyasu, K.; Kinoshita, S.; Suzuki, C.; Kawaguchi, A.; Round, P.; et al. Tolerability, pharmacokinetics and pharmacodynamics of TA-8995, a selective cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 2014, 78, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.F.; Heinig, R.; Schmeck, C.; Kohlsdorfer, C.; Ludwig, M.; Schaefer, A.; Gelfert-Peukert, S.; Wensing, G.; Weber, O. Single dose pharmacokinetics, pharmacodynamics, tolerability and safety of BAY 60–5521, a potent inhibitor of cholesteryl ester transfer protein. Br. J. Clin. Pharmacol. 2012, 73, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Bell, T.R.; Graham, M.J.; Lee, R.G.; Mullick, A.E.; Fu, W.; Norris, D.; Crooke, R.M. Antisense oligonucleotide inhibition of cholesteryl ester transfer protein enhances RCT in hyperlipidemic, CETP transgenic, LDLr–/– mice. J. Lipid Res. 2013, 54, 2647–2657. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Shen, W.J.; Kraemer, F.B.; Azhar, S. MicroRNAs 125a and 455 repress lipoprotein-supported steroidogenesis by targeting scavenger receptor class B type I in steroidogenic cells. Mol. Cell. Biol. 2012, 32, 5035–5045. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, X.J.; Jiang, H.J.; Du, Y.; Yang, F.; Si, S.Y.; Hong, B. MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol. Cell. Biol. 2013, 33, 1956–1964. [Google Scholar] [CrossRef] [PubMed]
- Sugano, M.; Fujikawa, T.; Hiratsuji, Y.; Nakashima, K.; Fukuda, N.; Hasegawa, Y. A novel use of chitosan as a hypocholesterolemic agent in rats. Am. J. Clin. Nutr. 1980, 33, 787–793. [Google Scholar] [PubMed]
- Baker, W.L.; Tercius, A.; Anglade, M.; White, C.M.; Coleman, C.I. A meta-analysis evaluating the impact of chitosan on serum lipids in hypercholesterolemic patients. Ann. Nutr. Metab. 2009, 55, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.Q.; Wu, S.H.; Zhang, H.L.; Feng, Y.F. Development and validation of an improved Bradford method for determination of insulin from chitosan nanoparticulate systems. Pharm. Biol. 2010, 48, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Gao, B.; Tao, Y.; Guo, J.; Su, Z.Q. Antiobese effects of capsaicin-chitosan microsphere (CCMS) in obese rats induced by high fat diet. J. Agric. Food Chem. 2014, 62, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, G.D.; Tan, S.R.; Guo, J.; Su, Z.Q. The preparation of capsaicin-chitosan microspheres (CCMS) enteric coated tablets. Int. J. Mol. Sci. 2013, 14, 24305–24319. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, H.L.; Hu, Y.M.; Wan, S.; Su, Z.Q. Preparation of chitosan and water-soluble chitosan microspheres via spray-drying method to lower blood lipids in rats fed with high-fat diets. Int. J. Mol. Sci. 2013, 14, 4174–4184. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Guo, J.; Su, Z. Advances in understanding the interrelations between leptin resistance and obesity. Physiol. Behav. 2014, 130, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Tao, Y.; Guo, J.; Hu, Y.M.; Su, Z.Q. Hypolipidemic effects of chitosan nanoparticles in hyperlipidemia rats induced by high fat diet. Int. Immunopharmacol. 2011, 11, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Q.; Wei, P.; Cheng, L.; Peng, Q.; Li, S.; Yin, H.; Du, Y. Chitosan oligosaccharides downregulate the expression of E-selectin and ICAM-1 induced by LPS in endothelial cells by inhibiting MAP kinase signaling. Int. J. Mol. Med. 2014, 33, 392–400. [Google Scholar] [PubMed]
- Liu, H.T.; Huang, P.; Ma, P.; Liu, Q.S.; Yu, C.; Du, Y.G. Chitosan oligosaccharides suppress LPS-induced IL-8 expression in human umbilical vein endothelial cells through blockade of p38 and Akt protein kinases. Acta Pharmacol. Sin. 2011, 32, 478–486. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, P.; Pan, H.; Xiao, T.; Zhou, T.; Guo, J.; Su, Z. Advances in the Study of the Antiatherogenic Function and Novel Therapies for HDL. Int. J. Mol. Sci. 2015, 16, 17245-17272. https://doi.org/10.3390/ijms160817245
Cao P, Pan H, Xiao T, Zhou T, Guo J, Su Z. Advances in the Study of the Antiatherogenic Function and Novel Therapies for HDL. International Journal of Molecular Sciences. 2015; 16(8):17245-17272. https://doi.org/10.3390/ijms160817245
Chicago/Turabian StyleCao, Peiqiu, Haitao Pan, Tiancun Xiao, Ting Zhou, Jiao Guo, and Zhengquan Su. 2015. "Advances in the Study of the Antiatherogenic Function and Novel Therapies for HDL" International Journal of Molecular Sciences 16, no. 8: 17245-17272. https://doi.org/10.3390/ijms160817245
APA StyleCao, P., Pan, H., Xiao, T., Zhou, T., Guo, J., & Su, Z. (2015). Advances in the Study of the Antiatherogenic Function and Novel Therapies for HDL. International Journal of Molecular Sciences, 16(8), 17245-17272. https://doi.org/10.3390/ijms160817245