
De Novo Characterization of Flower Bud Transcriptomes and the Development of EST-SSR Markers for the Endangered Tree Tapiscia sinensis


Abstract
:1. Introduction
2. Results and Discussion
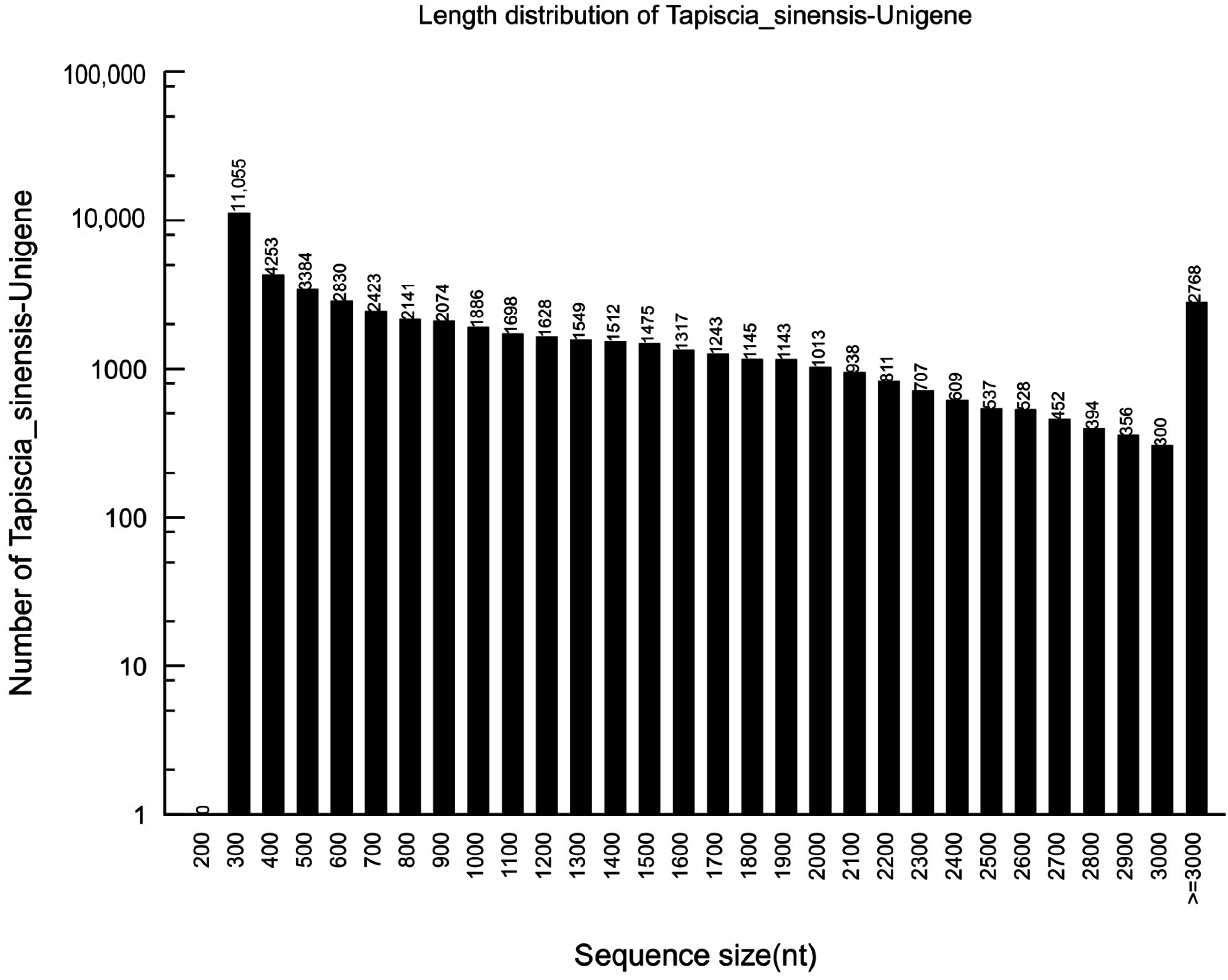
2.1. Sequencing and de novo Assembly
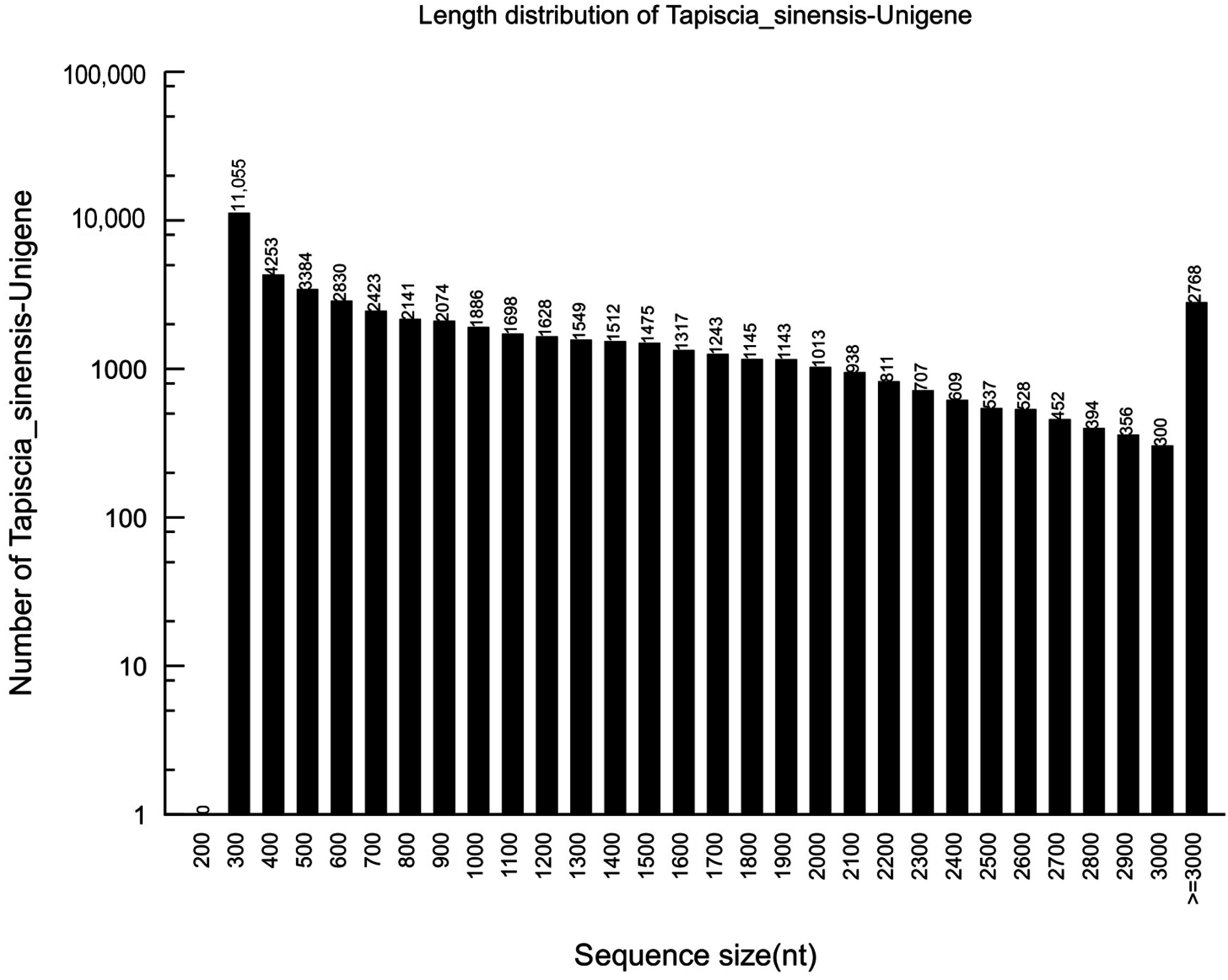
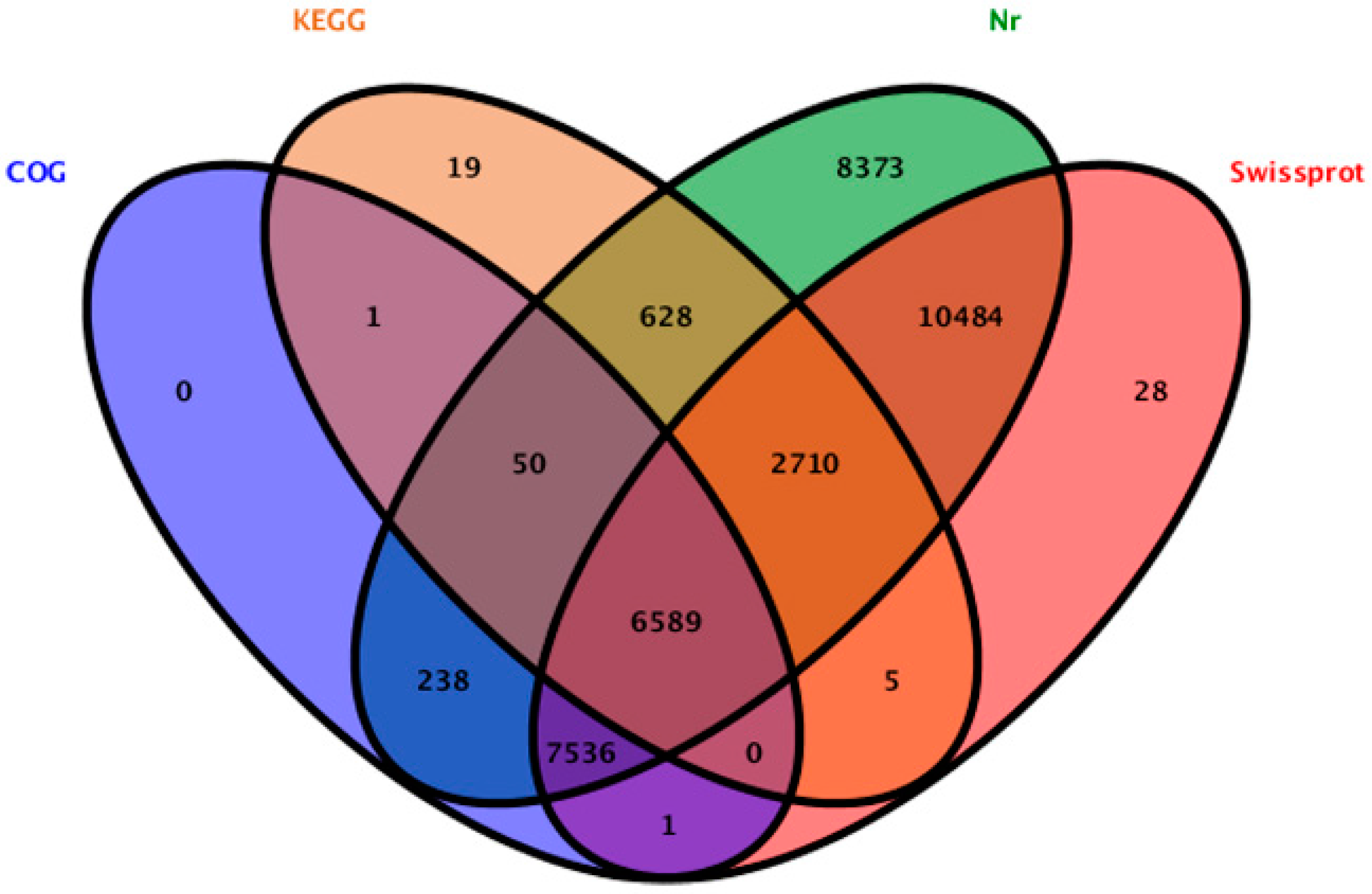
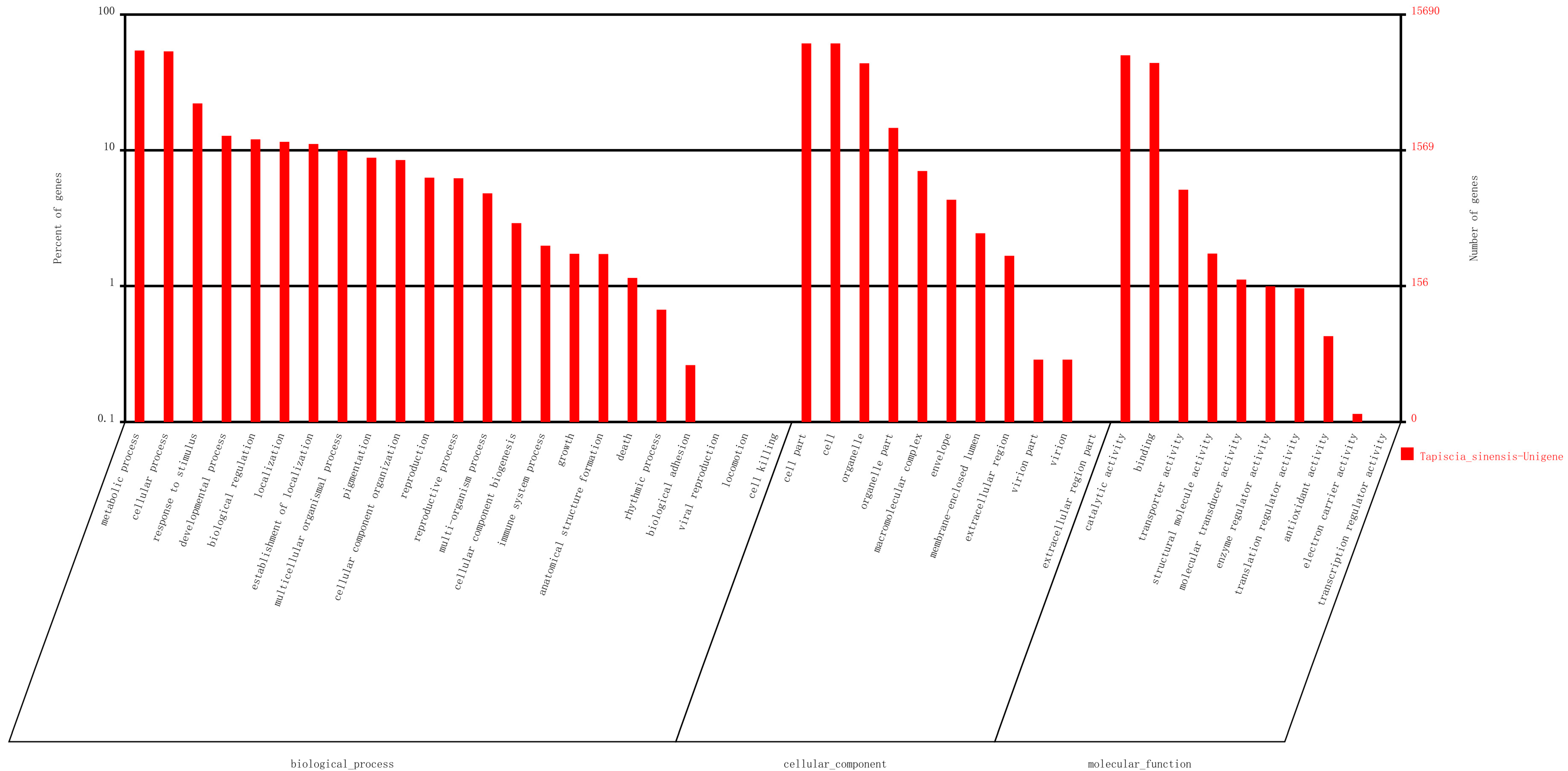
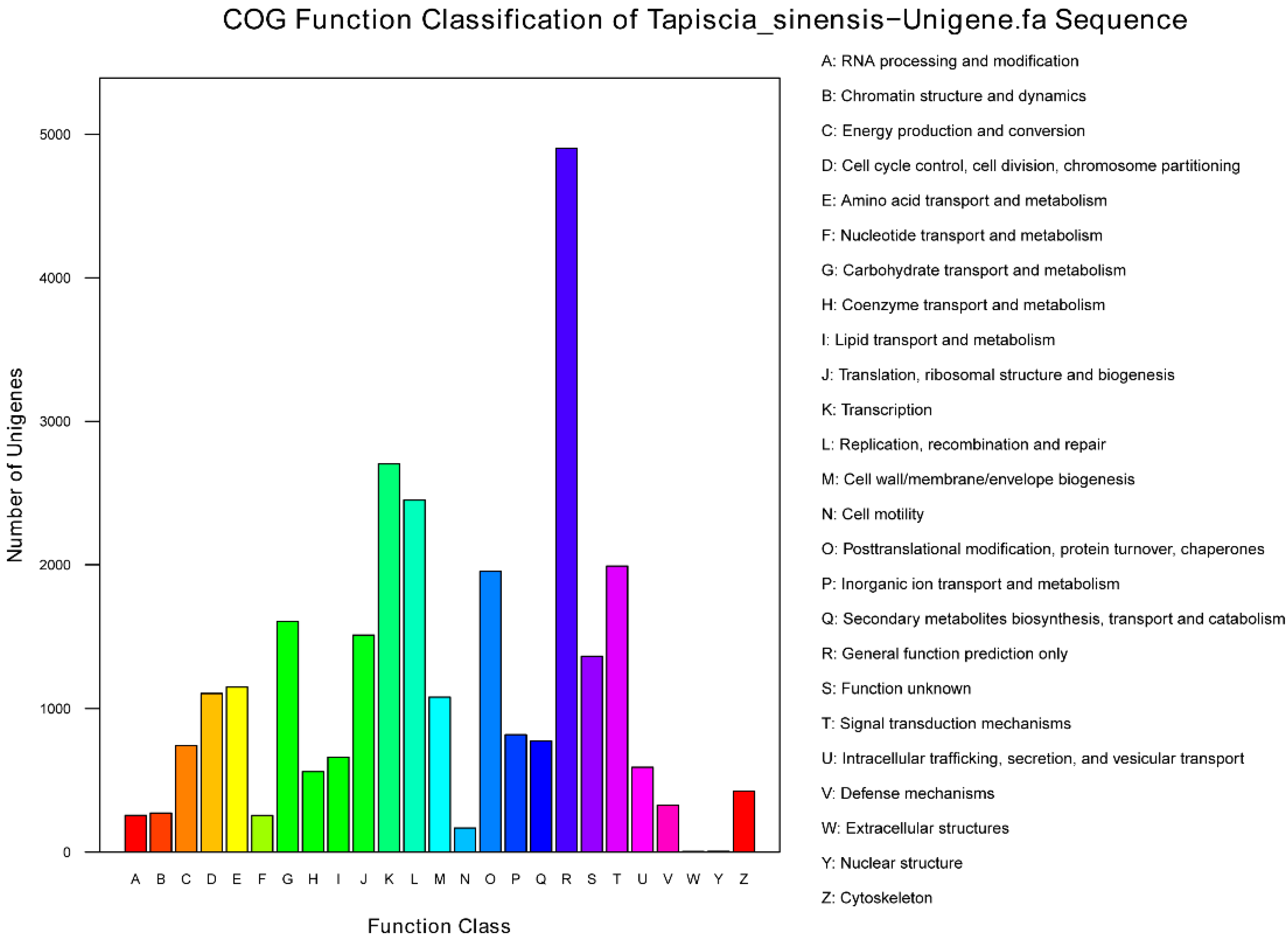
2.2. Sequence Annotation and Classification
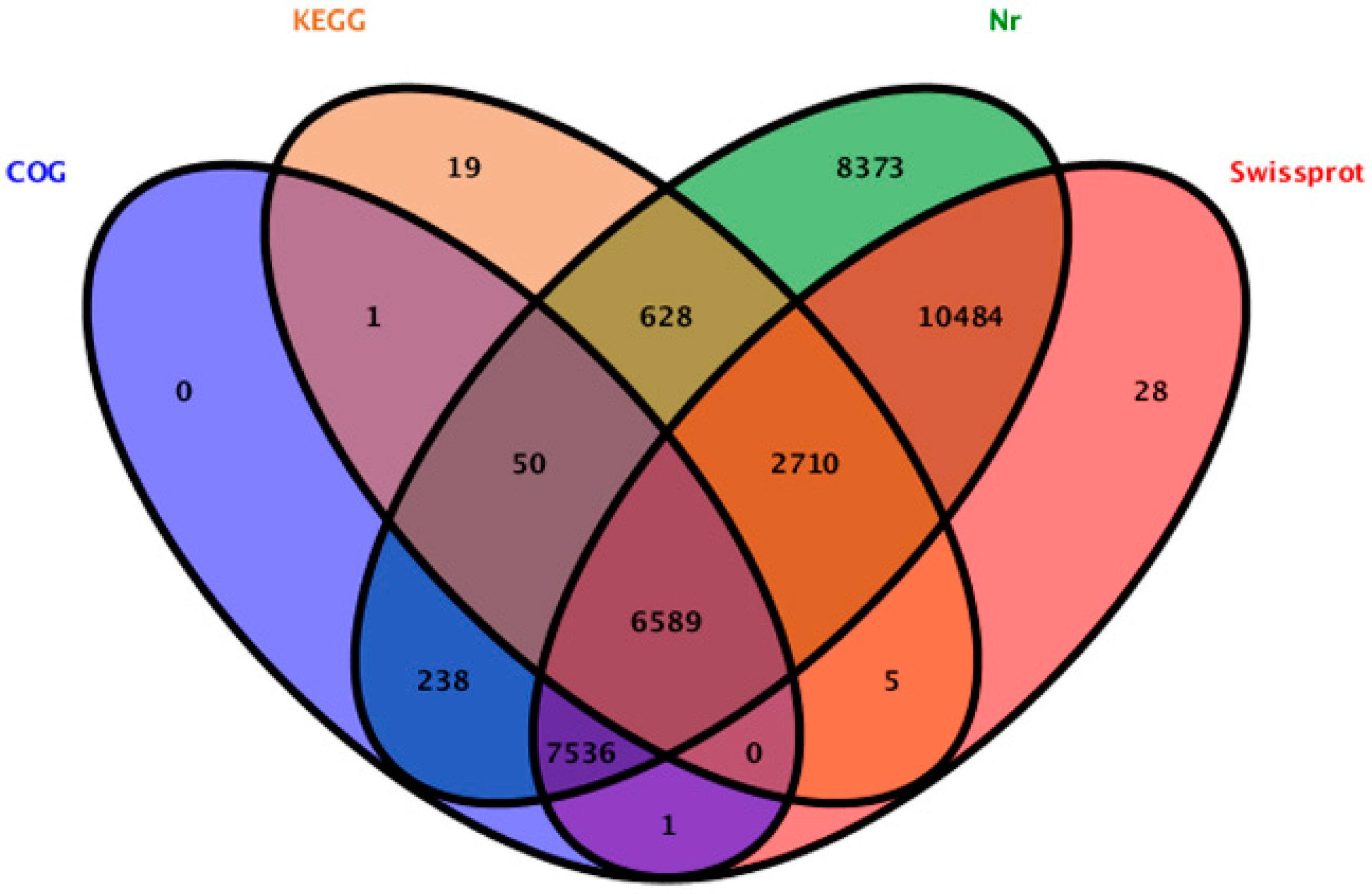
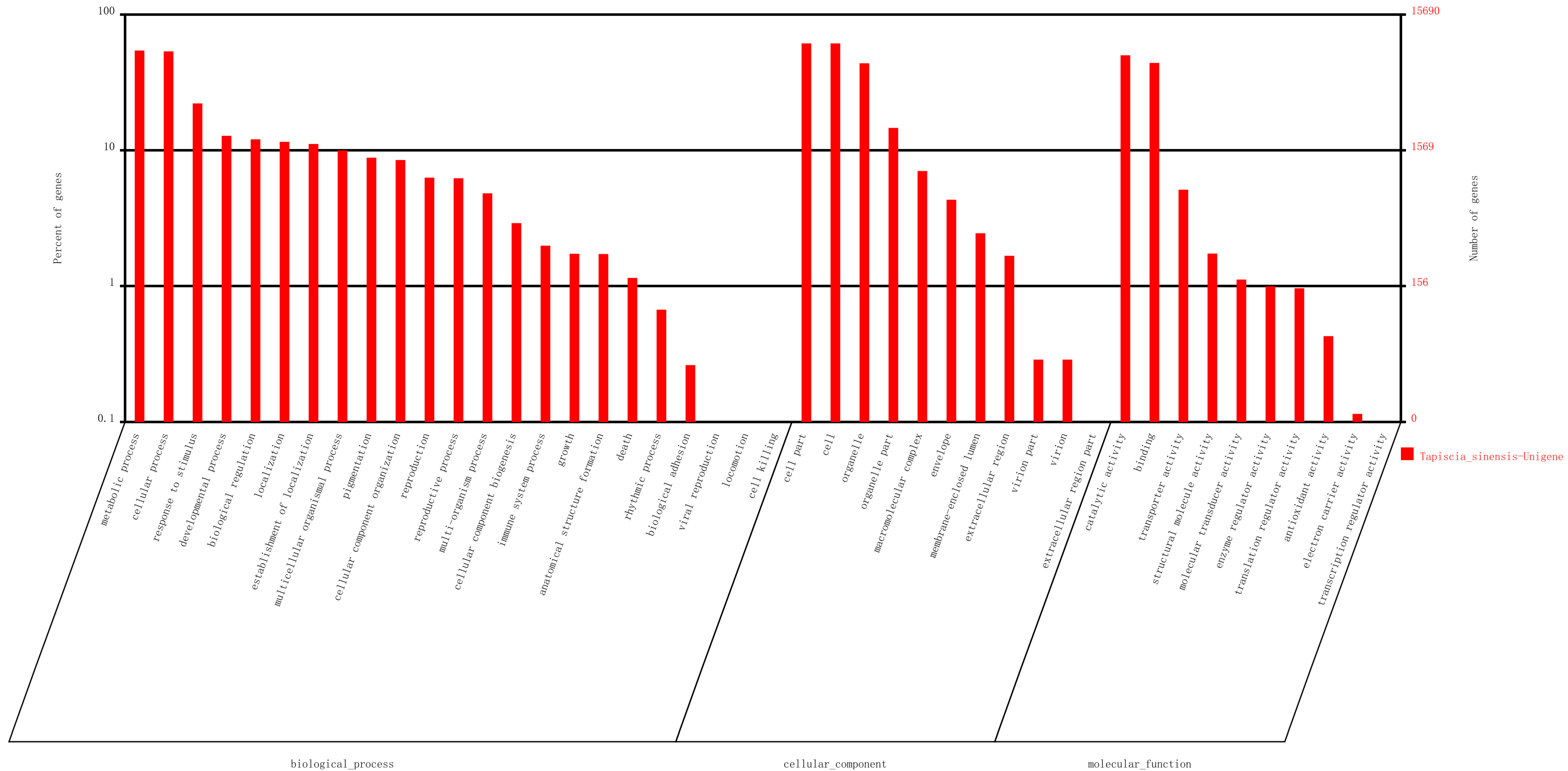
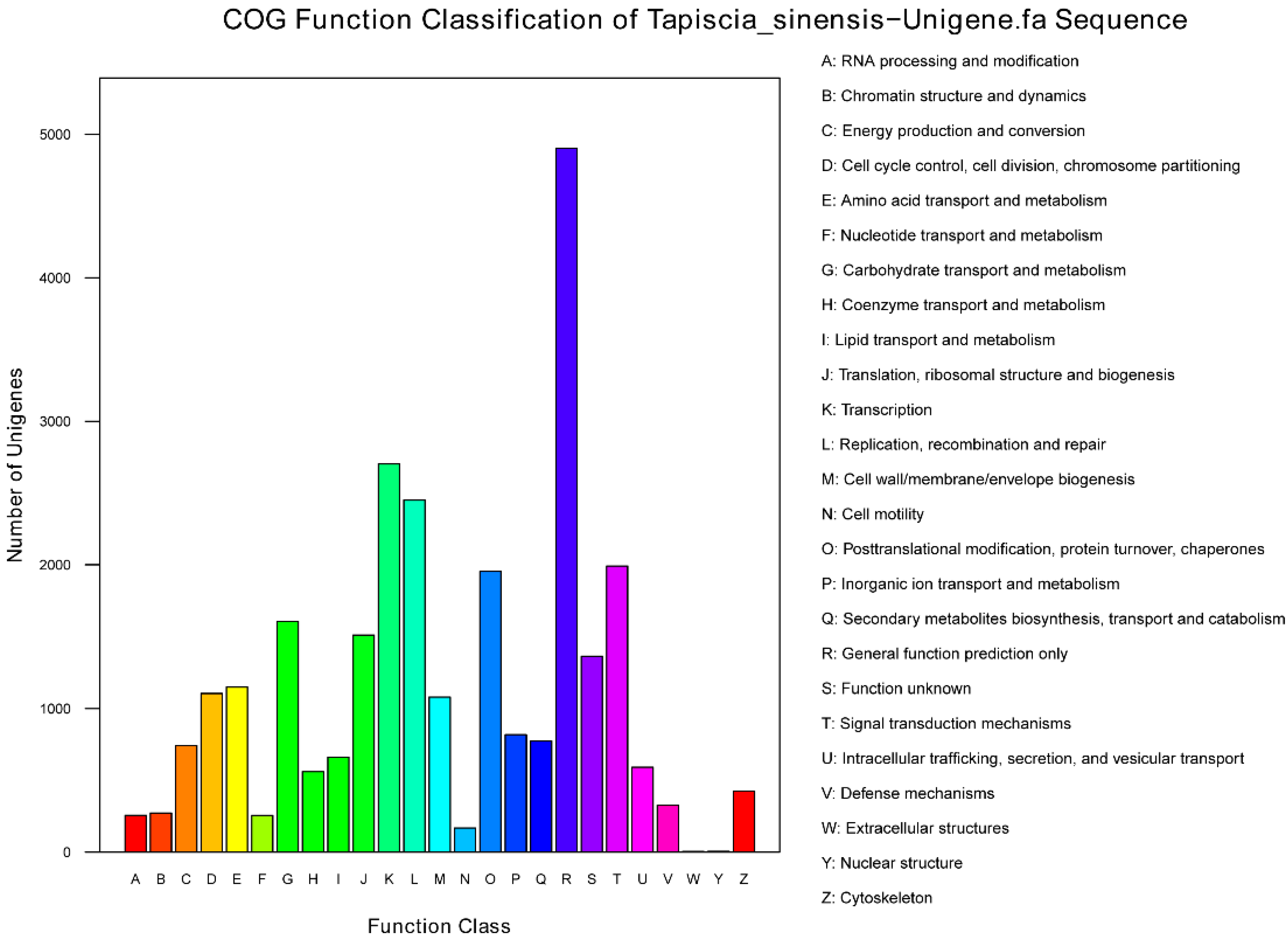
2.3. Genes Putatively Related to Flower Development

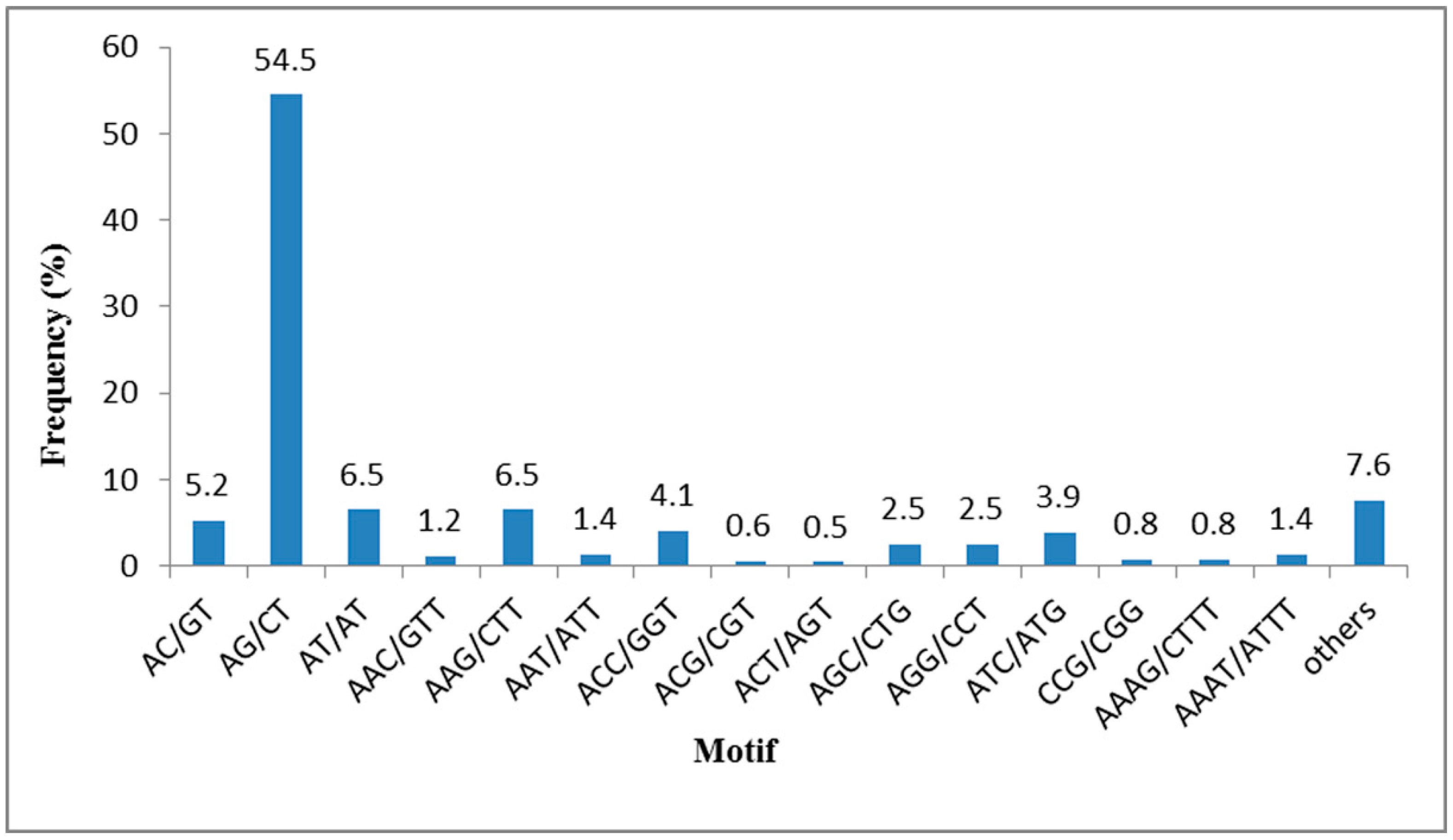
2.4. SSR Mining from the T. sinensis Transcriptome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Number |
|---|---|
| Total number of sequences examined | 52,169 |
| Total size of examined sequences (bp) | 58,253,075 |
| Total number of identified SSRs | 10,371 |
| Number of SSR containing sequences | 8608 |
| Number of sequences containing more than 1 SSR | 1436 |
| Number of SSRs present in compound formation | 733 |
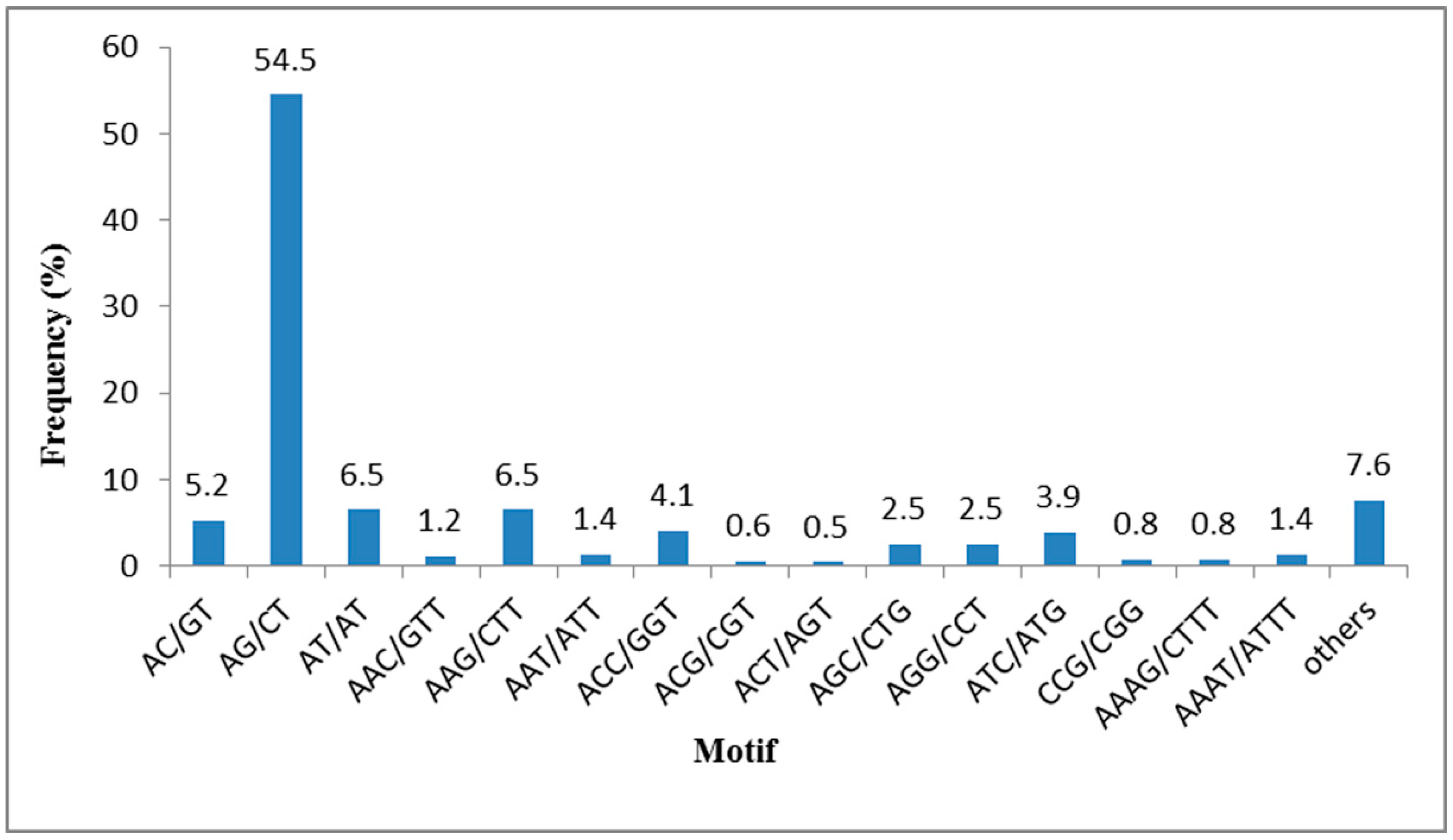
| Repeat Numbers | Motif | Total | Repeat Number (%) | ||||
|---|---|---|---|---|---|---|---|
| Di- | Tri- | Tetra- | Penta- | Hexa | |||
| 4 | 0 | 0 | 368 | 158 | 266 | 792 | 7.64 |
| 5 | 0 | 1319 | 101 | 15 | 12 | 1447 | 13.95 |
| 6 | 2026 | 703 | 52 | 5 | 11 | 2797 | 26.97 |
| 7 | 1416 | 411 | 0 | 0 | 4 | 1831 | 17.66 |
| 8 | 1164 | 49 | 1 | 1 | 2 | 1217 | 11.73 |
| 9 | 1256 | 0 | 0 | 0 | 0 | 1256 | 12.11 |
| 10 | 823 | 1 | 0 | 0 | 0 | 824 | 7.95 |
| 11 | 195 | 1 | 0 | 0 | 0 | 196 | 1.89 |
| ˃11 | 8 | 2 | 0 | 0 | 1 | 11 | 0.11 |
| Total | 6888 | 2486 | 522 | 179 | 296 | 10,371 | 100 |
| Motif (%) | 66.42 | 23.97 | 5.03 | 1.73 | 2.85 | ||
2.5. SSR Marker Assessment and Application
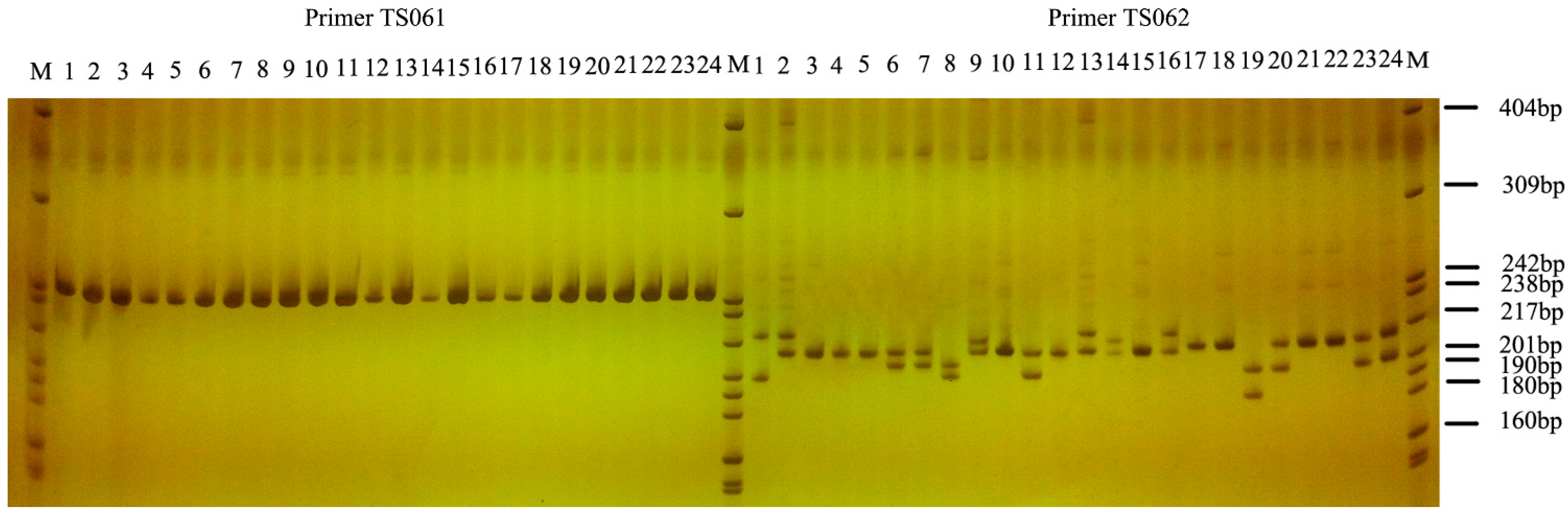
| Primer | Motif | K | HO | HE | PIC | HW |
|---|---|---|---|---|---|---|
| TS053 | (TCA)6 | 9 | 0.706 | 0.835 | 0.804 | ND |
| TS126 | (TAC)7 | 6 | 0.706 | 0.768 | 0.723 | NS |
| TS103 | (AAGA)5 | 5 | 0.549 | 0.631 | 0.573 | NS |
| TS149 | (TTG)11 | 6 | 0.608 | 0.769 | 0.725 | NS |
| TS060 | (TGT)6 | 6 | 0.529 | 0.501 | 0.464 | NS |
| TS013 | (AGA)8 | 5 | 0.51 | 0.653 | 0.592 | NS |
3. Materials and Methods
3.1. Plant Material and RNA Extraction
3.2. cDNA Library Construction and Illumina Sequencing
3.3. De Novo Assembly and Annotation
3.4. SSR Detection and Primer Design
3.5. SSR Validation and Assessment
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bremer, B.; Bremer, K.; Chase, M.; Fay, M.; Reveal, J.; Soltis, D.; Soltis, P.; Stevens, P. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar]
- Di, W.Z.; Yu, Z. The First Countries to Protect Rare and Endangered Plants in Shanxi Province; Northwest University Press: Xi’an, China, 1989; pp. 200–203. [Google Scholar]
- Li, D.Z.; Cai, J.; Wen, J. Flora of China; Science Press: Beijing, China, 1996; p. 496. [Google Scholar]
- Liu, W.Z.; Kang, H.Q.; Zheng, H.C.; Feng, Y.Z. An investigation on the sexual reproductive cycle in Tapiscia sinensis. J. Syst. Evol. 2008, 46, 175–182. [Google Scholar]
- Lü, W.; Liu, W.Z. Pollination biology in androdioecious species Tapiscia sinensis. Chin. Bull. Bot. 2010, 45, 713–722. (In Chinese) [Google Scholar]
- Krause, J. Staphyleaceae. In Die natürlichen Pflanzenfamilien, 2nd ed.; Duncker & Humblot 20b: Berlin, Germany, 1942; pp. 255–321. [Google Scholar]
- Matthews, M.L.; Endress, P.K. Comparative floral structure and systematics in Crossosomatales (Crossosomataceae, Stachyuraceae, Staphyleaceae, Aphloiaceae, Geissolomataceae, Ixerbaceae, Strasburgeriaceae). Bot. J. Linn. Soc. 2005, 147, 1–46. [Google Scholar] [CrossRef]
- Tang, Y.; Li, L. On historical elements and tertiary precursors of eastern Asiatic angiospermous flora-based on phytogeography of staphyleaceae, morinaceae and caprifoliaceae. Acta. Phytotaxon. Sin. 1996, 34, 453–478. (In Chinese) [Google Scholar]
- Xie, C.P. A review of research advances in rare and endangered plant Tapiscia sinensis. Subtrop. Plant Sci. 2006, 35, 71–74. (In Chinese) [Google Scholar]
- Tapiscia sinensis. The IUCN red list of threatened species. Available Online: http://www.iucnredlist.org. (accessed on 14 April 2015).
- Zhang, P.F.; Wang, Z.G.; Liu, Y.L.; Tian, H.; Yao, X.H.; Zhang, J.J. Development and characterization of 11 polymorphic microsatellite markers in Tapiscia sinensis (Staphyleaceae). Appl. Plant Sci. 2014, 1, 1300051. [Google Scholar]
- Zhang, J.; Liang, S.; Duan, J.; Wang, J.; Chen, S.; Cheng, Z.; Zhang, Q.; Liang, X.; Li, Y. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 2012, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Lu, G.; Fan, D.; Zhu, C.; Li, W.; Zhao, Q.; Feng, Q.; Zhao, Y.; Guo, Y.; Li, W.; et al. Function annotation of the rice transcriptome at singlenucleotide resolution by RNA-seq. Genome Res. 2010, 20, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Wang, Q.; Shen, H.L. De novo assembly, gene annotation and marker development using Illumina paired-end transcriptome sequences in celery (Apium graveolens L.). PLoS ONE 2013, 8, e57686. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, M.; Luo, Q.; Wang, J.; Li, H. De novo transcriptome analysis of Liriodendron chinense petals and leaves by Illumina sequencing. Gene 2014, 534, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Luo, S.; Wang, R.; Zhong, Y.; Xu, X.; Lin, Y.E.; He, X.; Sun, B.; Huang, H. The first Illumina-based de novo transcriptome sequencing and analysis of pumpkin (Cucurbita moschata Duch.) and SSR marker development. Mol. Breed. 2014, 34, 1437–1447. [Google Scholar] [CrossRef]
- Wei, L.; Li, S.; Liu, S.; He, A.; Wang, D.; Wang, J.; Tang, Y.; Wu, X. Transcriptome analysis of Houttuynia cordata Thunb. by illumina paired-end RNA sequencing and SSR marker discovery. PLoS ONE 2014, 9, e84105. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, C.; Lin, X.; Cui, B.; Wu, R.; Pang, X. De novo assembly and characterization of the fruit transcriptome of Chinese jujube (Ziziphus jujuba Mill.) using 454 pyrosequencing and the development of novel tri-nucleotide SSR Markers. PLoS ONE 2014, 9, e106438. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, Y.; Niu, J.; Ning, Y.; Lin, S.; Zhang, Z. De novo transcriptome analysis of the Siberian apricot (Prunus sibirica L.) and search for potential SSR markers by 454 pyrosequencing. Gene 2014, 544, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. ISMB 1999, 99, 138–148. [Google Scholar]
- Onouchi, H.; Igeño, M.I.; Périlleux, C.; Graves, K.; Coupland, G. Mutagenesis of plants overexpressing CONSTANS demonstrates novel interactions among arabidopsis flowering-time genes. Plant Cell 2000, 12, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.R.; Davis, S.J.; Bastow, R.M.; McWatters, H.G.; Kozma-Bognár, L.; Nagy, F.; Millar, A.J.; Amasino, R.M. The ELF4 gene controls circadian rhythms and flowering time in Arabidopsis thaliana. Nature 2002, 419, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, X.; Hu, R.; Wu, F.; Ma, J.; Meng, Y.; Fu, Y. Identification and molecular characterization of FKF1 and GI homologous genes in soybean. PLoS ONE 2013, 8, e79036. [Google Scholar] [CrossRef] [PubMed]
- Kinney, M.S.; Columbus, J.T.; Friar, E.A. Molecular evolution of the maize sex-determining gene TASSELSEED2 in Bouteloua (Poaceae). Mol. Phylogenet. Evol. 2003, 29, 519–528. [Google Scholar] [CrossRef] [PubMed]
- DeLong, A.; Calderon-Urrea, A.; Dellaporta, S.L. Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 1993, 74, 757–768. [Google Scholar] [CrossRef]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Triwitayakorn, K.; Chatkulkawin, P.; Kanjanawattanawong, S.; Sraphet, S.; Yoocha, T.; Sangsrakru, D.; Chanprasert, J.; Ngamphiw, C.; Jomchai, N.; Therawattanasuk, K.; et al. Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res. 2011, 18, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Jia, J.Q.; Zhang, X.H.; La, H.X.; Yang, W.D. Development and application of EST-SSR markers in oat. Plant Sci. J. 2014, 32, 240–250. [Google Scholar]
- Wang, M.L.; Mosjidis, J.A.; Morris, J.B.; Dean, R.E.; Jenkins, T.M.; Pederson, G.A. Genetic diversity of Crotalaria germplasm assessed through phylogenetic analysis of EST-SSR markers. Genome 2006, 49, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Soren, K.R.; Yadav, A.; Pandey, G.; Gangwar, P.; Parihar, A.K.; Bohra, A.; Dixit, G.P.; Datta, S.; Singh, N.P. EST-SSR analysis provides insights about genetic relatedness, population structure and gene flow in grass pea (Lathyrus sativus). Plant Breed. 2015. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. In Bioinformatics Methods and Protocols; Humana Press: New Jersey, NJ, USA, 1999; pp. 365–386. [Google Scholar]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genepop’ 007: A complete re-implementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Documentation for Structure Software. Available online: http://pritchardlab.stanford.edu/structure.html (accessed on 2 February 2010).
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.-J.; Wang, Y.-Y.; Xu, Y.-N.; Yan, R.-S.; Zhao, P.; Liu, W.-Z. De Novo Characterization of Flower Bud Transcriptomes and the Development of EST-SSR Markers for the Endangered Tree Tapiscia sinensis. Int. J. Mol. Sci. 2015, 16, 12855-12870. https://doi.org/10.3390/ijms160612855
Zhou X-J, Wang Y-Y, Xu Y-N, Yan R-S, Zhao P, Liu W-Z. De Novo Characterization of Flower Bud Transcriptomes and the Development of EST-SSR Markers for the Endangered Tree Tapiscia sinensis. International Journal of Molecular Sciences. 2015; 16(6):12855-12870. https://doi.org/10.3390/ijms160612855
Chicago/Turabian StyleZhou, Xiao-Jun, Yue-Yue Wang, Ya-Nan Xu, Rong-Shan Yan, Peng Zhao, and Wen-Zhe Liu. 2015. "De Novo Characterization of Flower Bud Transcriptomes and the Development of EST-SSR Markers for the Endangered Tree Tapiscia sinensis" International Journal of Molecular Sciences 16, no. 6: 12855-12870. https://doi.org/10.3390/ijms160612855
APA StyleZhou, X.-J., Wang, Y.-Y., Xu, Y.-N., Yan, R.-S., Zhao, P., & Liu, W.-Z. (2015). De Novo Characterization of Flower Bud Transcriptomes and the Development of EST-SSR Markers for the Endangered Tree Tapiscia sinensis. International Journal of Molecular Sciences, 16(6), 12855-12870. https://doi.org/10.3390/ijms160612855