
Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach


Abstract
:1. Introduction
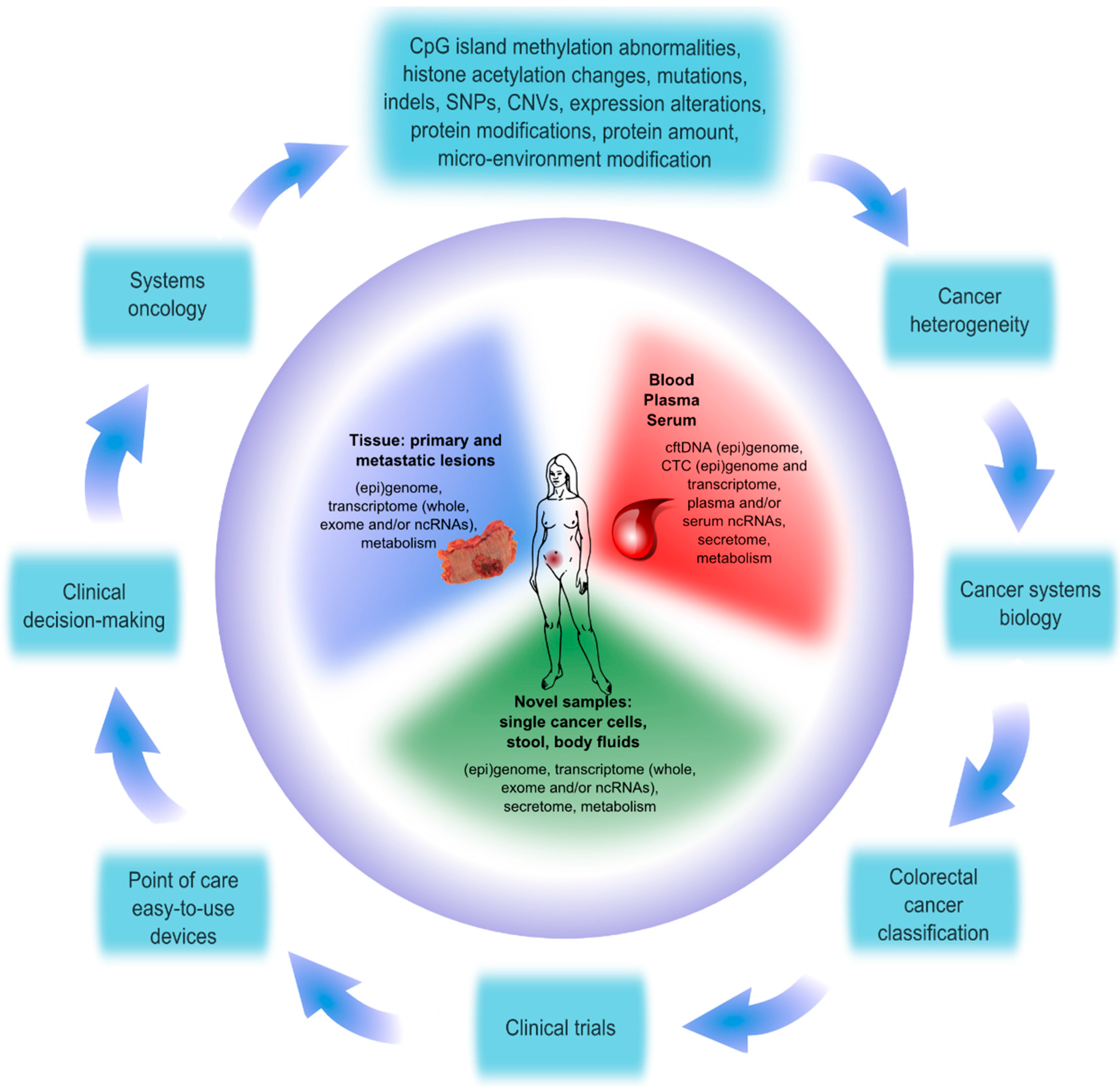
2. Sources of Heterogeneity in Colorectal Cancer
3. Novel Molecular Classifications of Colorectal Cancer
4. Cell-to-Cell Heterogeneity in Colorectal Cancer
5. Immunological Heterogeneity in Colorectal Cancer
6. Heterogeneity in Disseminated and Metastatic Disease
7. Clinical Implications of Heterogeneity in Metastatic Disease

{kind=link}
| Genetic Source | Heterogeneity | Drug | Clinical Significance | Sample Source | Analysis | References |
|---|---|---|---|---|---|---|
| RAS (KRAS, NRAS) | Mutations | Anti-EGFR antibodies | Predictive | Primary and metastatic tissue, CTC, cfDNA | Next-generation and Sanger sequencing, BEAMing®, high-performance liquid chromatography, dropled dPCR, qPCR | [61,62,63,78,79,89,95,96,97,98,99] |
| BRAF | Mutations | Chemotherapy and targeted agents | Prognostic, possible predictive (anti-EGFR antibodies) | Primary and metastatic tissue, cfDNA | Next-generation and Sanger sequencing, high-performance liquid chromatography, BEAMing®, qPCR | [62,78,89,96,97,100,101,102,103] |
| MMR system (e.g., MLH1 gene) | Mutations (hereditary CRC) or CpG island methylation (sporadic CRC) | Chemotherapy in adjuvant setting | Prognostic, possible predictive to adjuvant 5-FU-based regimens | Primary tissue | IHC, (q)PCR | [9,103,104,105,106,107,108,109,110,111] |
| PI3K | Mutations | Anti-EGFR antibodies | Possible predictive | Primary and metastatic tissue, cfDNA | Next-generation and Sanger sequencing, BEAMing®, qPCR | [78,96,112] |
| cMET | Expression | Anti-EGFR antibodies | Possible prognostic and predictive | Primary and metastatic tissue | Expression microarrays, IHC | [43,113,114] |
| EGFR | Mutations, amplifications | Anti-EGFR antibodies | Possible predictive | Primary and metastatic tissue, cfDNA | Next-generation and Sanger sequencing, BEAMing®, qPCR, FISH | [62,92,115] |
8. Future Directions
| Challenge | Possible Solution |
|---|---|
| Inter-patient variability | Achieve full knowledge and understanding about the variability between individuals and how this variability contributes to disease |
| Inter-tumor variability | Classification of the diverse types of tumors from the point of view of common phenotypic, clinical and molecular features |
| Intra-tumor heterogeneity | Novel analytical techniques and devices must be developed in order to increase the resolution of current high-throughput technologies and make possible the entire analysis of all cells within tumors |
| Design of precise/personalized anticancer drugs | Anticancer therapies must be designed based on deep analysis of tumors and their intrinsic heterogeneity |
| Novel design of clinical trials | Clinical trials must include multi-level high-throughput analysis to define the responsiveness of different patients, tumors, and even cells within tumors |
| Technological barrier | Design of affordable and simple technologies to make possible their clinical implementation |
| Analysis and integration of data | Development of cancer systems biology in order to generate models to obtain understandable and useful data for clinicians and patients |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aparicio, S.; Caldas, C. The implications of clonal genome evolution for cancer medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Salazar, R.; Tabernero, J. The evolution of our molecular understanding of colorectal cancer: What we are doing now, what the future holds, and how tumor profiling is just the beginning. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Arnold, C.N.; Niedzwiecki, D.; Chang, D.K.; Ricciardiello, L.; Carethers, J.M.; Dowell, J.M.; Wasserman, L.; Compton, C.; Mayer, R.J.; et al. Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res. 2003, 63, 1608–1614. [Google Scholar] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular pathways: Microsatellite instability in colorectal cancer: Prognostic, predictive, and therapeutic implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; d’ Ario, G.; Lamb, J.R.; Martin, E.; Wang, K.; Tejpar, S.; Delorenzi, M.; Bosman, F.T.; Roth, A.D.; Yan, P.; et al. A comprehensive characterization of genome-wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns of alterations. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Liu, K. Epigenetics and colorectal cancer pathogenesis. Cancers 2013, 5, 676–713. [Google Scholar] [CrossRef] [PubMed]
- Barrow, T.M.; Michels, K.B. Epigenetic epidemiology of cancer. Biochem. Biophys. Res. Commun. 2014, 455, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; de Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Islam, A.B.M.M.K.; Herrera, A.; Martín, P.; García, V.; Silva, J.; Garcia, J.M.; Salas, C.; Casal, I.; de Herreros, A.G.; et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clin. Cancer Res. 2013, 19, 5914–5926. [Google Scholar] [CrossRef] [PubMed]
- Berdiel-Acer, M.; Sanz-Pamplona, R.; Calon, A.; Cuadras, D.; Berenguer, A.; Sanjuan, X.; Paules, M.J.; Salazar, R.; Moreno, V.; Batlle, E.; et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol. Oncol. 2014, 8, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Norat, T.; Ferrari, P.; Jenab, M.; Bueno-de-Mesquita, B.; Skeie, G.; Dahm, C.C.; Overvad, K.; Olsen, A.; Tjønneland, A.; et al. Dietary fibre intake and risks of cancers of the colon and rectum in the European prospective investigation into cancer and nutrition (EPIC). PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Jenab, M.; Bueno-de-Mesquita, H.B.; Ferrari, P.; van Duijnhoven, F.J.B.; Norat, T.; Pischon, T.; Jansen, E.H.J.M.; Slimani, N.; Byrnes, G.; Rinaldi, S.; et al. Association between pre-diagnostic circulating vitamin D concentration and risk of colorectal cancer in European populations: A nested case-control study. BMJ 2010, 340, b5500. [Google Scholar] [CrossRef] [PubMed]
- Van Duijnhoven, F.J.B.; Bueno-De-Mesquita, H.B.; Calligaro, M.; Jenab, M.; Pischon, T.; Jansen, E.H.J.M.; Frohlich, J.; Ayyobi, A.; Overvad, K.; Toft-Petersen, A.P.; et al. Blood lipid and lipoprotein concentrations and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition. Gut 2011, 60, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, K.; Pischon, T.; Buijsse, B.; May, A.M.; Peeters, P.H.; Bueno-de-Mesquita, H.B.; Jenab, M.; Fedirko, V.; Dahm, C.C.; Siersema, P.D.; et al. Adult weight change and risk of colorectal cancer in the European Prospective Investigation into Cancer and Nutrition. Eur. J. Cancer 2013, 49, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Leitzmann, M.F. Television viewing and time spent sedentary in relation to cancer risk: A meta-analysis. J. Natl. Cancer Inst. 2014, 106, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Fuchs, C.S.; Giovannucci, E. How many molecular subtypes? Implications of the unique tumor principle in personalized medicine. Expert Rev. Mol. Diagn. 2012, 12, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Lascorz, J.; Chen, B.; Hemminki, K.; Försti, A. Consensus pathways implicated in prognosis of colorectal cancer identified through systematic enrichment analysis of gene expression profiling studies. PLoS ONE 2011, 6, e18867. [Google Scholar] [CrossRef] [PubMed]
- Popovici, V.; Budinska, E.; Tejpar, S.; Weinrich, S.; Estrella, H.; Hodgson, G.; van Cutsem, E.; Xie, T.; Bosman, F.T.; Roth, A.D.; et al. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J. Clin. Oncol. 2012, 30, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Creighton, C.J.; Schultz, N.; Shinbrot, E.; Chang, K.; Gunaratne, P.H.; Muzny, D.; Sander, C.; Hamilton, S.R.; Gibbs, R.A.; et al. MLH1-silenced and non-silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. J. Pathol. 2013, 229, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, B.; Kerick, M.; Roehr, C.; Fischer, A.; Isau, M.; Boerno, S.T.; Wunderlich, A.; Barmeyer, C.; Seemann, P.; Koenig, J.; et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS ONE 2010, 5, e15661. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.C.; Park, Y.-Y.; Park, E.S.; Lim, J.Y.; Kim, S.M.; Kim, S.-B.; Kim, J.; Kim, S.C.; Chu, I.-S.; Smith, J.J.; et al. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut 2012, 61, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Griffith, O.L.; Tai, I.T.; Jones, S.J.M. Meta-analysis of colorectal cancer gene expression profiling studies identifies consistently reported candidate biomarkers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; de Reyniès, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.-C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, A.; Beran, G.; Chresta, C.M.; McWalter, G.; Pritchard, A.; Weston, S.; Runswick, S.; Davenport, S.; Heathcote, K.; Castro, D.A.; et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med. Genomics 2012, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Perez-Villamil, B.; Romera-Lopez, A.; Hernandez-Prieto, S.; Lopez-Campos, G.; Calles, A.; Lopez-Asenjo, J.-A.; Sanz-Ortega, J.; Fernandez-Perez, C.; Sastre, J.; Alfonso, R.; et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer 2012, 12, 260. [Google Scholar] [CrossRef] [PubMed]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; di Narzo, A.F.; Yan, P.; Graeme Hodgson, J.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Tian, S.; Moreno, V.; Snel, M.H.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int. J. Cancer 2013, 134, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J.; et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 2011, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Niedzwiecki, D.; Lopatin, M.; Ye, X.; Lee, M.; Friedman, P.N.; Frankel, W.; Clark-Langone, K.; Millward, C.; Shak, S.; et al. Biologic determinants of tumor recurrence in stage II colon cancer: Validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J. Clin. Oncol. 2013, 31, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- De Sousa E Melo, F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.M.H.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.G.; Lannon, W.A.; Grotzinger, C.; del Rio, M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Guinney, J.; Delorenzi, M.; de Reynies, A.; Roepman, P.; Sadanandam, A.; Vermeulen, L.; Schlicker, A.; Missiaglia, E.; Soneson, C.; et al. Colorectal Cancer Subtyping Consortium Colorectal cancer subtyping consortium (CRCSC) identification of a consensus of molecular subtypes. J. Clin. Oncol. 2014, 32, 3511. [Google Scholar]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.W.; Lopez-Diaz, F.J.; Khan, S.Y.; Tariq, M.A.; Dayn, Y.; Vaske, C.J.; Radenbaugh, A.J.; Kim, H.J.; Emerson, B.M.; Pourmand, N. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, E4726–E4735. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Concha, A.; Glew, S.; Ruiz-Cabello, F.; Stern, P.L. Natural history of HLA expression during tumour development. Immunol. Today 1993, 14, 491–499. [Google Scholar] [CrossRef]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Pérez-Villar, J.J.; López-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.; Concha, A.; Sáenz-López, P.; Rodríguez, A.I.; Cabrera, T.; Garrido, F.; Ruiz-Cabello, F. Leukocyte infiltrate in gastrointestinal adenocarcinomas is strongly associated with tumor microsatellite instability but not with tumor immunogenicity. Cancer Immunol. Immunother. 2011, 60, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.; García-Alcalde, F.; Concha, A.; Cano, C.; Blanco, A.; Garrido, F.; Ruiz-Cabello, F. Genome-wide differential genetic profiling characterizes colorectal cancers with genetic instability and specific routes to HLA class I loss and immune escape. Cancer Immunol. Immunother. 2012, 61, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the “Immunoscore” in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.H.; Pagès, F.; et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Broussard, E.K.; Disis, M.L. TNM staging in colorectal cancer: T is for T Cell and M is for memory. J. Clin. Oncol. 2011, 29, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res.-Rev. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Kalikaki, A.; Politaki, H.; Souglakos, J.; Apostolaki, S.; Papadimitraki, E.; Georgoulia, N.; Tzardi, M.; Mavroudis, D.; Georgoulias, V.; Voutsina, A. KRAS genotypic changes of circulating tumor cells during treatment of patients with metastatic colorectal cancer. PLoS ONE 2014, 9, e104902. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–576. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Arena, S.; Lamba, S.; Siravegna, G.; Lallo, A.; Hobor, S.; Russo, M.; Buscarino, M.; Lazzari, L.; Sartore-Bianchi, A.; et al. Blockade of EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci. Transl. Med. 2014, 6, 224ra26. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Felicioni, L.; Buscarino, M.; de Dosso, S.; Buttitta, F.; Malatesta, S.; Movilia, A.; Luoni, M.; Boldorini, R.; Alabiso, O.; et al. Increased detection sensitivity for KRAS mutations enhances the prediction of anti-EGFR monoclonal antibody resistance in metastatic colorectal cancer. Clin. Cancer Res. 2011, 17, 4901–4914. [Google Scholar] [CrossRef] [PubMed]
- Tougeron, D.; Lecomte, T.; Pagès, J.C.; Villalva, C.; Collin, C.; Ferru, A.; Tourani, J.M.; Silvain, C.; Levillain, P.; Karayan-tapon, L. Effect of low-frequency KRAS mutations on the response to anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2013, 24, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Xiao, X.; Lu, J.; Qian, X.; Liu, B.; Feng, J. Colorectal cancer patients with low abundance of KRAS mutation may benefit from EGFR antibody therapy. PLoS ONE 2013, 8, e68022. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hanash, S.M.; Pitteri, S.J.; Faca, V.M. Mining the plasma proteome for cancer biomarkers. Nature 2008, 452, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, C.; Nice, E.C. Proteomics, genomics and transcriptomics: Their emerging roles in the discovery and validation of colorectal cancer biomarkers. Expert Rev. Proteomics 2014, 11, 179–205. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.M.; Scherl, A.; Nguyen, A.; Man, F.Y.; Weinberg, E.; Zeng, Z.; Saltz, L.; Paty, P.B.; Tavazoie, S.F. Extracellular metabolic energetics can promote cancer progression. Cell 2015, 160, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Artale, S.; Sartore-Bianchi, A.; Veronese, S.M.; Gambi, V.; Sarnataro, C.S.; Gambacorta, M.; Lauricella, C.; Siena, S. Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J. Clin. Oncol. 2008, 26, 4217–4219. [Google Scholar] [CrossRef] [PubMed]
- Knijn, N.; Mekenkamp, L.J.M.; Klomp, M.; Vink-Börger, M.E.; Tol, J.; Teerenstra, S.; Meijer, J.W.R.; Tebar, M.; Riemersma, S.; van Krieken, J.H.J.M.; et al. KRAS mutation analysis: A comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br. J. Cancer 2011, 104, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Wu, X.; Yang, Z.; Threapleton, D.E.; Yuan, J.; Yu, Y.; Tang, J. Concordant analysis of KRAS, BRAF, PIK3CA mutations, and PTEN expression between primary colorectal cancer and matched metastases. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef] [PubMed]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed]
- Vignot, S.; Lefebvre, C.; Frampton, G.M.; Meurice, G.; Yelensky, R.; Palmer, G.; Capron, F.; Lazar, V.; Hannoun, L.; Miller, V.A.; et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: Evaluation of concordance between genomic and transcriptional profiles. Eur. J. Cancer 2015, 51, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Jesinghaus, M.; Wolf, T.; Pfarr, N.; Muckenhuber, A.; Ahadova, A.; Warth, A.; Goeppert, B.; Sers, C.; Kloor, M.; Endris, V.; et al. Distinctive spatiotemporal stability of somatic mutations in metastasized microsatellite-stable colorectal cancer. Am. J. Surg. Pathol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Blomme, A.; Debois, D.; Somja, J.; Delvaux, D.; Patsos, G.; di Valentin, E.; Peulen, O.; Mutijima, E.N.; de Pauw, E.; et al. Organized proteomic heterogeneity in colorectal cancer liver metastases and implications for therapies. Hepatology 2014, 59, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Paterson, E.L.; Kazenwadel, J.; Bert, A.G.; Khew-Goodall, Y.; Ruszkiewicz, A.; Goodall, G.J. Down-regulation of the miRNA-200 family at the invasive front of colorectal cancers with degraded basement membrane indicates EMT is involved in cancer progression. Neoplasia 2013, 15, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Siena, S.; Sartore-Bianchi, A.; di Nicolantonio, F.; Balfour, J.; Bardelli, A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J. Natl. Cancer Inst. 2009, 101, 1308–1324. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; de Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Mao, C.; Liao, R.-Y.; Qiu, L.-X.; Wang, X.-W.; Ding, H.; Chen, Q. BRAF V600E mutation and resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer: A meta-analysis. Mol. Biol. Rep. 2011, 38, 2219–2223. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Rubio, E.; Gómez-España, A.; Massutí, B.; Sastre, J.; Reboredo, M.; Manzano, J.L.; Rivera, F.; Safont, M.J.; Montagut, C.; González, E.; et al. Role of kras status in patients with metastatic colorectal cancer receiving first-line chemotherapy plus bevacizumab: A TTD group cooperative study. PLoS ONE 2012, 7, e47345. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.-J.; Kohne, C.-H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Nowak, M.A. Timing and heterogeneity of mutations associated with drug resistance in metastatic cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15964–15968. [Google Scholar] [CrossRef] [PubMed]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 1445–1445. [Google Scholar] [CrossRef]
- Jeffers, M.; van Cutsem, E.; Sobrero, A.F.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Mutational analysis of biomarker samples from the CORRECT study: Correlating mutation status with clinical response to regorafenib. J. Clin. Oncol. 2013, 31, 381. [Google Scholar]
- Bedard, P.L.; Hansen, A.R.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, C.; van Cutsem, E.; Rougier, P.; Ciardiello, F.; Heeger, S.; Schlichting, M.; Celik, I.; Köhne, C.-H. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur. J. Cancer 2012, 48, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Normanno, N.; Maiello, E.; Martinelli, E.; Troiani, T.; Pisconti, S.; Giuliani, F.; Barone, C.; Cartenì, G.; Rachiglio, A.M.; et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next generation sequencing: Findings from the CAPRI-GOIM trial. Ann. Oncol. 2014, 25, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.; et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: A pooled analysis of the CAIRO, CAIRO2, COIN and FOCUS studies. Clin. Cancer Res. 2014, 20, 5322–5331. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; K Kotsopoulos, S.; Nizard, P.; Perez Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical relevance of KRAS-mutated sub-clones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized controlled trials. Ann. Oncol. 2014, 26, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Tejpar, S.; Delorenzi, M.; Yan, P.; Fiocca, R.; Klingbiel, D.; Dietrich, D.; Biesmans, B.; Bodoky, G.; Barone, C.; et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: Results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J. Clin. Oncol. 2010, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Shima, K.; Meyerhardt, J.A.; McCleary, N.J.; Ng, K.; Hollis, D.; Saltz, L.B.; Mayer, R.J.; Schaefer, P.; Whittom, R.; et al. Predictive and prognostic roles of BRAF mutation in stage III colon cancer: Results from intergroup trial CALGB 89803. Clin Cancer Res 2012, 18, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: Results from the MRC FOCUS trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J.; Richman, S.; Chambers, P.; Seymour, M.; Kerr, D.; et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Des Guetz, G.; Schischmanoff, O.; Nicolas, P.; Perret, G.-Y.; Morere, J.-F.; Uzzan, B. Does microsatellite instability predict the efficacy of adjuvant chemotherapy in colorectal cancer? A systematic review with meta-analysis. Eur. J. Cancer 2009, 45, 1890–1896. [Google Scholar] [CrossRef] [PubMed]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Foster, N.R.; Thibodeau, S.N.; Marsoni, S.; Monges, G.; Labianca, R.; Kim, G.P.; Yothers, G.; Allegra, C.; Moore, M.J.; et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J. Natl. Cancer Inst. 2011, 103, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; Kortman, G.A.M.; Mekenkamp, L.; Ligtenberg, M.J.L.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.A.; van Krieken, J.H.J.M. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, M.M.; Niedzwiecki, D.; Compton, C.C.; Hahn, H.P.; Hall, M.; Damas, B.; Jewell, S.D.; Mayer, R.J.; Goldberg, R.M.; Saltz, L.B.; et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J. Clin. Oncol. 2009, 27, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Kishiki, T.; Ohnishi, H.; Masaki, T.; Ohtsuka, K.; Ohkura, Y.; Furuse, J.; Watanabe, T.; Sugiyama, M. Overexpression of MET is a new predictive marker for anti-EGFR therapy in metastatic colorectal cancer with wild-type KRAS. Cancer Chemother. Pharmacol. 2014, 73, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Yamada, Y.; Taniguchi, H.; Nagashima, K.; Okita, N.; Takashima, A.; Honma, Y.; Iwasa, S.; Kato, K.; Hamaguchi, T.; et al. Clinical impact of c-MET expression and genetic mutational status in colorectal cancer patients after liver resection. Cancer Sci 2014, 105, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, C.; Li, F.; Wang, X. EGFR gene copy number as a prognostic marker in colorectal cancer patients treated with cetuximab or panitumumab: A systematic review and meta analysis. PLoS ONE 2013, 8, e56205. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, V.N.; Lingjærde, O.C.; Russnes, H.G.; Vollan, H.K.M.; Frigessi, A.; Børresen-Dale, A.-L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer systems biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Yan, Z.; Wang, X.; Cao, Q.; Munshi, N.C.; Li, C.; Shah, P.K. canEvolve: A web portal for integrative oncogenomics. PLoS ONE 2013, 8, e56228. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Calvo, M.; Concha, Á.; Figueroa, A.; Garrido, F.; Valladares-Ayerbes, M. Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. Int. J. Mol. Sci. 2015, 16, 13610-13632. https://doi.org/10.3390/ijms160613610
Blanco-Calvo M, Concha Á, Figueroa A, Garrido F, Valladares-Ayerbes M. Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. International Journal of Molecular Sciences. 2015; 16(6):13610-13632. https://doi.org/10.3390/ijms160613610
Chicago/Turabian StyleBlanco-Calvo, Moisés, Ángel Concha, Angélica Figueroa, Federico Garrido, and Manuel Valladares-Ayerbes. 2015. "Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach" International Journal of Molecular Sciences 16, no. 6: 13610-13632. https://doi.org/10.3390/ijms160613610
APA StyleBlanco-Calvo, M., Concha, Á., Figueroa, A., Garrido, F., & Valladares-Ayerbes, M. (2015). Colorectal Cancer Classification and Cell Heterogeneity: A Systems Oncology Approach. International Journal of Molecular Sciences, 16(6), 13610-13632. https://doi.org/10.3390/ijms160613610