
Impaired Focal Adhesion Kinase-Grb2 Interaction during Elevated Activity in Hippocampal Neurons

Abstract
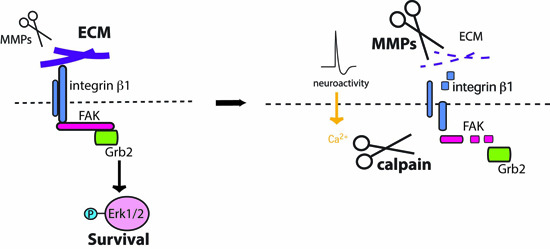
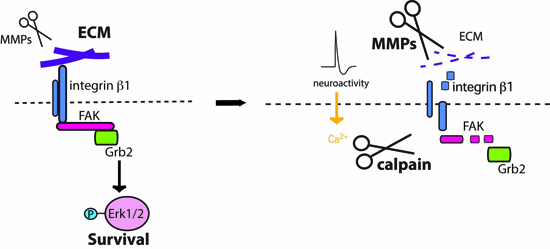
:
{kind=link}
{kind=link}
{kind=link}
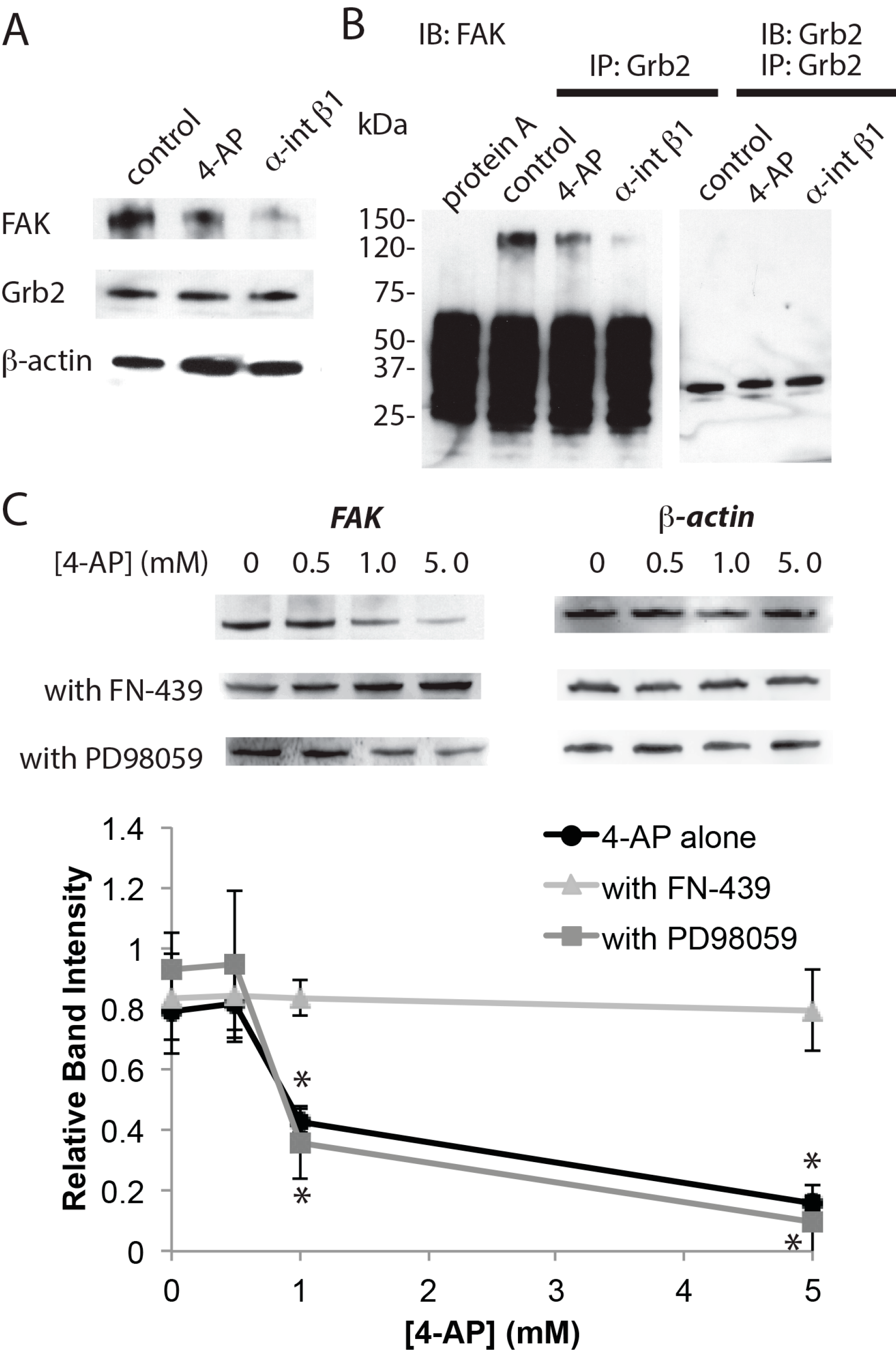{kind=link}
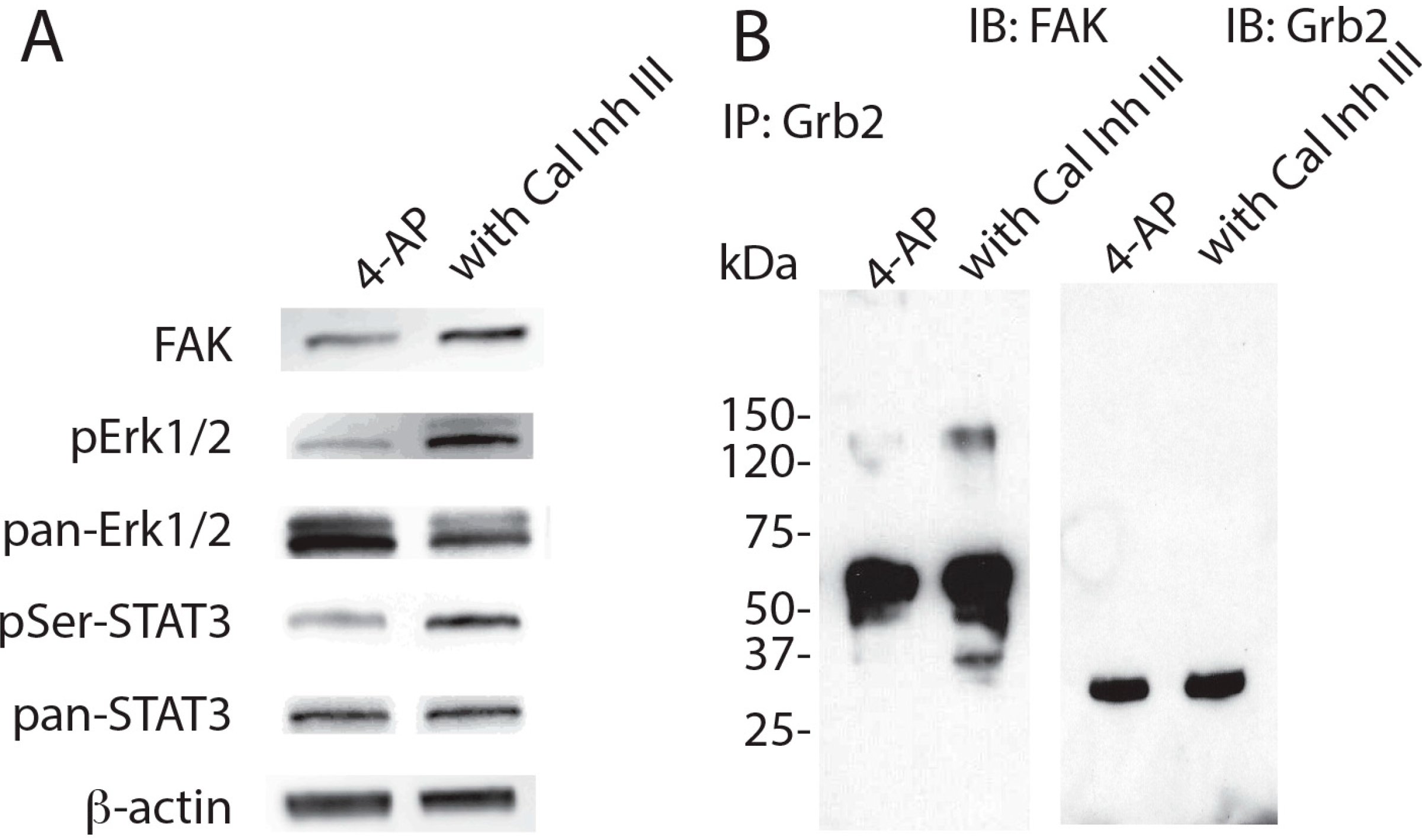{kind=link}
1. Introduction
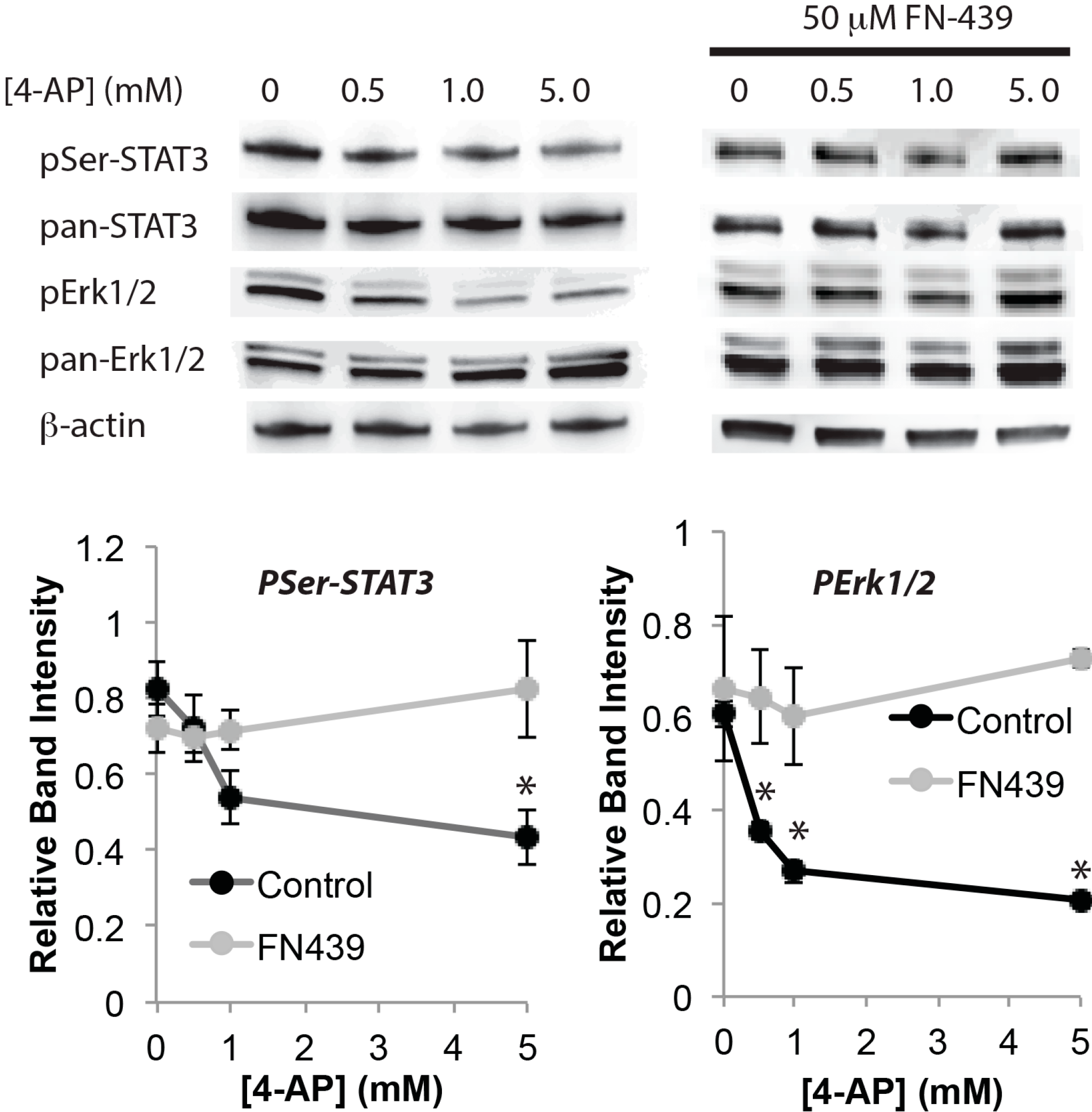
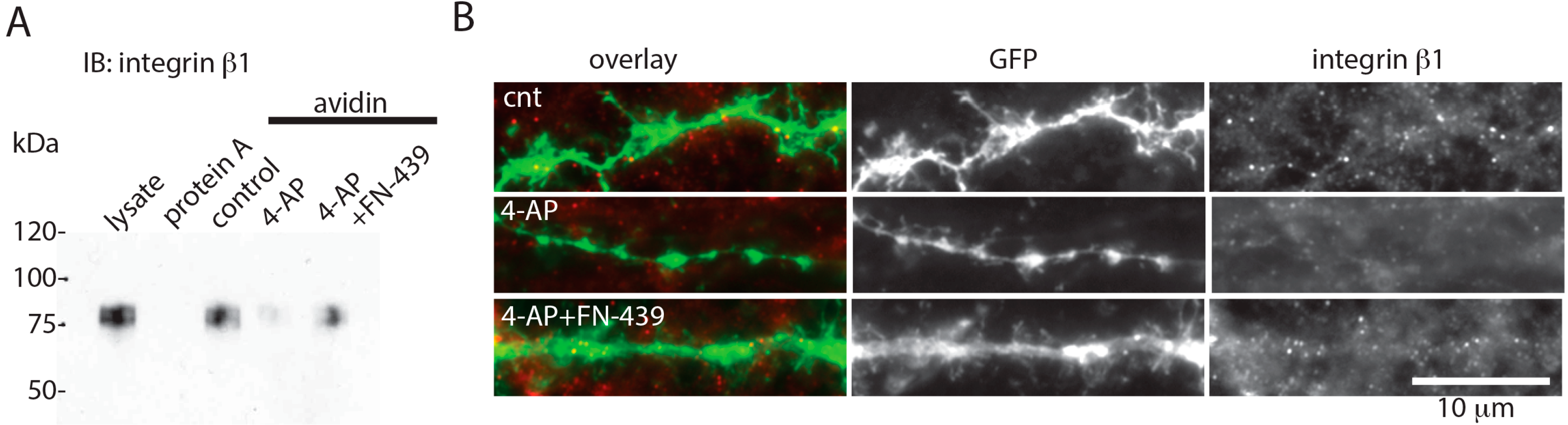
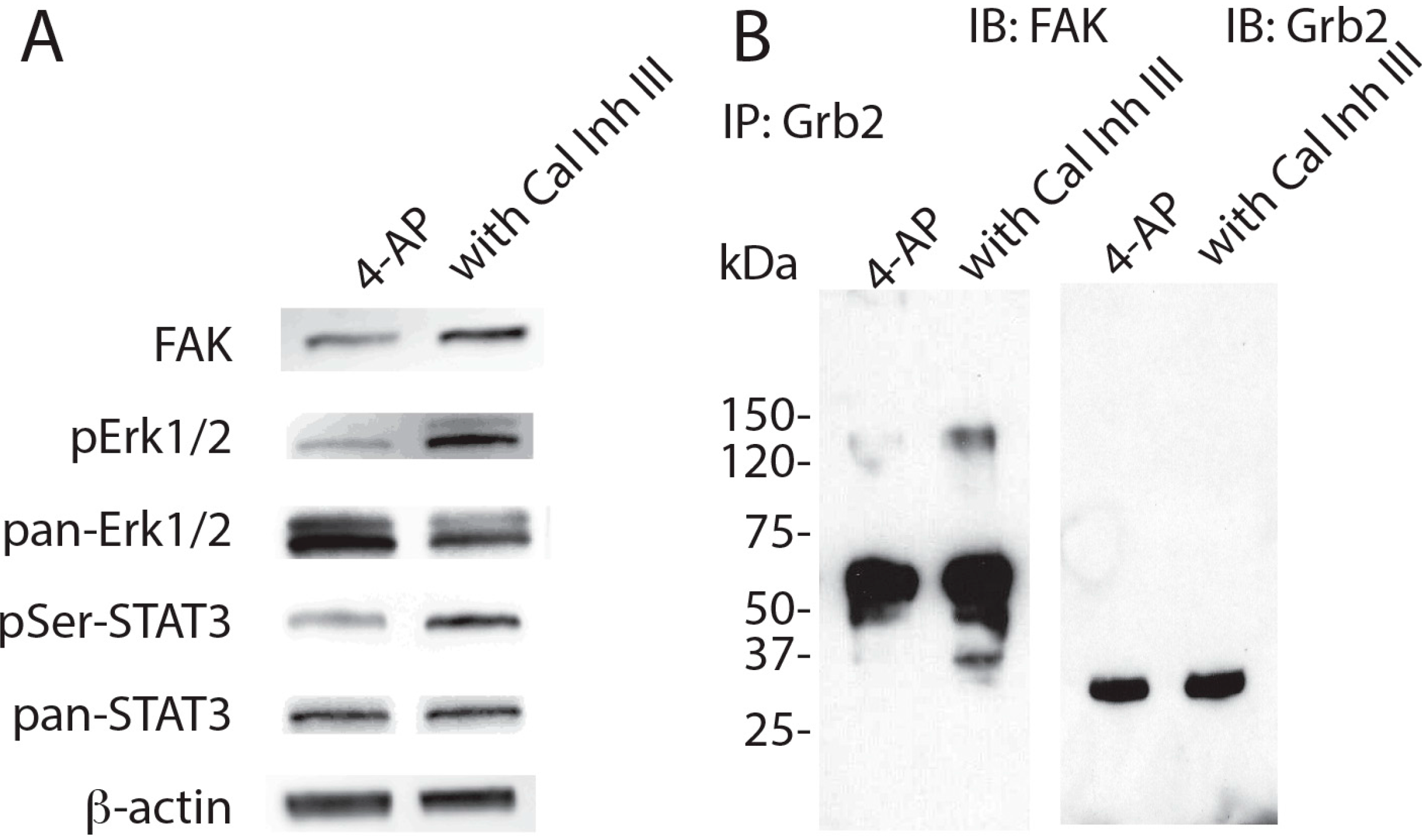
2. Results
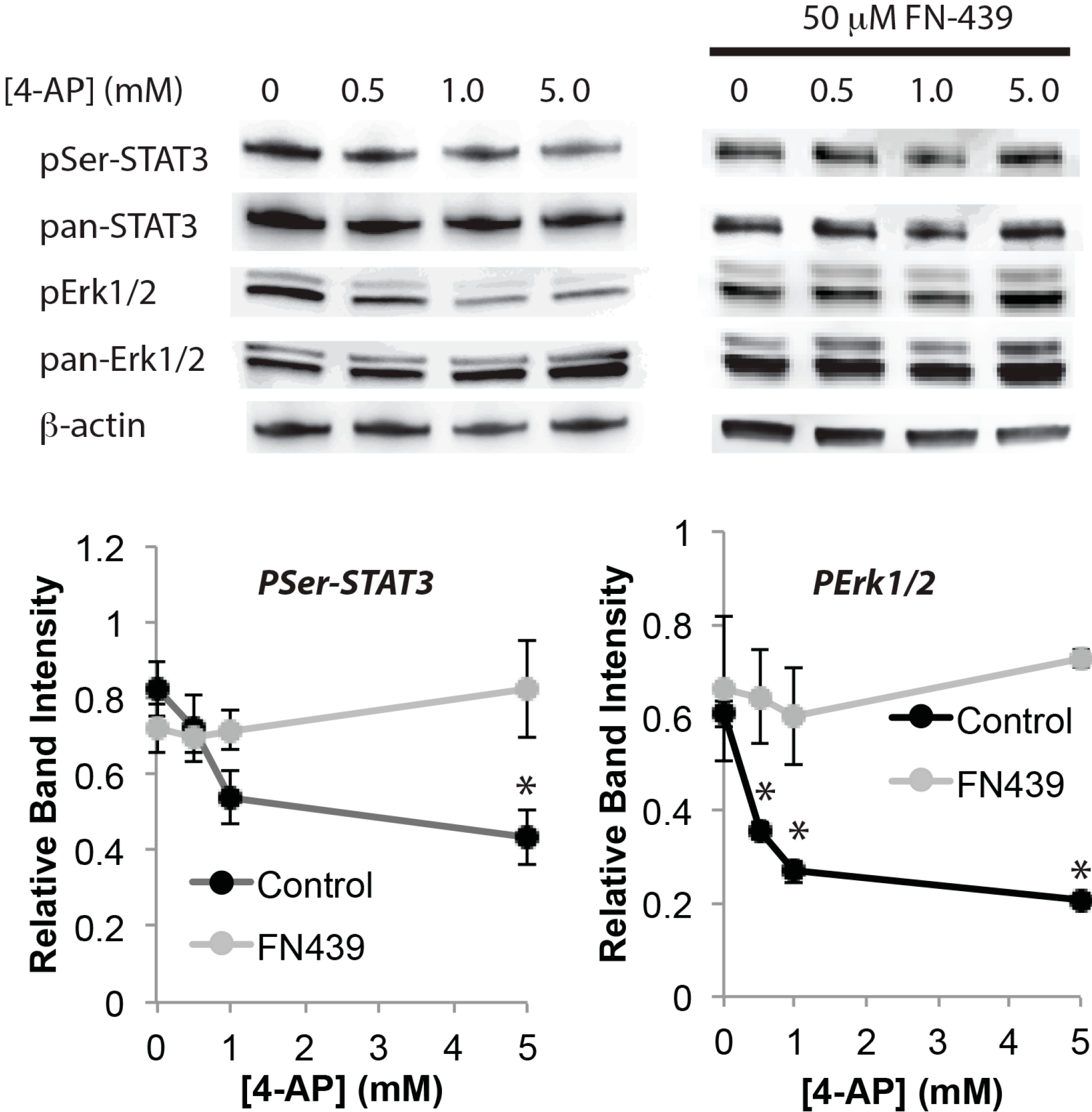
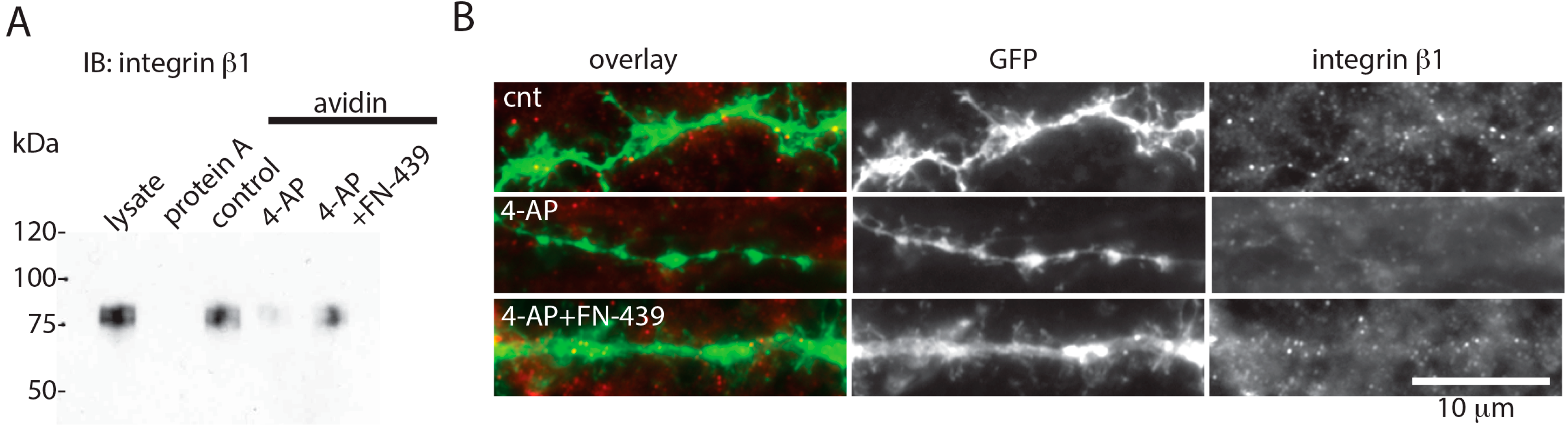

3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Antibodies
4.3. Dissociated Primary Hippocampal Culture
4.4. Transfection
4.5. Western Blot
4.6. Immunocytochemistry
4.7. Cell Surface Biotinylation
4.8. Immuno-Precipitation Assay
4.9. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davis, G.W. Homeostatic control of neural activity: From phenomenology to molecular design. Annu. Rev. Neurosci. 2006, 29, 307–323. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G. Homeostatic signaling: The positive side of negative feedback. Curr. Opin. Neurobiol. 2007, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, U. Basic mechanisms of partial epilepsies. Curr. Opin. Neurol. 2004, 17, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Belforte, J.E.; Zsiros, V.; Sklar, E.R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E.M.; Nakazawa, K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2009, 13, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr. Opin. Neurol. 2010, 23, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.A.; Fried, I.; Watanabe, K.; Forsythe, A.B.; Scheibel, A.B. Structural correlates of seizure behavior in the mongolian gerbil. Science 1981, 213, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Garey, L.J.; Ong, W.Y.; Patel, T.S.; Kanani, M.; Davis, A.; Mortimer, A.M.; Barnes, T.R.; Hirsch, S.R. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 1998, 65, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Boda, B.; Dubos, A.; Muller, D. Signaling mechanisms regulating synapse formation and function in mental retardation. Curr. Opin. Neurobiol. 2010, 20, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Murase, S.; Kim, E.; Lin, L.; Hoffman, D.A.; McKay, R.D. Loss of signal transducer and activator of transcription 3 (STAT3) signaling during elevated activity causes vulnerability in hippocampal neurons. J. Neurosci. 2012, 32, 15511–15520. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Lukashev, M.E.; Werb, Z. ECM signalling: Orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998, 8, 437–441. [Google Scholar] [CrossRef]
- Werb, Z. ECM and cell surface proteolysis: Regulating cellular ecology. Cell 1997, 91, 439–442. [Google Scholar] [CrossRef]
- Burridge, K.; Fath, K.; Kelly, T.; Nuckolls, G.; Turner, C. Focal adhesions: Transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 1988, 4, 487–525. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hanks, S.K.; Hunter, T.; van der Geer, P. Integrin-mediated signal transduction linked to Ras pathway by Grb2 binding to focal adhesion kinase. Nature 1994, 372, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Bozdagi, O.; Matynia, A.; Balcerzyk, M.; Okulski, P.; Dzwonek, J.; Costa, R.M.; Silva, A.J.; Kaczmarek, L.; Huntley, G.W. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci. 2006, 26, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Spolidoro, M.; Putignano, E.; Munafo, C.; Maffei, L.; Pizzorusso, T. Inhibition of matrix metalloproteinases prevents the potentiation of nondeprived-eye responses after monocular deprivation in juvenile rats. Cereb. Cortex 2012, 22, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Kaliszewska, A.; Bijata, M.; Kaczmarek, L.; Kossut, M. Experience-dependent plasticity of the barrel cortex in mice observed with 2-DG brain mapping and c-Fos: Effects of MMP-9 KO. Cereb. Cortex 2012, 22, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, A.; Lapinska, J.; Rylski, M.; McKay, R.D.; Kaczmarek, L. Matrix metalloproteinase-9 undergoes expression and activation during dendritic remodeling in adult hippocampus. J. Neurosci. 2002, 22, 920–930. [Google Scholar] [PubMed]
- Wilczynski, G.M.; Konopacki, F.A.; Wilczek, E.; Lasiecka, Z.; Gorlewicz, A.; Michaluk, P.; Wawrzyniak, M.; Malinowska, M.; Okulski, P.; Kolodziej, L.R.; et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol. 2008, 180, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Kupchik, Y.M.; Scofield, M.D.; Gipson, C.D.; Wiggins, A.; Thomas, C.A.; Kalivas, P.W. Synaptic plasticity mediating cocaine relapse requires matrix metalloproteinases. Nat. Neurosci. 2014, 17, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Huntley, G.W. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 2012, 13, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Murase, S.; McKay, R.D. Matrix metalloproteinase-9 regulates survival of neurons in newborn hippocampus. J. Biol. Chem. 2012, 287, 12184–12194. [Google Scholar] [CrossRef] [PubMed]
- Yarmola, E.G.; Somasundaram, T.; Boring, T.A.; Spector, I.; Bubb, M.R. Actin-latrunculin A structure and function. Differential modulation of actin-binding protein function by latrunculin A. J. Biol. Chem. 2000, 275, 28120–28127. [Google Scholar] [PubMed]
- Murachi, T.; Tanaka, K.; Hatanaka, M.; Murakami, T. Intracellular Ca2+-dependent protease (calpain) and its high-molecular-weight endogenous inhibitor (calpastatin). Adv. Enzym. Regul. 1980, 19, 407–424. [Google Scholar] [CrossRef]
- Carragher, N.O.; Levkau, B.; Ross, R.; Raines, E.W. Degraded collagen fragments promote rapid disassembly of smooth muscle focal adhesions that correlates with cleavage of pp125FAK, paxillin, and talin. J. Cell Biol. 1999, 147, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Rozakis-Adcock, M.; McGlade, J.; Mbamalu, G.; Pelicci, G.; Daly, R.; Li, W.; Batzer, A.; Thomas, S.; Brugge, J.; Pelicci, P.G.; et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 1992, 360, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein Grb2 links receptor tyrosine kinases to Ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Kryczka, J.; Stasiak, M.; Dziki, L.; Mik, M.; Dziki, A.; Cierniewski, C. Matrix metalloproteinase-2 cleavage of the β1 integrin ectodomain facilitates colon cancer cell motility. J. Biol. Chem. 2012, 287, 36556–36566. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, L.A.; Thalhammer, A.; Yu, L.M.; Catalano, M.; Ramos, T.; Colicos, M.A.; Goda, Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by β3 integrins. Neuron 2008, 58, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Staubli, U.; Chun, D.; Lynch, G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J. Neurosci. 1998, 18, 3460–3469. [Google Scholar] [PubMed]
- Chan, C.S.; Weeber, E.J.; Kurup, S.; Sweatt, J.D.; Davis, R.L. Integrin requirement for hippocampal synaptic plasticity and spatial memory. J. Neurosci. 2003, 23, 7107–7116. [Google Scholar] [PubMed]
- Murase, S.; Owens, D.F.; McKay, R.D. In the newborn hippocampus, neurotrophin-dependent survival requires spontaneous activity and integrin signaling. J. Neurosci. 2011, 31, 7791–7800. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.P.; Fahrni, J.A.; Troie, S.; Guan, J.L.; Orth, K.; Rosen, G.D. Cleavage of focal adhesion kinase by caspases during apoptosis. J. Biol. Chem. 1997, 272, 26056–26061. [Google Scholar] [CrossRef] [PubMed]
- Cicala, C.; Arthos, J.; Rubbert, A.; Selig, S.; Wildt, K.; Cohen, O.J.; Fauci, A.S. HIV-1 envelope induces activation of caspase-3 and cleavage of focal adhesion kinase in primary human CD4+ T cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Cooray, P.; Yuan, Y.; Schoenwaelder, S.M.; Mitchell, C.A.; Salem, H.H.; Jackson, S.P. Focal adhesion kinase (pp125FAK) cleavage and regulation by calpain. Biochem. J. 1996, 318, 41–47. [Google Scholar] [PubMed]
- Zadran, S.; Jourdi, H.; Rostamiani, K.; Qin, Q.; Bi, X.; Baudry, M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 2010, 30, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.T.; Bennin, D.A.; Huttenlocher, A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 2010, 285, 11418–11426. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Saleem, A.; Yuan, Z.; Emoto, Y.; Prasad, K.V.; Kufe, D. Stimulation of human monocytes with macrophage colony-stimulating factor induces a Grb2-mediated association of the focal adhesion kinase pp125FAK and dynamin. Proc. Natl. Acad. Sci. USA 1995, 92, 6132–6136. [Google Scholar] [CrossRef] [PubMed]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E. The structural basis of localization and signaling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Fujikawa, D.G. Prolonged seizures and cellular injury: Understanding the connection. Epilepsy Behav. 2005, 7, S3–S11. [Google Scholar] [CrossRef] [PubMed]
- Murase, S.; McKay, R.D. A specific survival response in dopamine neurons at most risk in Parkinson’s disease. J. Neurosci. 2006, 26, 9750–9760. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murase, S. Impaired Focal Adhesion Kinase-Grb2 Interaction during Elevated Activity in Hippocampal Neurons. Int. J. Mol. Sci. 2015, 16, 15659-15669. https://doi.org/10.3390/ijms160715659
Murase S. Impaired Focal Adhesion Kinase-Grb2 Interaction during Elevated Activity in Hippocampal Neurons. International Journal of Molecular Sciences. 2015; 16(7):15659-15669. https://doi.org/10.3390/ijms160715659
Chicago/Turabian StyleMurase, Sachiko. 2015. "Impaired Focal Adhesion Kinase-Grb2 Interaction during Elevated Activity in Hippocampal Neurons" International Journal of Molecular Sciences 16, no. 7: 15659-15669. https://doi.org/10.3390/ijms160715659
APA StyleMurase, S. (2015). Impaired Focal Adhesion Kinase-Grb2 Interaction during Elevated Activity in Hippocampal Neurons. International Journal of Molecular Sciences, 16(7), 15659-15669. https://doi.org/10.3390/ijms160715659