
Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells

Abstract
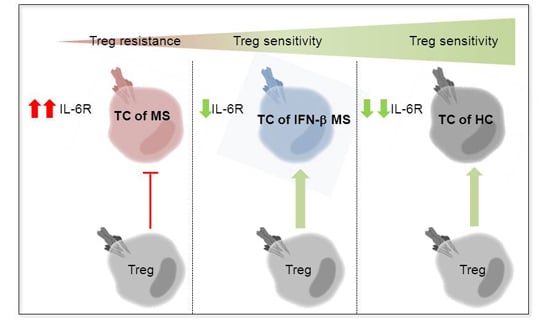
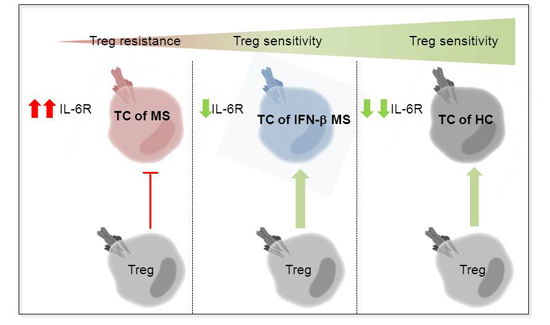
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
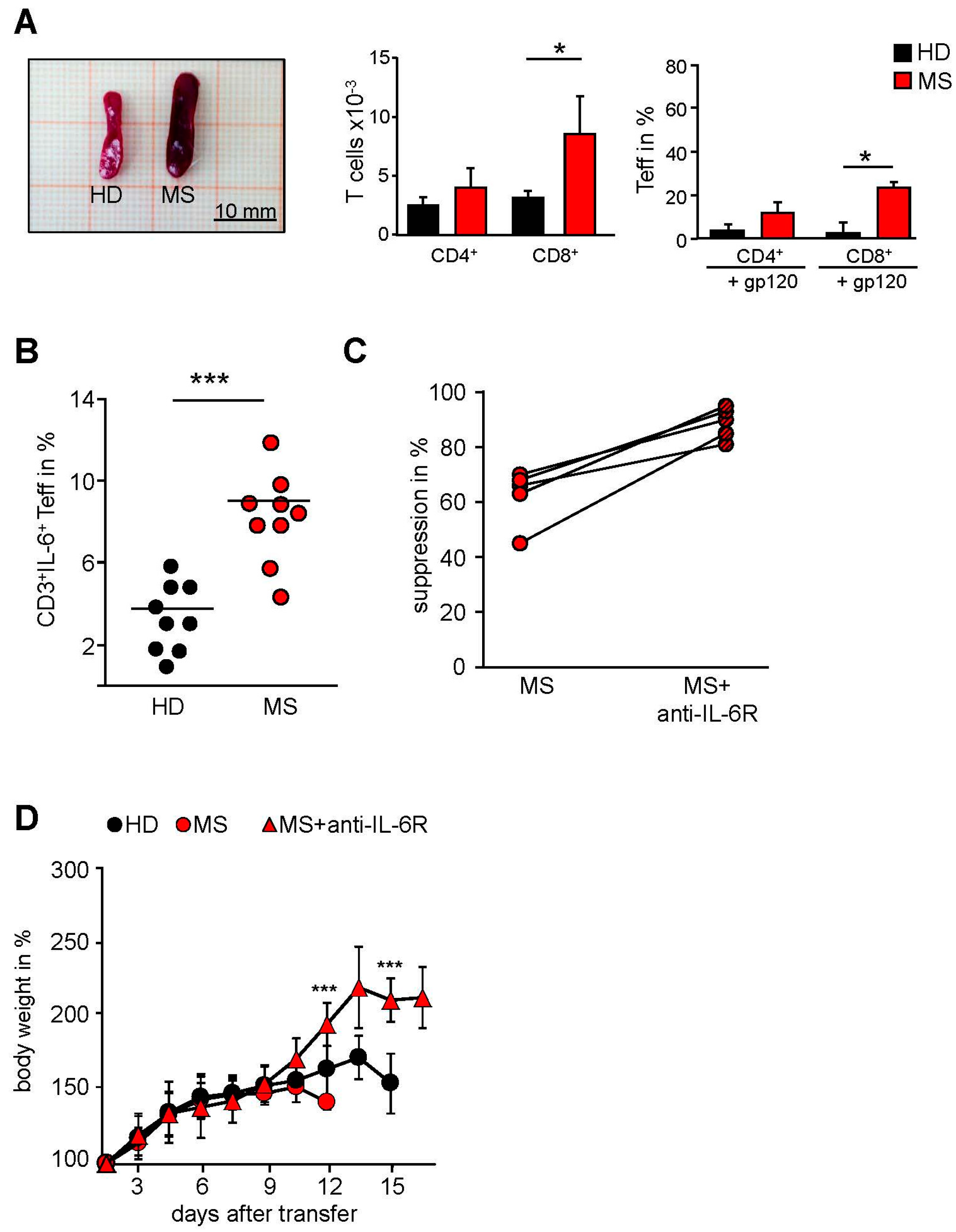
2.1. T Cells in the Peripheral Blood of MS Patients Are Treg-Resistant

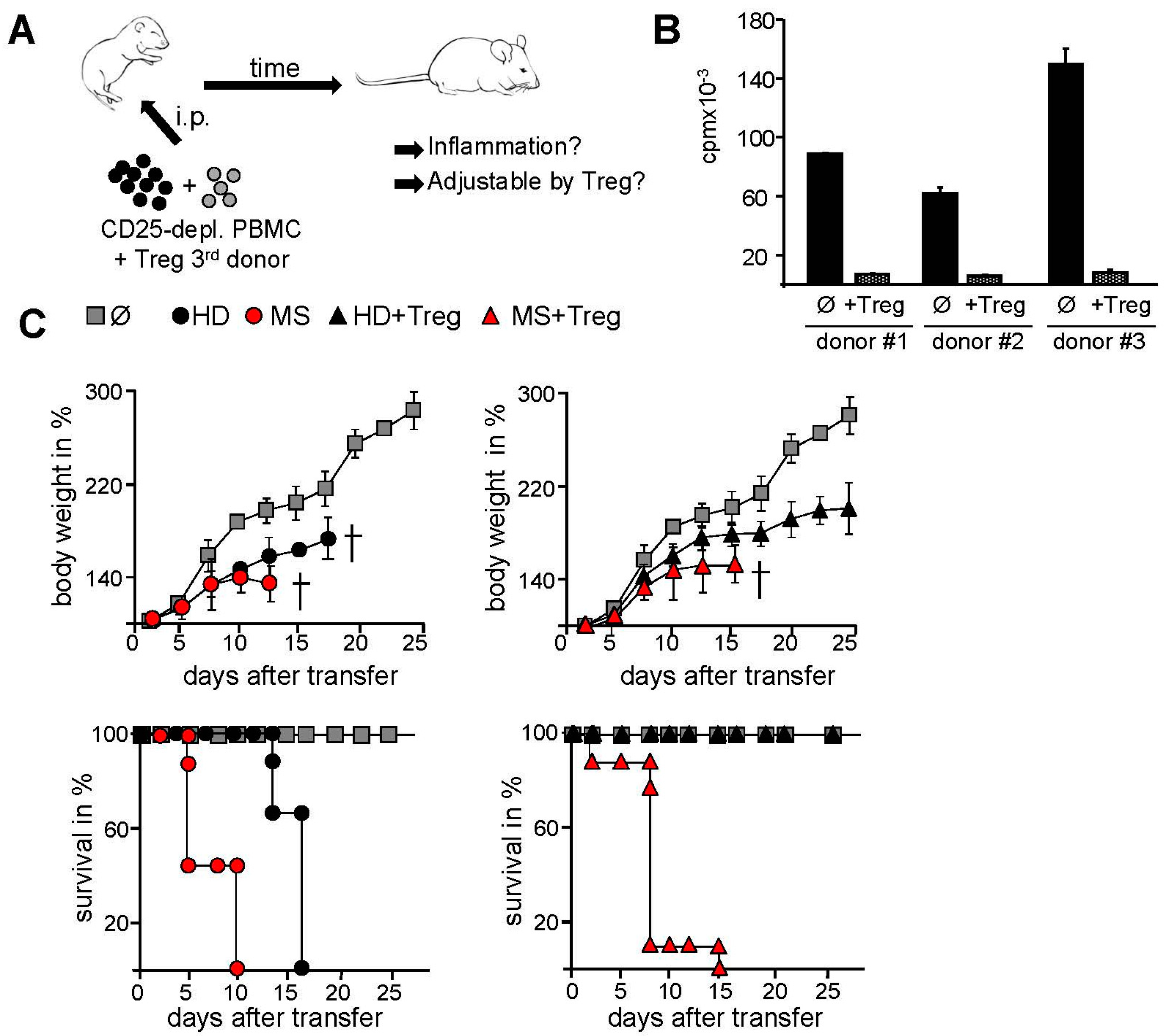
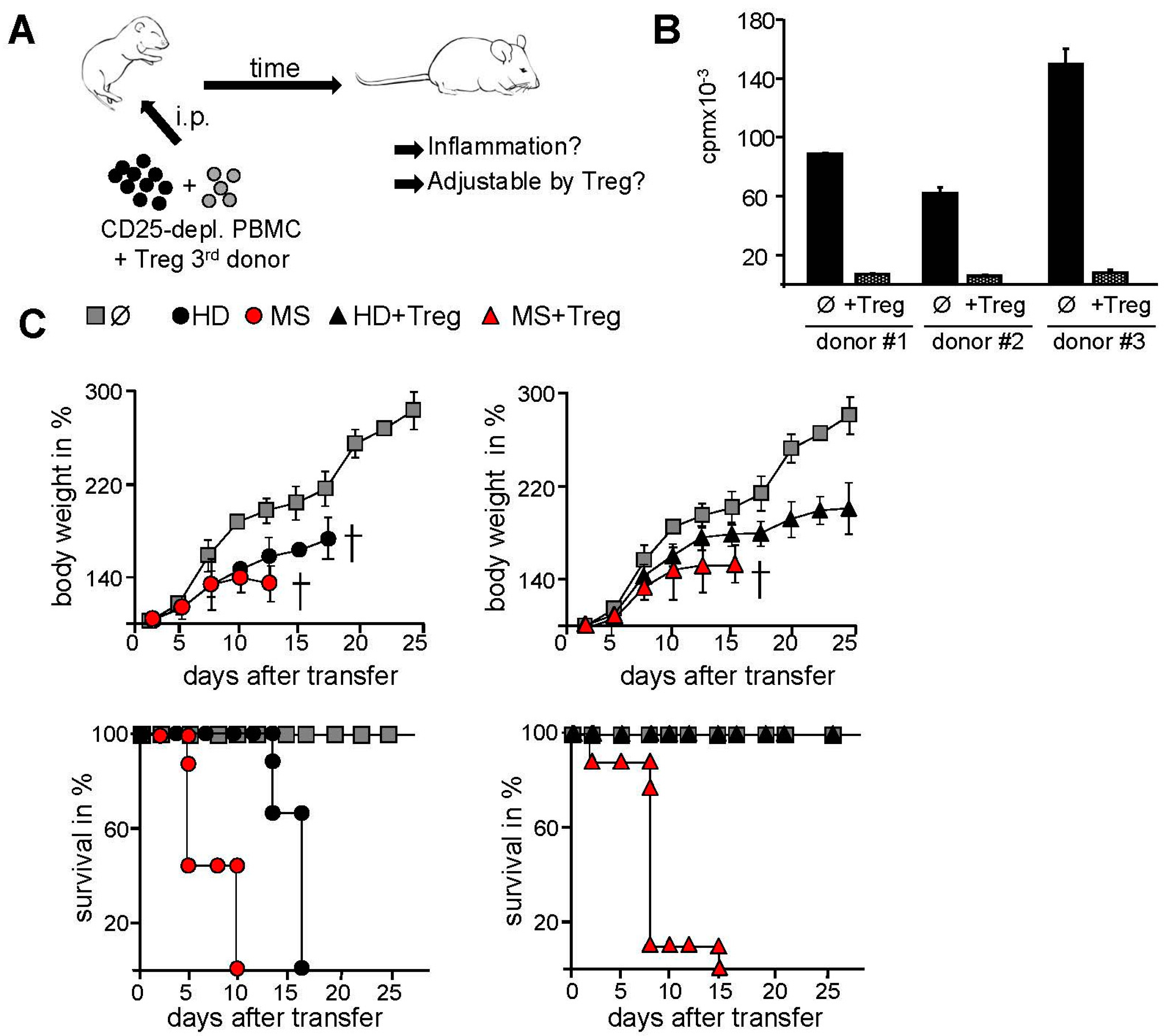
2.2. Transfer of PBMC from Therapy-Naive MS Patients into Immunodeficient Mice Resulted in an Accelerated Systemic Inflammation that Is Not Controlled by Activated Treg
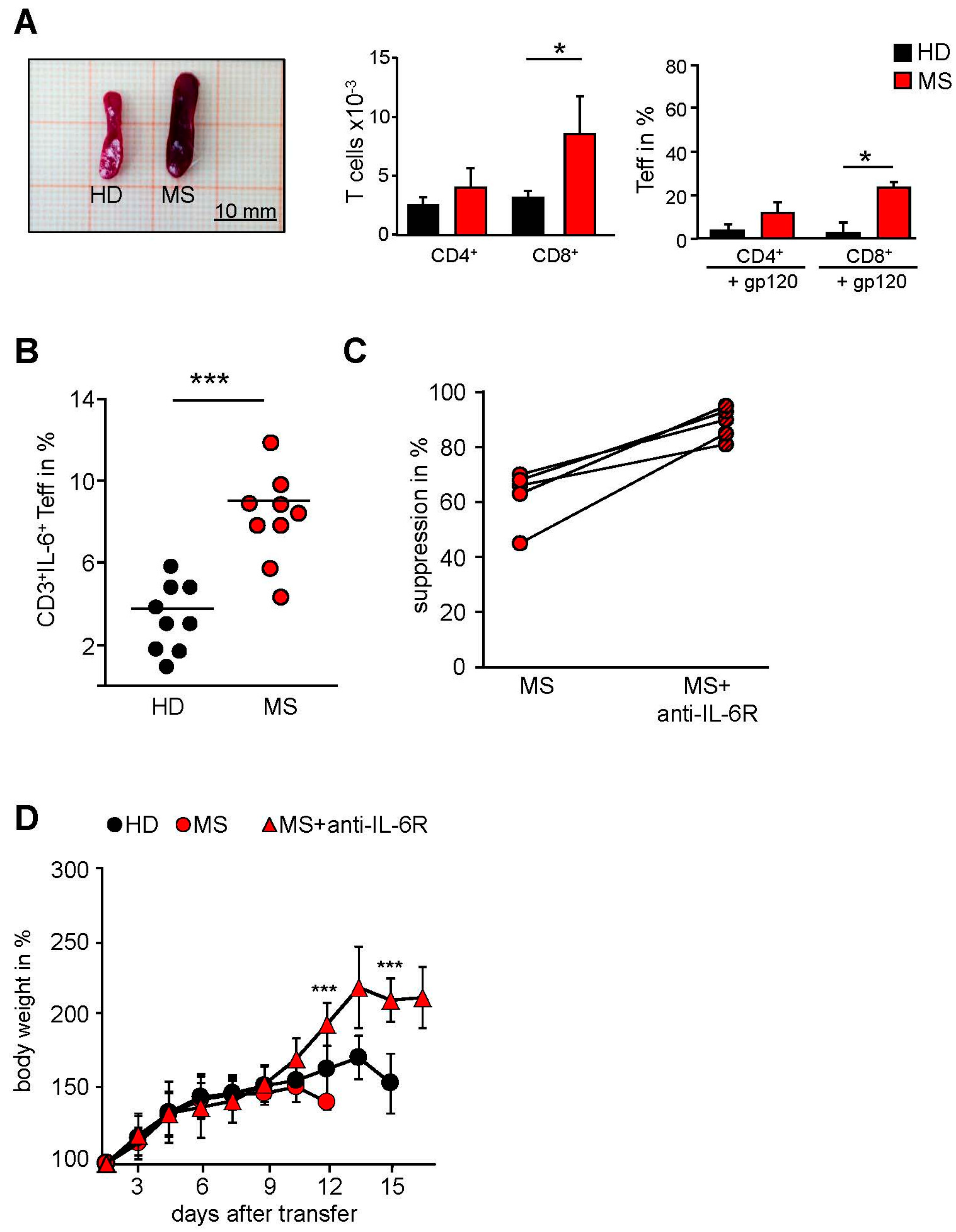
2.3. MS-Related Treg Resistance Was Mediated by IL-6
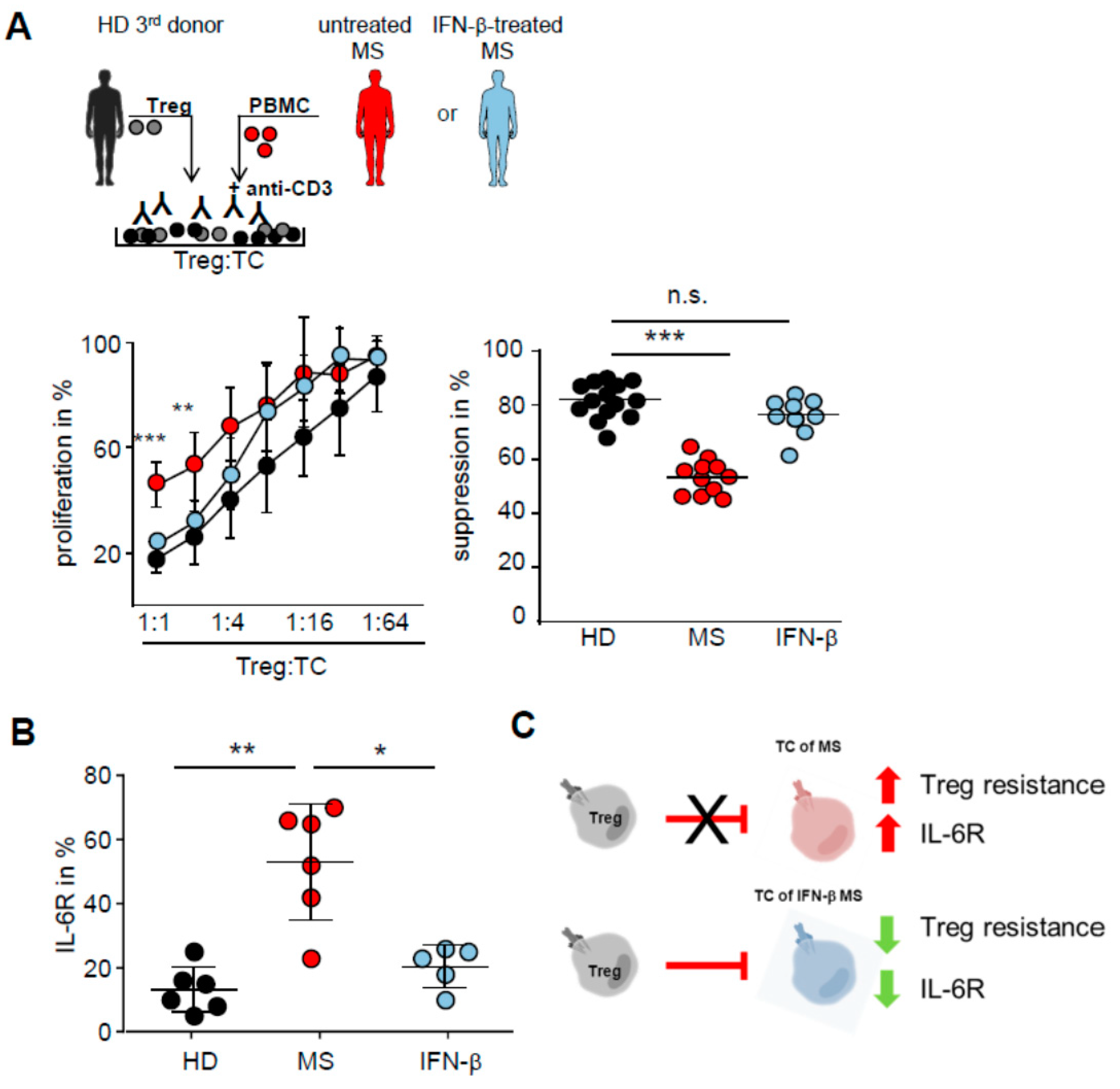
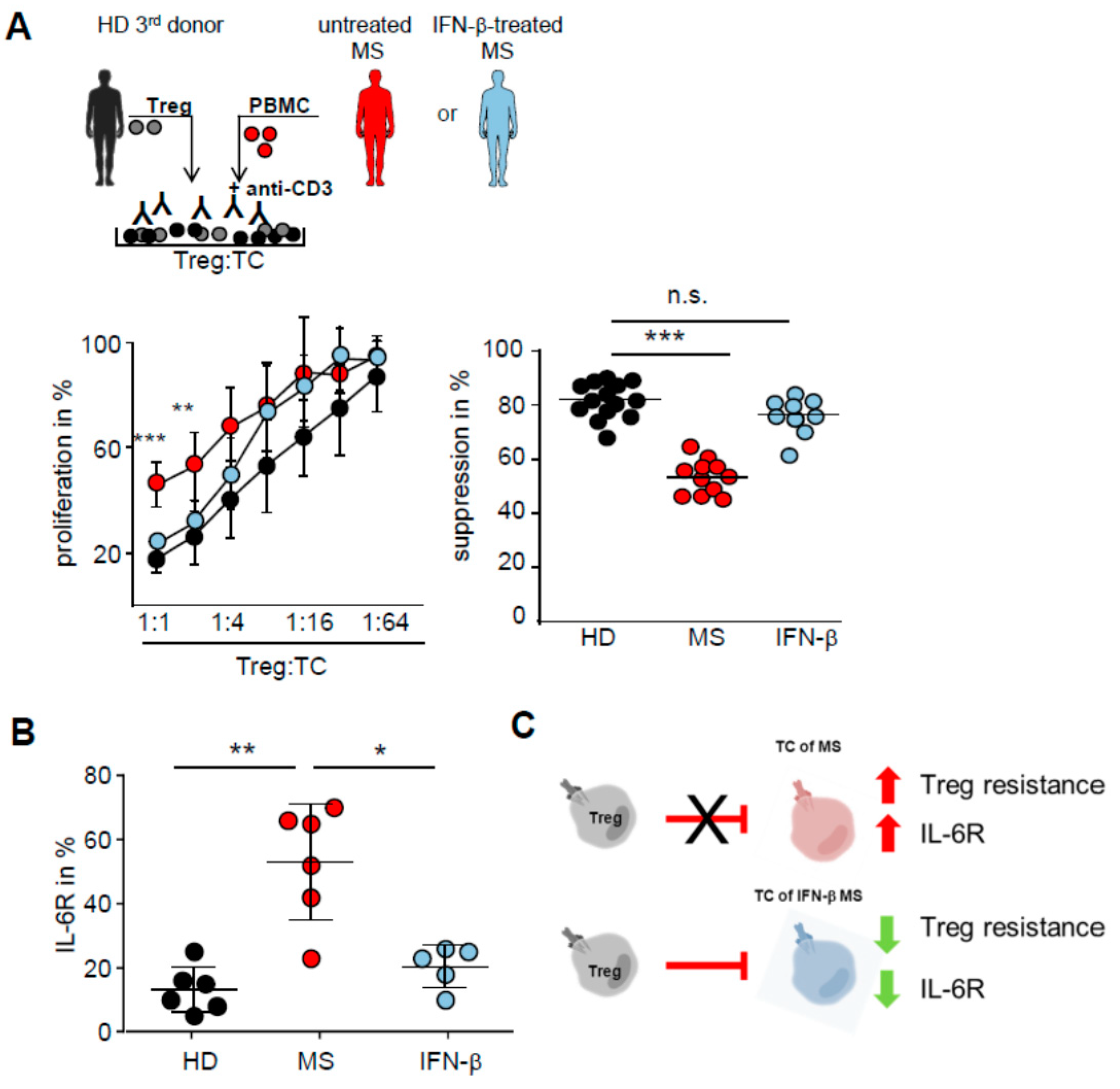
2.4. IFN-β Therapy Normalizes IL-6R Expression and Restores T Tell Responsiveness to Treg
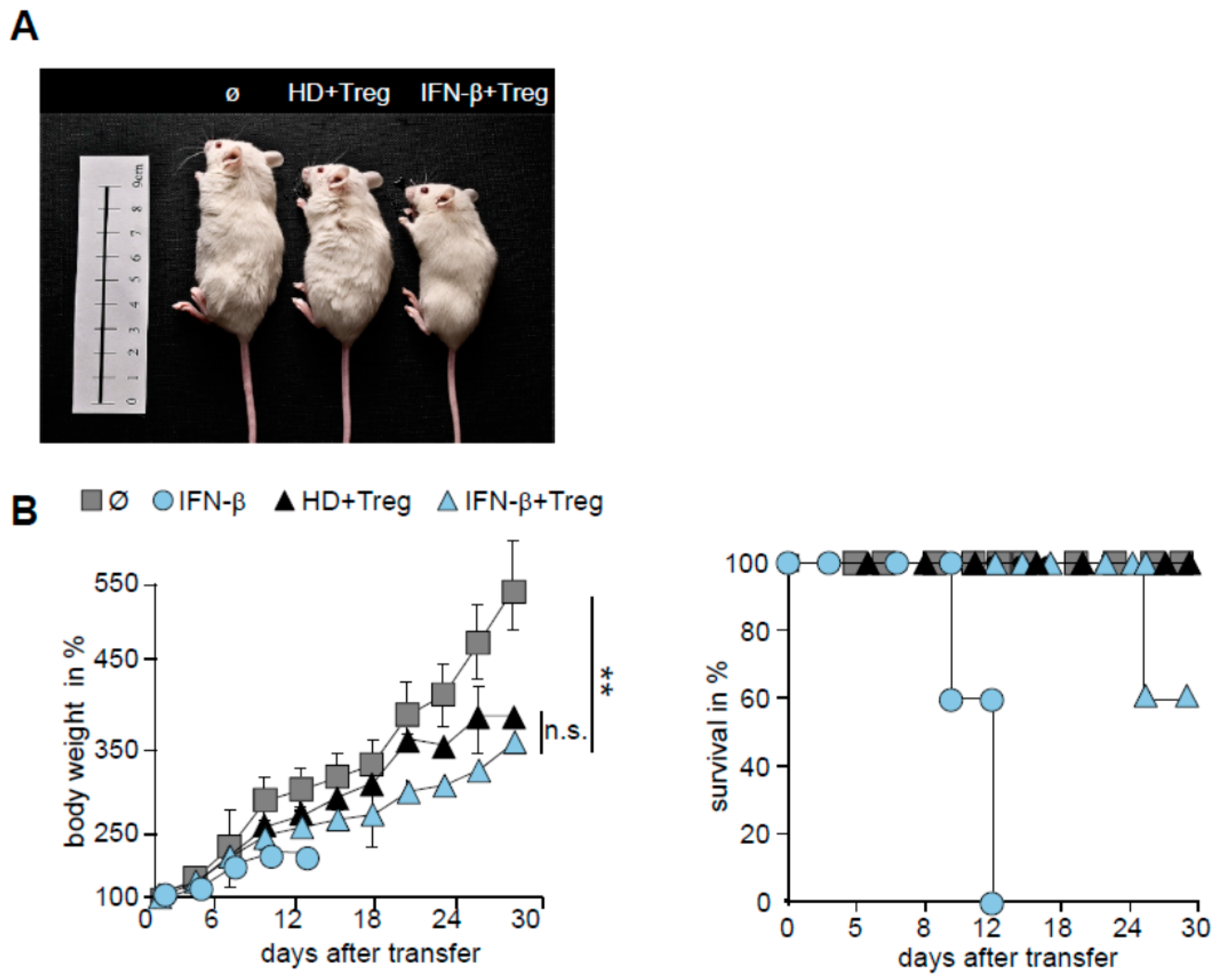
2.5. T Cells from IFN-β Treated MS Patients Showed an Improved Responsiveness for Immune Suppression in Vivo
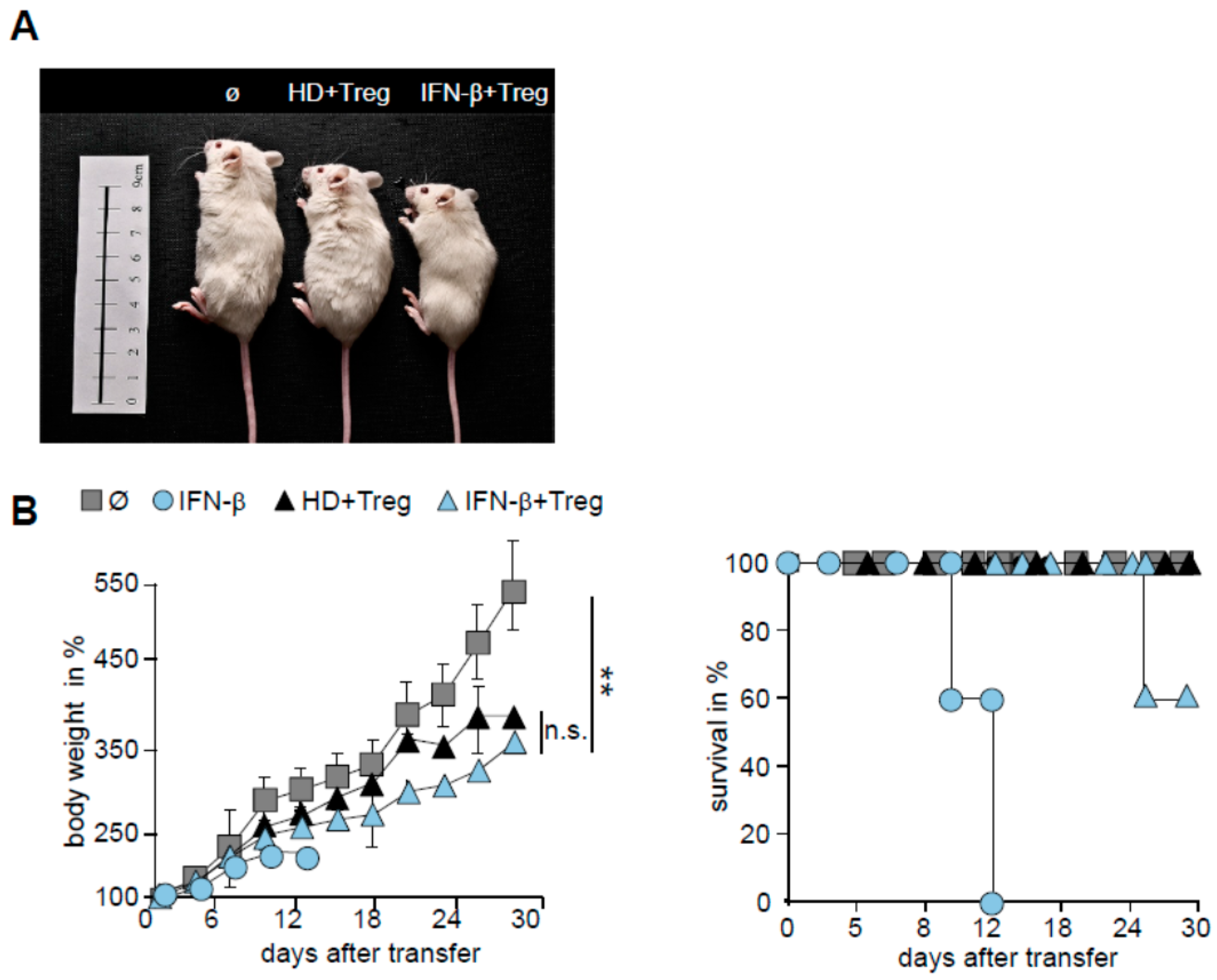
3. Discussion
4. Material and Methods
4.1. Patients and Healthy Controls
4.2. Transfer of Human Immune Cells
4.3. Culture Medium and Antibodies
4.4. Flow Cytometry
4.5. Isolation of T Cell Subsets
4.6. Cytokine Analysis
4.7. Suppressor Assays
4.8. Cell Isolation from Different Tissues of Humanized Mice and MLR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zozulya, A.L.; Wiendl, H. The role of regulatory T cells in multiple sclerosis. Nat. Clin. Pract. Neurol. 2008, 4, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Hafler, D.A. Suppressor t cells in human diseases. J. Exp. Med. 2004, 200, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Costantino, C.M.; Baecher-Allan, C.; Hafler, D.A. Multiple sclerosis and regulatory T cells. J. Clin. Immunol. 2008, 28, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Trinschek, B.; Luessi, F.; Haas, J.; Wildemann, B.; Zipp, F.; Wiendl, H.; Becker, C.; Jonuleit, H. Kinetics of il-6 production defines T effector cell responsiveness to regulatory T cells in multiple sclerosis. PLoS ONE 2013, 8, e77634. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Long, S.A.; Cerosaletti, K.; Ni, C.T.; Samuels, P.; Kita, M.; Buckner, J.H. In active relapsing-remitting multiple sclerosis, effector T cell resistance to adaptive T(regs) involves il-6-mediated signaling. Sci. Transl. Med. 2013, 5, 170ra115. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.D.; Keenan, E.; Porter, B.E.; Kapoor, R.; Rudge, P.; Thompson, A.J.; Miller, D.H.; Giovannoni, G. Interferon beta in multiple sclerosis: Experience in a British specialist multiple sclerosis centre. J. Neurol. Neurosurg. Psychiatry 2003, 74, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Walther, E.U.; Hohlfeld, R. Multiple sclerosis: Side effects of interferon beta therapy and their management. Neurology 1999, 53, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Palace, J.; Duddy, M.; Bregenzer, T.; Lawton, M.; Zhu, F.; Boggild, M.; Piske, B.; Robertson, N.P.; Oger, J.; Tremlett, H.; et al. Effectiveness and cost-effectiveness of interferon beta and glatiramer acetate in the UK multiple sclerosis risk sharing scheme at 6 years: A clinical cohort study with natural history comparator. Lancet Neurol. 2015, 14, 497–505. [Google Scholar] [CrossRef]
- Armuzzi, A.; Lionetti, P.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; Lapadula, G.; Marchesoni, A.; et al. Anti-tnf agents as therapeutic choice in immune-mediated inflammatory diseases: Focus on adalimumab. Int. J. Immunopathol. Pharmacol. 2014, 27, 11–32. [Google Scholar] [PubMed]
- Lapadula, G.; Marchesoni, A.; Armuzzi, A.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimaz, R.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; et al. Adalimumab in the treatment of immune-mediated diseases. Int. J. Immunopathol. Pharmacol. 2014, 27, 33–48. [Google Scholar] [PubMed]
- Burmester, G.R.; Feist, E.; Kellner, H.; Braun, J.; Iking-Konert, C.; Rubbert-Roth, A. Effectiveness and safety of the interleukin 6-receptor antagonist tocilizumab after 4 and 24 weeks in patients with active rheumatoid arthritis: The first phase IIIb real-life study (tamara). Ann. Rheum. Dis. 2011, 70, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of il-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Winthrop, K.L. Serious infections with antirheumatic therapy: Are biologicals worse? Ann. Rheum. Dis. 2006, 65, iii54–iii57. [Google Scholar] [CrossRef] [PubMed]
- Chari, D.M. Remyelination in multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [PubMed]
- Medina-Rodriguez, E.M.; Arenzana, F.J.; Pastor, J.; Redondo, M.; Palomo, V.; Garcia de Sola, R.; Gil, C.; Martinez, A.; Bribian, A.; de Castro, F. Inhibition of endogenous phosphodiesterase 7 promotes oligodendrocyte precursor differentiation and survival. Cell. Mol. Life Sci. 2013, 70, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Skripuletz, T.; Manzel, A.; Gropengiesser, K.; Schafer, N.; Gudi, V.; Singh, V.; Salinas Tejedor, L.; Jorg, S.; Hammer, A.; Voss, E.; et al. Pivotal role of choline metabolites in remyelination. Brain 2015, 138, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Wiles, M.V.; Greiner, D.L.; Shultz, L.D. Generation of improved humanized mouse models for human infectious diseases. J. Immunol. Methods 2014, 410, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Facciponte, J.; Jin, M.; Shen, Q.; Lin, Q. Humanized nod-scid il2rg–/– mice as a preclinical model for cancer research and its potential use for individualized cancer therapies. Cancer Lett. 2014, 344, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Croxford, A.L.; Kurschus, F.C.; Waisman, A. Mouse models for multiple sclerosis: Historical facts and future implications. Biochim. Biophys. Acta 2011, 1812, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Roep, B.O.; Buckner, J.; Sawcer, S.; Toes, R.; Zipp, F. The problems and promises of research into human immunology and autoimmune disease. Nat. Med. 2012, 18, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Jouvet, N.; Greiner, D.L.; Shultz, L.D. Humanized mice for the study of infectious diseases. Curr. Opin. Immunol. 2013, 25, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Jensen, L.T.; Willcox, N.; Fugger, L. Humanized mouse models for organ-specific autoimmune diseases. Curr. Opin. Immunol. 2006, 18, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Zayoud, M.; El Malki, K.; Frauenknecht, K.; Trinschek, B.; Kloos, L.; Karram, K.; Wanke, F.; Georgescu, J.; Hartwig, U.F.; Sommer, C.; et al. Subclinical CNS inflammation as response to a myelin antigen in humanized mice. J. Neuroimmune Pharmacol. 2013, 8, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Viglietta, V.; Baecher-Allan, C.; Weiner, H.L.; Hafler, D.A. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 2004, 199, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Taube, C.; Bopp, T.; Becker, C.; Michel, K.; Kubach, J.; Reuter, S.; Dehzad, N.; Neurath, M.F.; Reifenberg, K.; et al. Protection from graft-versus-host disease by hiv-1 envelope protein gp120-mediated activation of human cd4+cd25+ regulatory T cells. Blood 2009, 114, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.; Reuter, S.; Dehzad, N.; Heinz, A.; Bellinghausen, I.; Saloga, J.; Haasler, I.; Korn, S.; Jonuleit, H.; Buhl, R.; et al. Cd4-mediated regulatory T-cell activation inhibits the development of disease in a humanized mouse model of allergic airway disease. J. Allergy Clin. Immunol. 2012, 129, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Stahl, H.F.; Becker, C.; Correll, A.; Schneider, F.J.; Tuettenberg, A.; Jonuleit, H. Soluble garp has potent antiinflammatory and immunomodulatory impact on human cd4(+) T cells. Blood 2013, 122, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Bopp, T.; Jonuleit, H. Boosting regulatory T cell function by cd4 stimulation enters the clinic. Front. Immunol. 2012, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Kubach, J.; Wijdenes, J.; Knop, J.; Jonuleit, H. Cd4-mediated functional activation of human CD4+CD25+ regulatory T cells. Eur. J. Immunol. 2007, 37, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Kocur, M.; Schneider, R.; Pulm, A.K.; Bauer, J.; Kropp, S.; Gliem, M.; Ingwersen, J.; Goebels, N.; Alferink, J.; Prozorovski, T.; et al. Ifnbeta secreted by microglia mediates clearance of myelin debris in cns autoimmunity. Acta Neuropathol. Commun. 2015, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Bermel, R.A.; Rudick, R.A. Interferon-beta treatment for multiple sclerosis. Neurotherapeutics 2007, 4, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, X.; Podolsky, R.; Hopkins, D.; Makala, L.H.; Muir, A.; She, J.X. Apc dysfunction is correlated with defective suppression of T cell proliferation in human type 1 diabetes. Clin. Immunol. 2009, 130, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, E.J.; Mijnheer, G.; Duurland, C.L.; Klein, M.; Meerding, J.; van Loosdregt, J.; de Jager, W.; Sawitzki, B.; Coffer, P.J.; Vastert, B.; et al. Functional human regulatory T cells fail to control autoimmune inflammation due to pkb/c-akt hyperactivation in effector cells. Blood 2011, 118, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Goodman, W.A.; Cooper, K.D.; McCormick, T.S. Regulation generation: The suppressive functions of human regulatory T cells. Crit. Rev. Immunol. 2012, 32, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Fritzsching, B.; Trubswetter, P.; Korporal, M.; Milkova, L.; Fritz, B.; Vobis, D.; Krammer, P.H.; Suri-Payer, E.; Wildemann, B. Prevalence of newly generated naive regulatory T cells (treg) is critical for treg suppressive function and determines treg dysfunction in multiple sclerosis. J. Immunol. 2007, 179, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Viglietta, V.; Hafler, D.A. Human CD4+CD25+ regulatory T cells. Semin. Immunol. 2004, 16, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.; Hellings, N.; Hensen, K.; Rummens, J.L.; Medaer, R.; D'hooghe, M.B.; Dubois, B.; Raus, J.; Stinissen, P. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4+CD25+ regulatory t-cell function and foxp3 expression. J. Neurosci. Res. 2006, 83, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Jolivel, V.; Luessi, F.; Masri, J.; Kraus, S.H.; Hubo, M.; Poisa-Beiro, L.; Klebow, S.; Paterka, M.; Yogev, N.; Tumani, H.; et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain 2013, 136, 1048–1066. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Haak, S.; Sisirak, V.; Reizis, B. The role of dendritic cells in autoimmunity. Nat. Rev. Immunol. 2013, 13, 566–577. [Google Scholar] [CrossRef] [PubMed]
- De Andres, C.; Aristimuno, C.; de, L.H., V.; Martinez-Gines, M.L.; Bartolome, M.; Arroyo, R.; Navarro, J.; Gimenez-Roldan, S.; Fernandez-Cruz, E.; Sanchez-Ramon, S. Interferon beta-1a therapy enhances CD4+ regulatory T-cell function: An ex vivo and in vitro longitudinal study in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2007, 182, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Paty, D.W.; Li, D.K. Interferon beta-lb is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 2001, 57, S10–S15. [Google Scholar] [PubMed]
- Korporal, M.; Haas, J.; Balint, B.; Fritzsching, B.; Schwarz, A.; Moeller, S.; Fritz, B.; Suri-Payer, E.; Wildemann, B. Interferon beta-induced restoration of regulatory T-cell function in multiple sclerosis is prompted by an increase in newly generated naive regulatory T cells. Arch. Neurol. 2008, 65, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of il-6-producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Toscas, A.; Jensen, M.A. Interferon beta decreases T cell activation and interferon gamma production in multiple sclerosis. J. Neuroimmunol. 1993, 46, 145–153. [Google Scholar] [CrossRef]
- Rudick, R.A.; Carpenter, C.S.; Cookfair, D.L.; Tuohy, V.K.; Ransohoff, R.M. In vitro and in vivo inhibition of mitogen-driven T-cell activation by recombinant interferon beta. Neurology 1993, 43, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. Il-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Malpass, K. Multiple sclerosis: T-cell resistance to regulation in rrms linked to il-6 pathway. Nat. Rev. Neurol. 2013, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Neurath, M.F. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C.; Stuve, O.; Dehmel, T.; Goebels, N.; Leussink, V.I.; Mausberg, A.K.; Ringelstein, M.; Turowski, B.; Aktas, O.; Antoch, G.; et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: Implication for cellular immune responses. JAMA Neurol. 2013, 70, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Baji, P.; Pentek, M.; Czirjak, L.; Szekanecz, Z.; Nagy, G.; Gulacsi, L.; Brodszky, V. Efficacy and safety of infliximab-biosimilar compared to other biological drugs in rheumatoid arthritis: A mixed treatment comparison. Eur. J. Health Econ. 2014, 15, S53–S64. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Tocilizumab: A review of its use in the treatment of juvenile idiopathic arthritis. Paediatr. Drugs 2013, 15, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting eae in the sjl/j mouse: Mog-reactive transgenic T cells recruit endogenous mog-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A., Jr. Surface expression of alpha 4 integrin by cd4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Wekerle, H.; Flugel, A.; Fugger, L.; Schett, G.; Serreze, D. Autoimmunity’s next top models. Nat. Med. 2012, 18, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Wiendl, H.; Hohlfeld, R. Therapeutic approaches in multiple sclerosis: Lessons from failed and interrupted treatment trials. BioDrugs 2002, 16, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Macchiarini, F.; Manz, M.G.; Palucka, A.K.; Shultz, L.D. Humanized mice: Are we there yet? J. Exp. Med. 2005, 202, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Kubach, J.; Hubo, M.; Amendt, C.; Stroh, C.; Jonuleit, H. IgG1 anti-epidermal growth factor receptor antibodies induce cd8-dependent antitumor activity. Int. J. Cancer 2015, 136, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. Nod/scid/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Lowry, P.A.; Shultz, L.D.; Greiner, D.L.; Hesselton, R.M.; Kittler, E.L.; Tiarks, C.Y.; Rao, S.S.; Reilly, J.; Leif, J.H.; Ramshaw, H.; et al. Improved engraftment of human cord blood stem cells in nod/ltsz-scid/scid mice after irradiation or multiple-day injections into unirradiated recipients. Biol. Blood Marrow Transplant. 1996, 2, 15–23. [Google Scholar] [PubMed]
- Jonuleit, H.; Schmitt, E.; Stassen, M.; Tuettenberg, A.; Knop, J.; Enk, A.H. Identification and functional characterization of human Cd4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J. Exp. Med. 2001, 193, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinschek, B.; Luessi, F.; Gross, C.C.; Wiendl, H.; Jonuleit, H. Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells. Int. J. Mol. Sci. 2015, 16, 16330-16346. https://doi.org/10.3390/ijms160716330
Trinschek B, Luessi F, Gross CC, Wiendl H, Jonuleit H. Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells. International Journal of Molecular Sciences. 2015; 16(7):16330-16346. https://doi.org/10.3390/ijms160716330
Chicago/Turabian StyleTrinschek, Bettina, Felix Luessi, Catharina C. Gross, Heinz Wiendl, and Helmut Jonuleit. 2015. "Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells" International Journal of Molecular Sciences 16, no. 7: 16330-16346. https://doi.org/10.3390/ijms160716330
APA StyleTrinschek, B., Luessi, F., Gross, C. C., Wiendl, H., & Jonuleit, H. (2015). Interferon-Beta Therapy of Multiple Sclerosis Patients Improves the Responsiveness of T Cells for Immune Suppression by Regulatory T Cells. International Journal of Molecular Sciences, 16(7), 16330-16346. https://doi.org/10.3390/ijms160716330