
Specific Colon Cancer Cell Cytotoxicity Induced by Bacteriophage E Gene Expression under Transcriptional Control of Carcinoembryonic Antigen Promoter

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
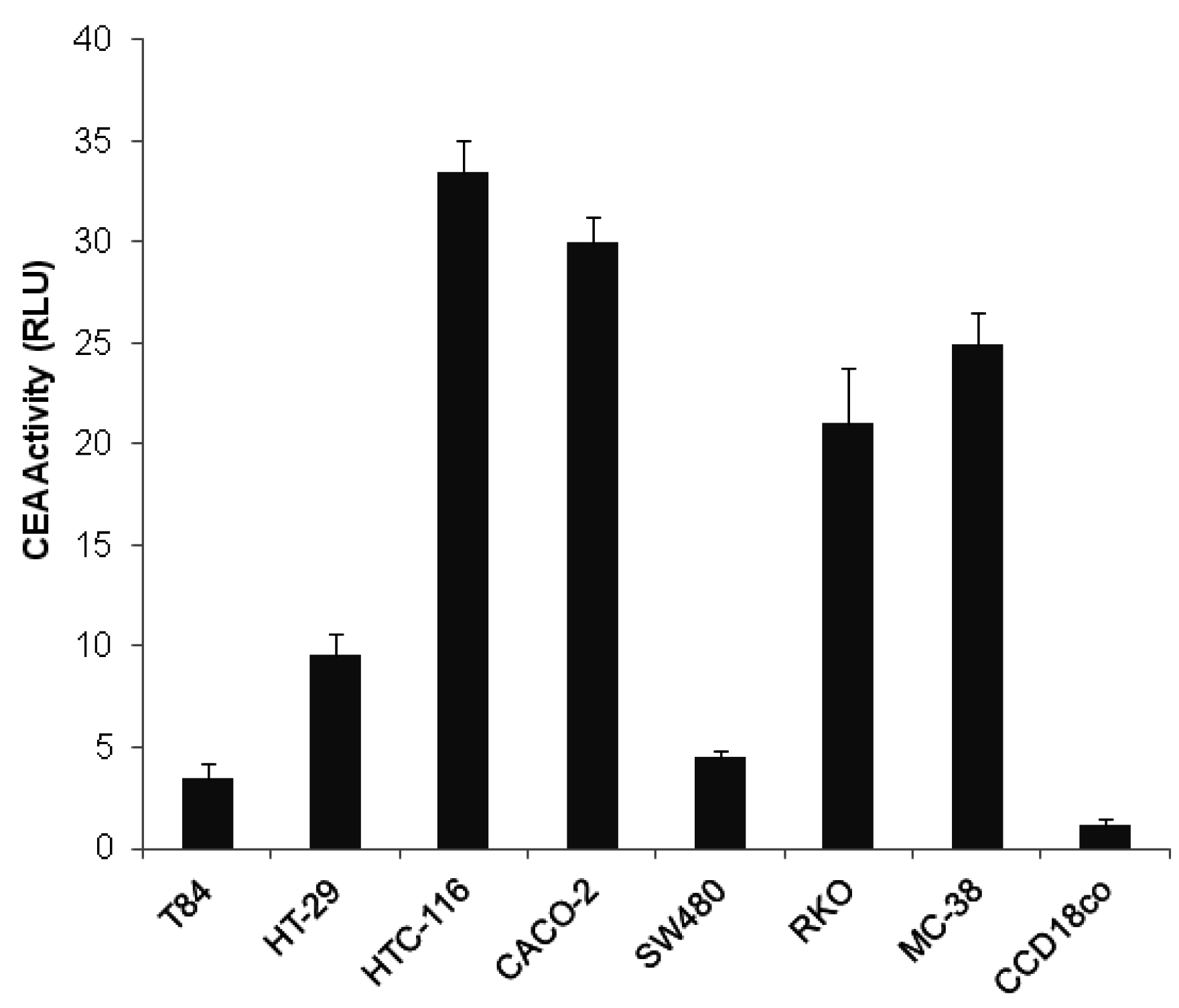
2.1. Transcriptional Activity of CEA Promoter
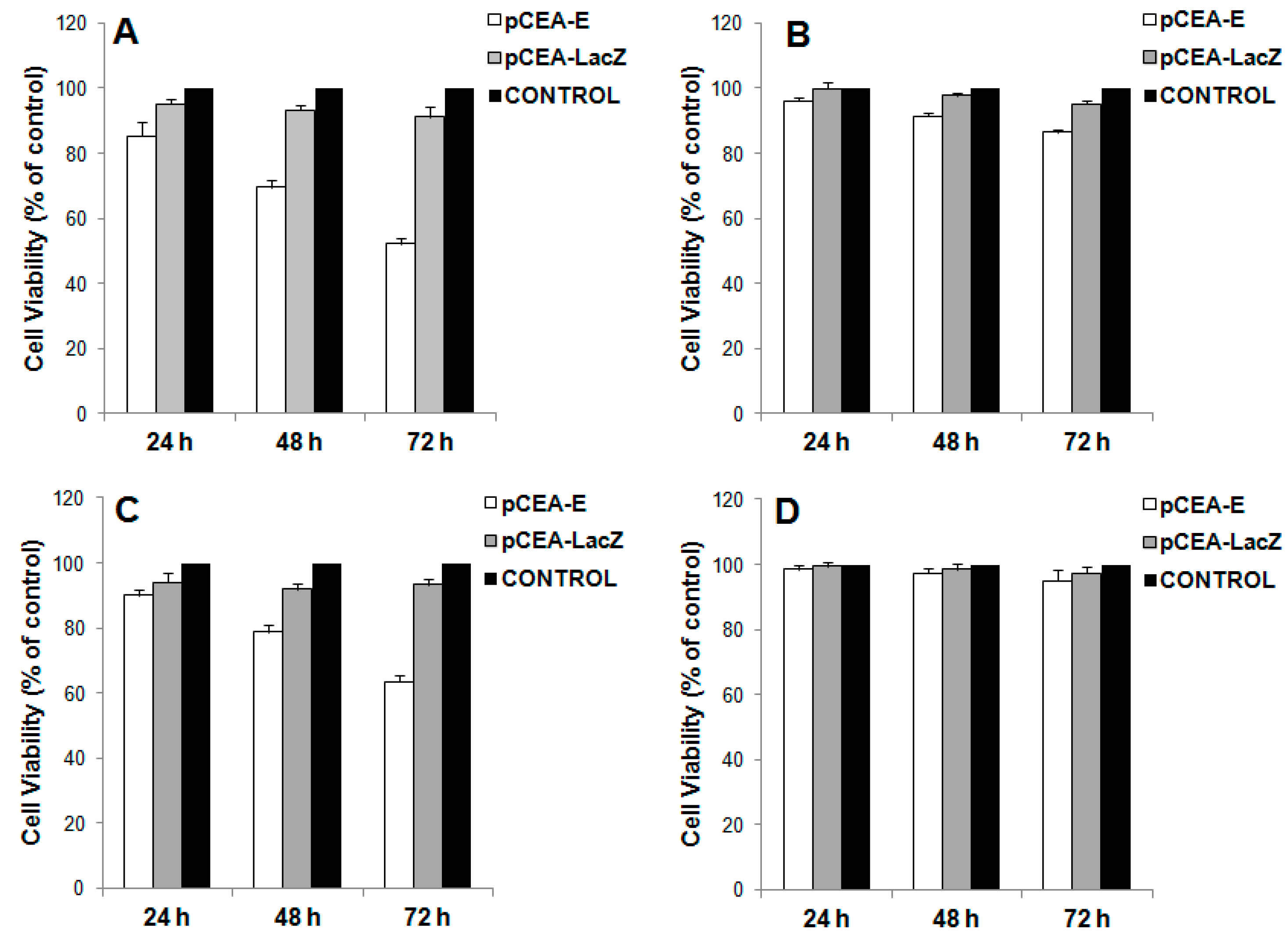
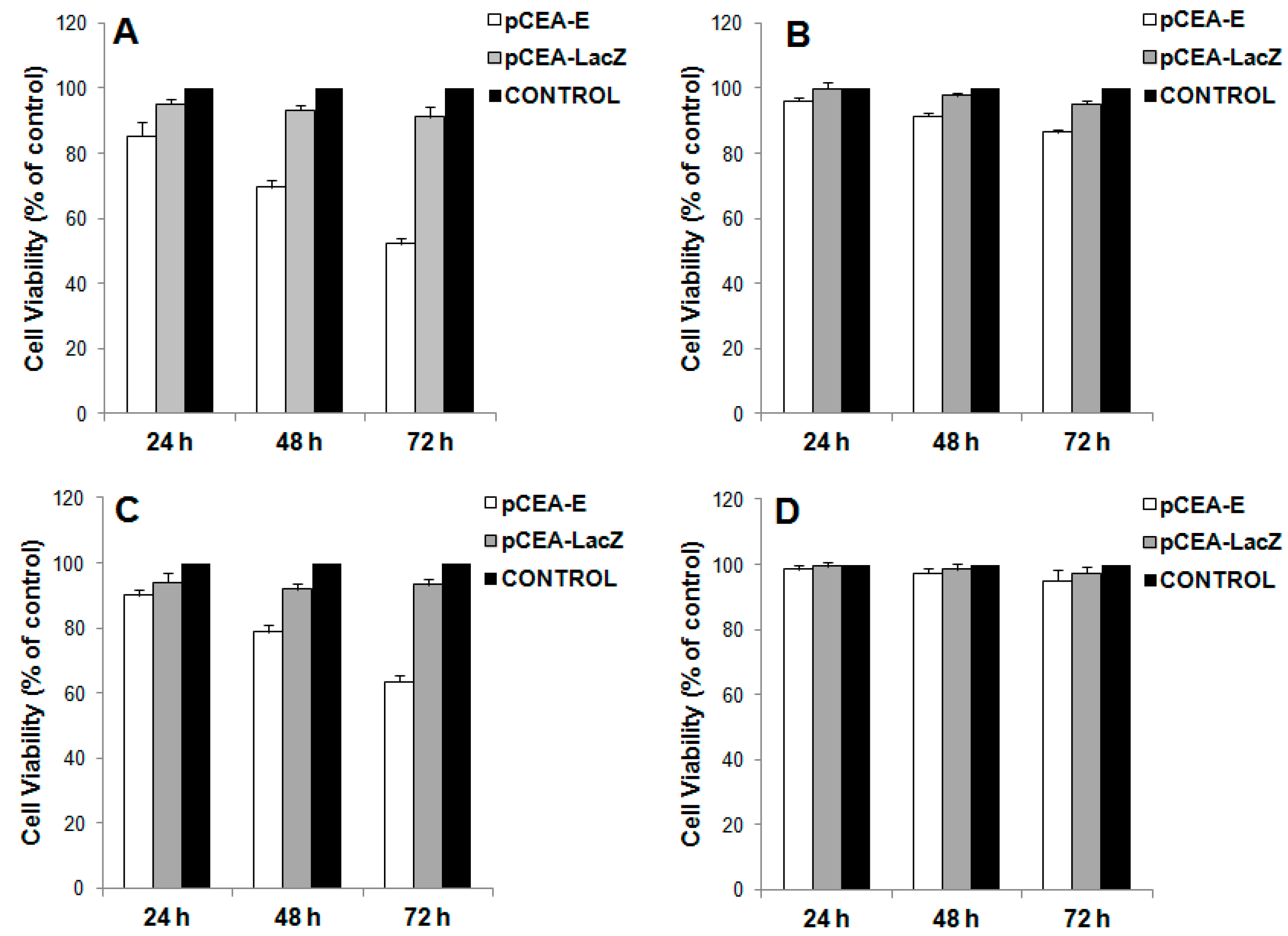
2.2. In Vitro Inhibition of Cell Growth by pCEA-E
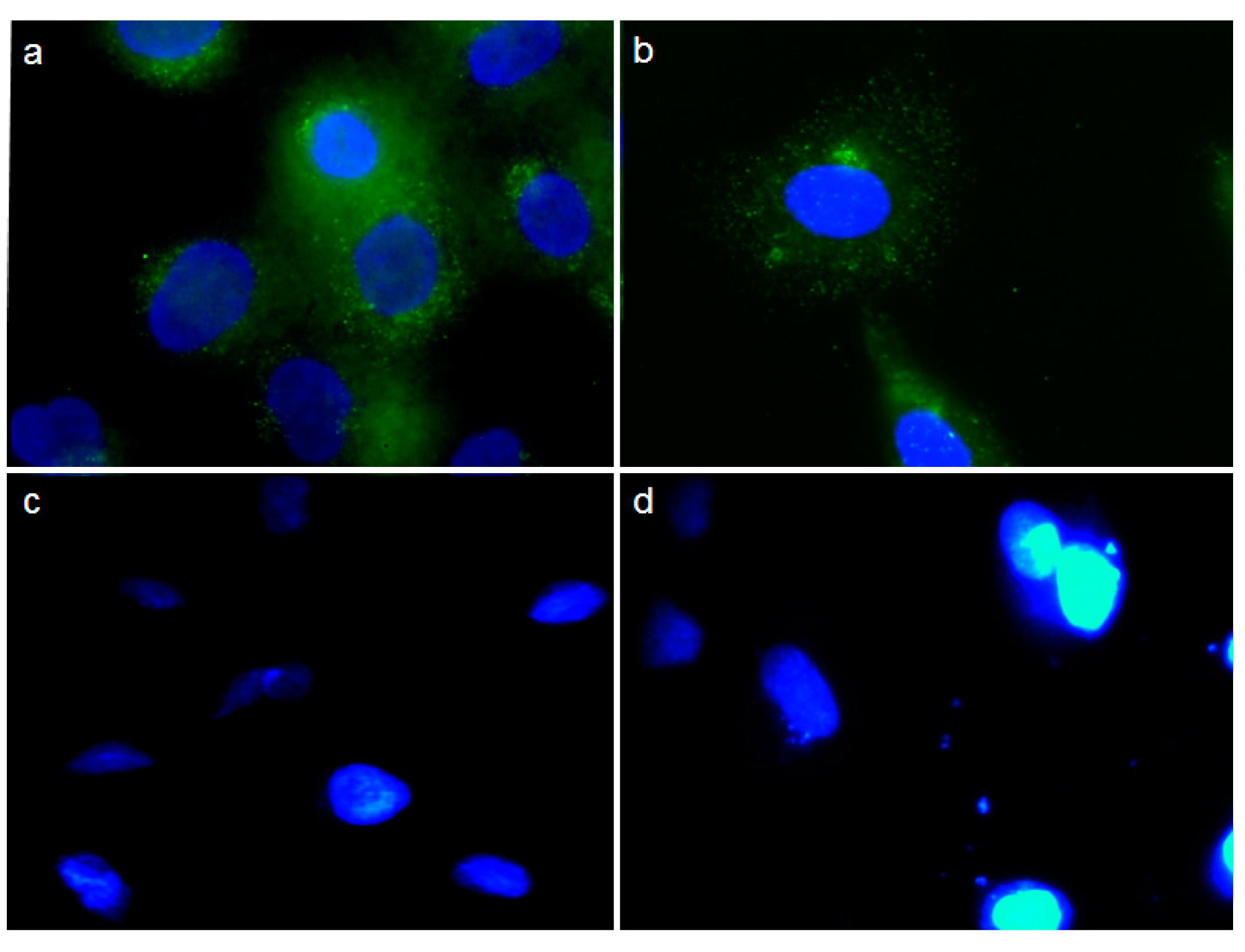
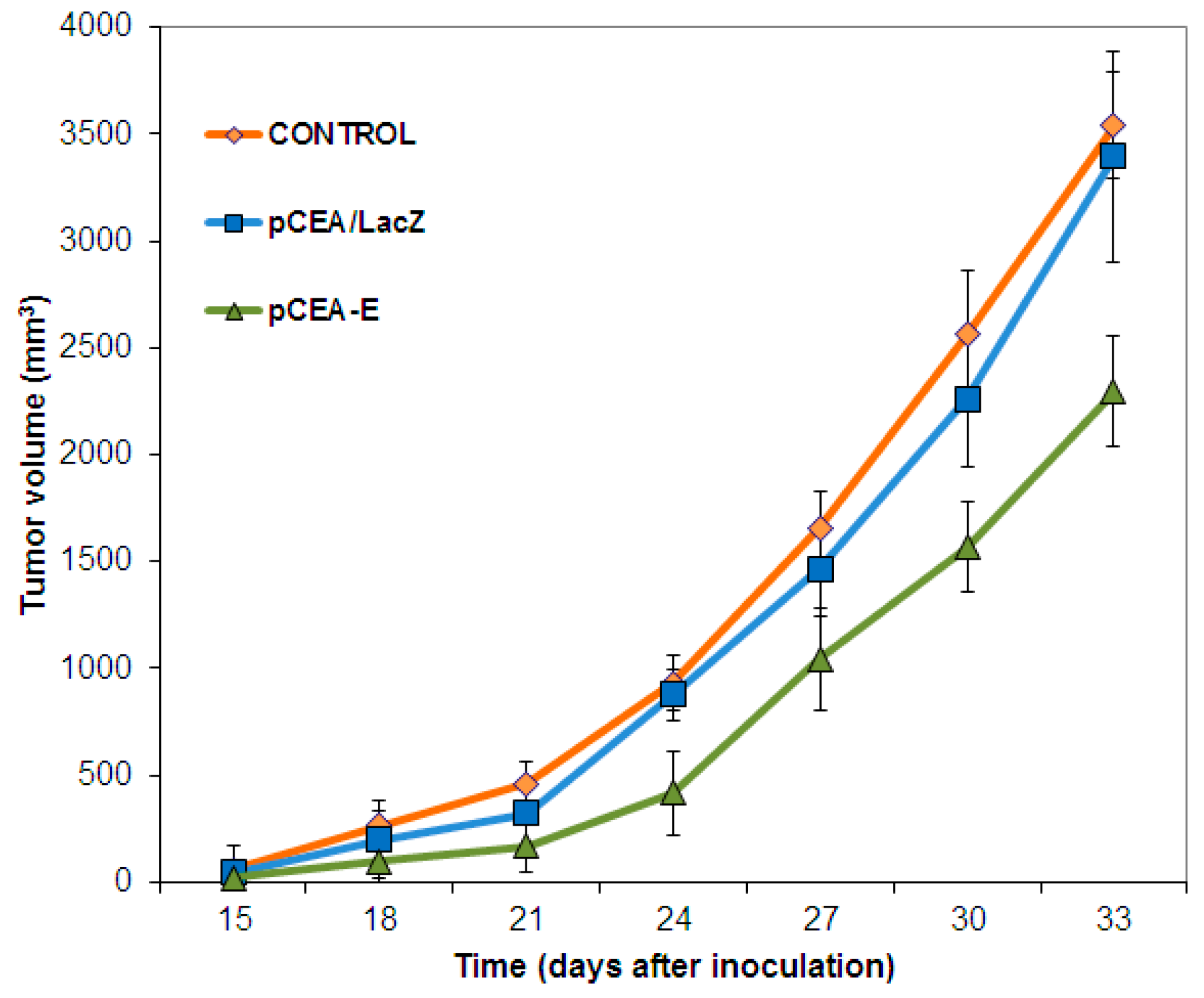
2.3. In Vivo Tumor Growth Inhibition and Survival Analysis

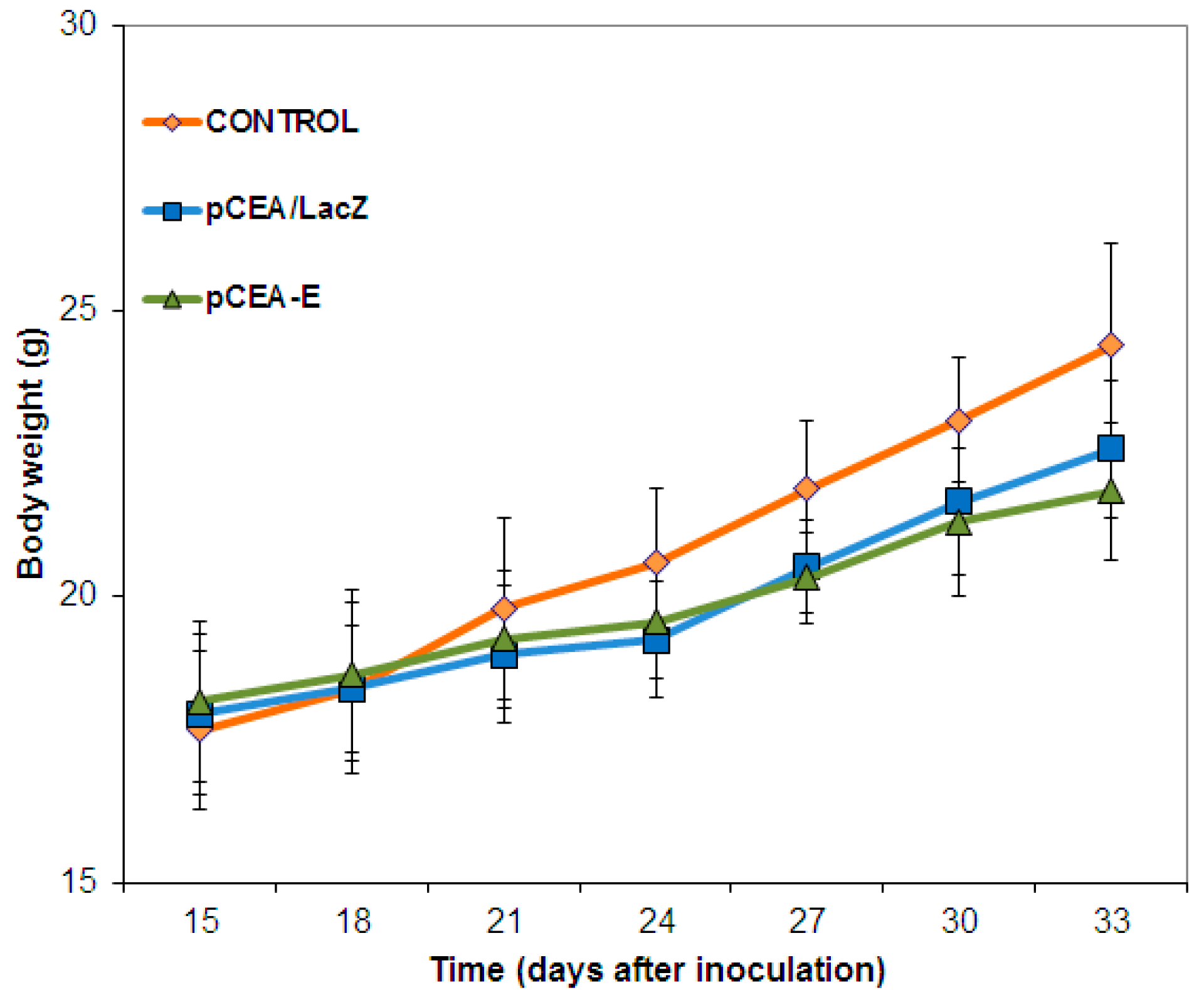
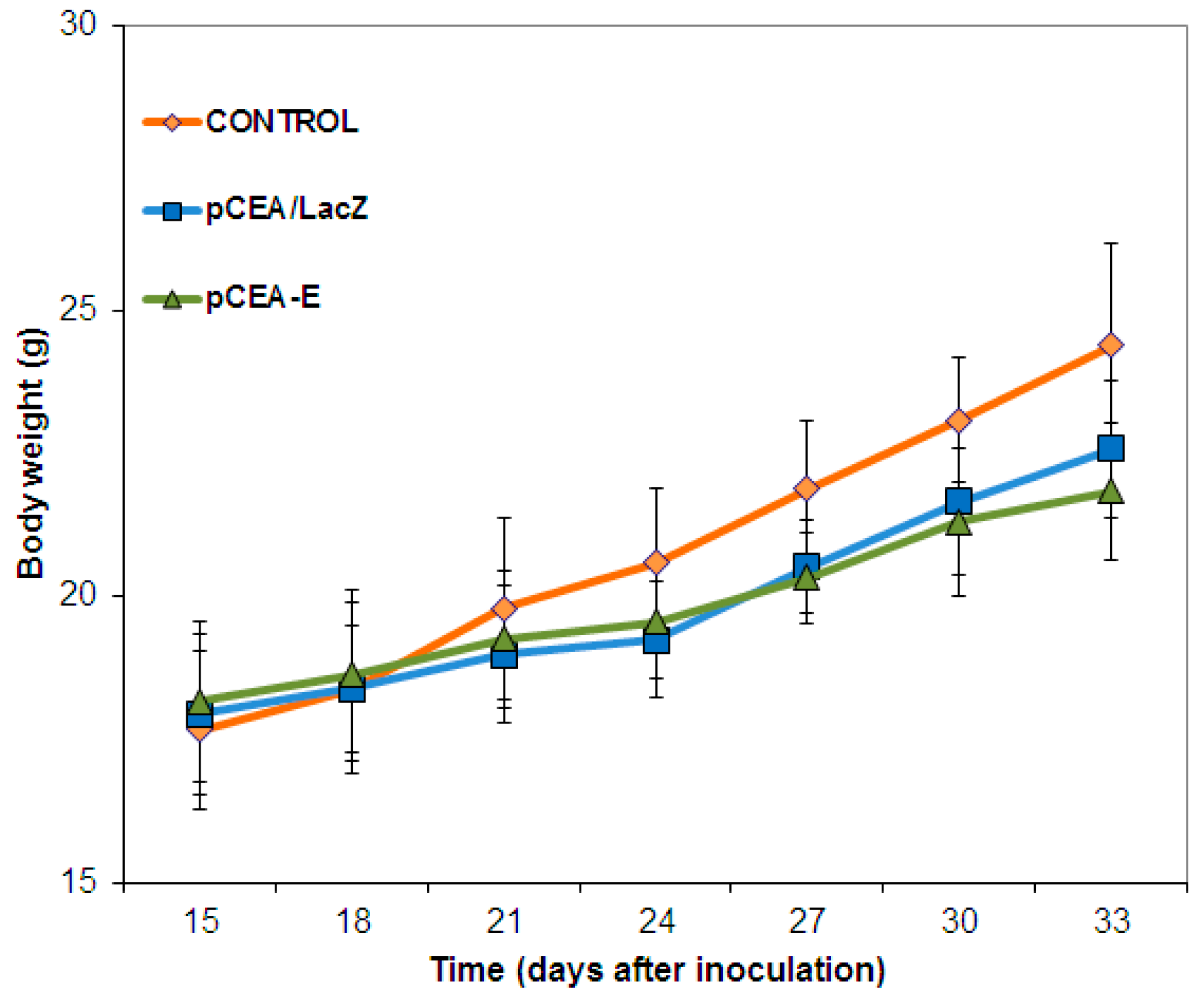
2.4. In Vivo Toxicity
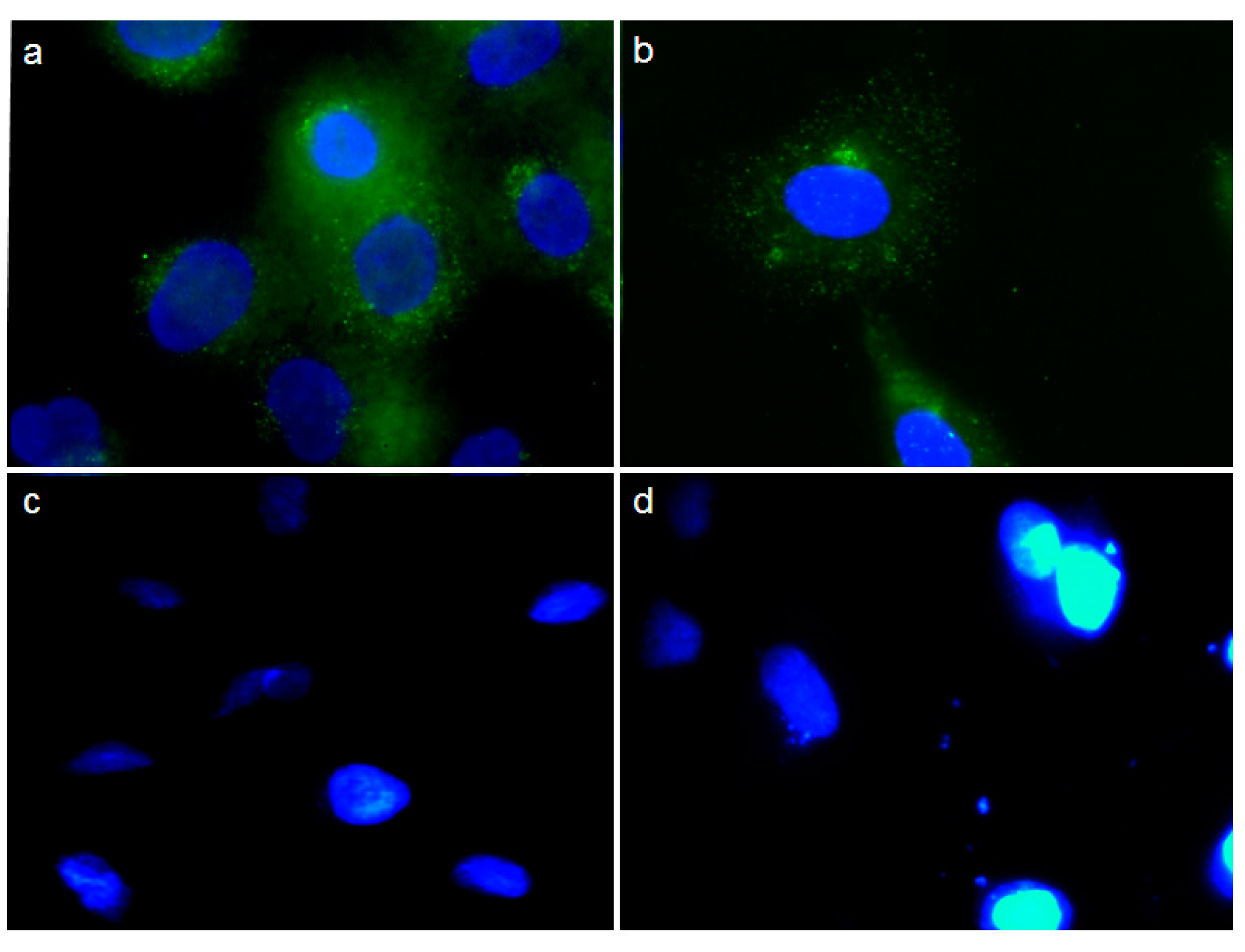
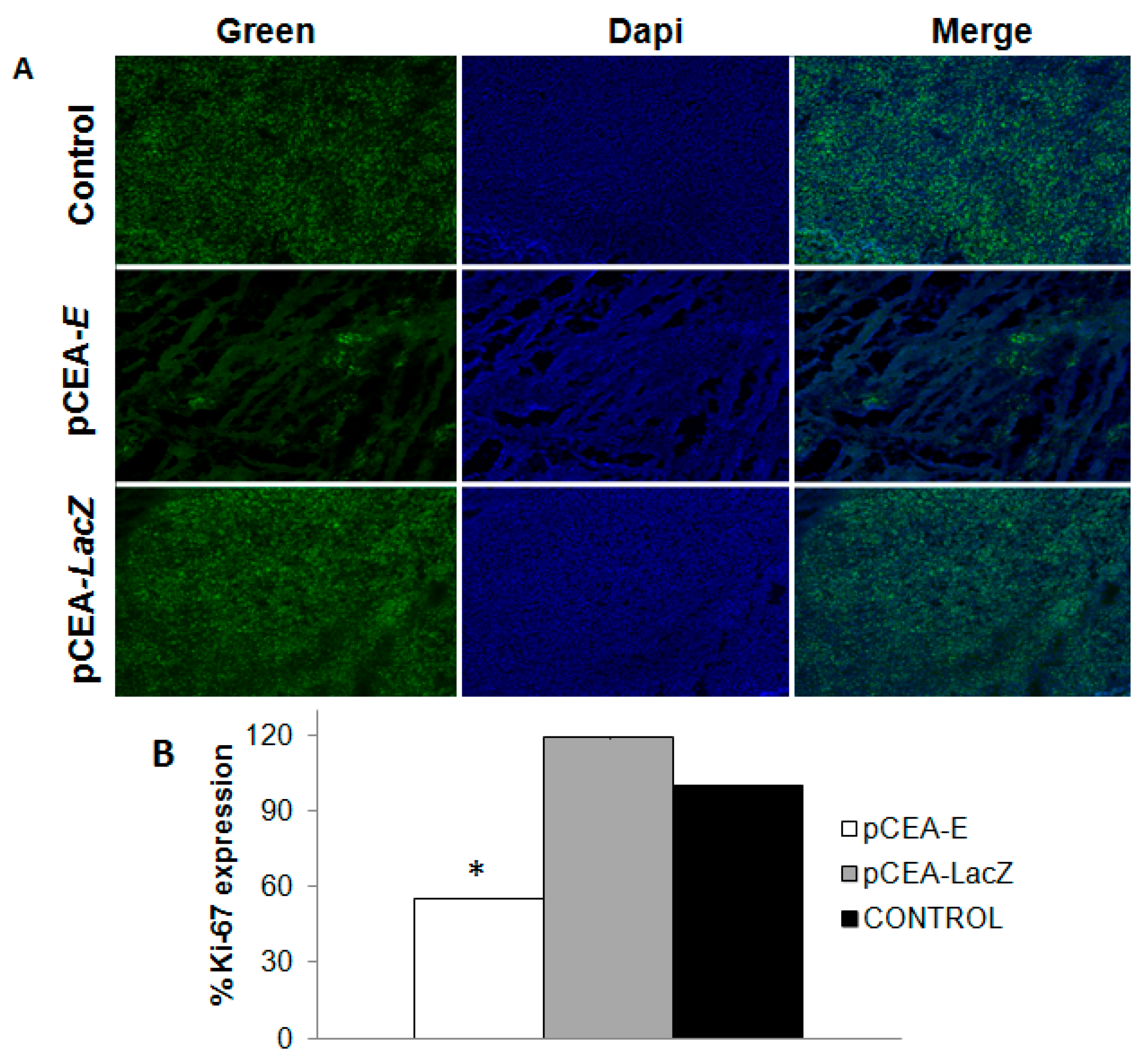
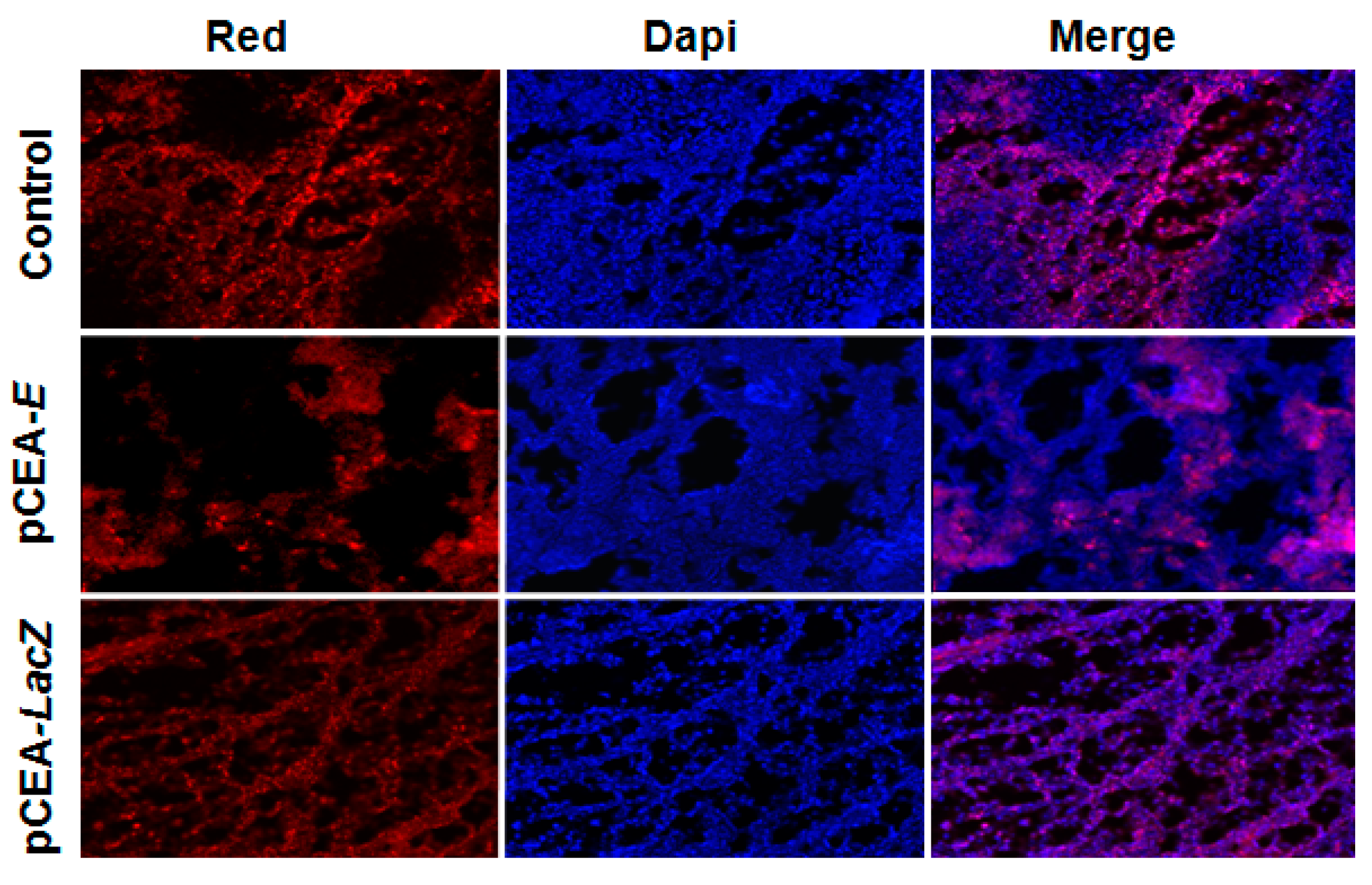
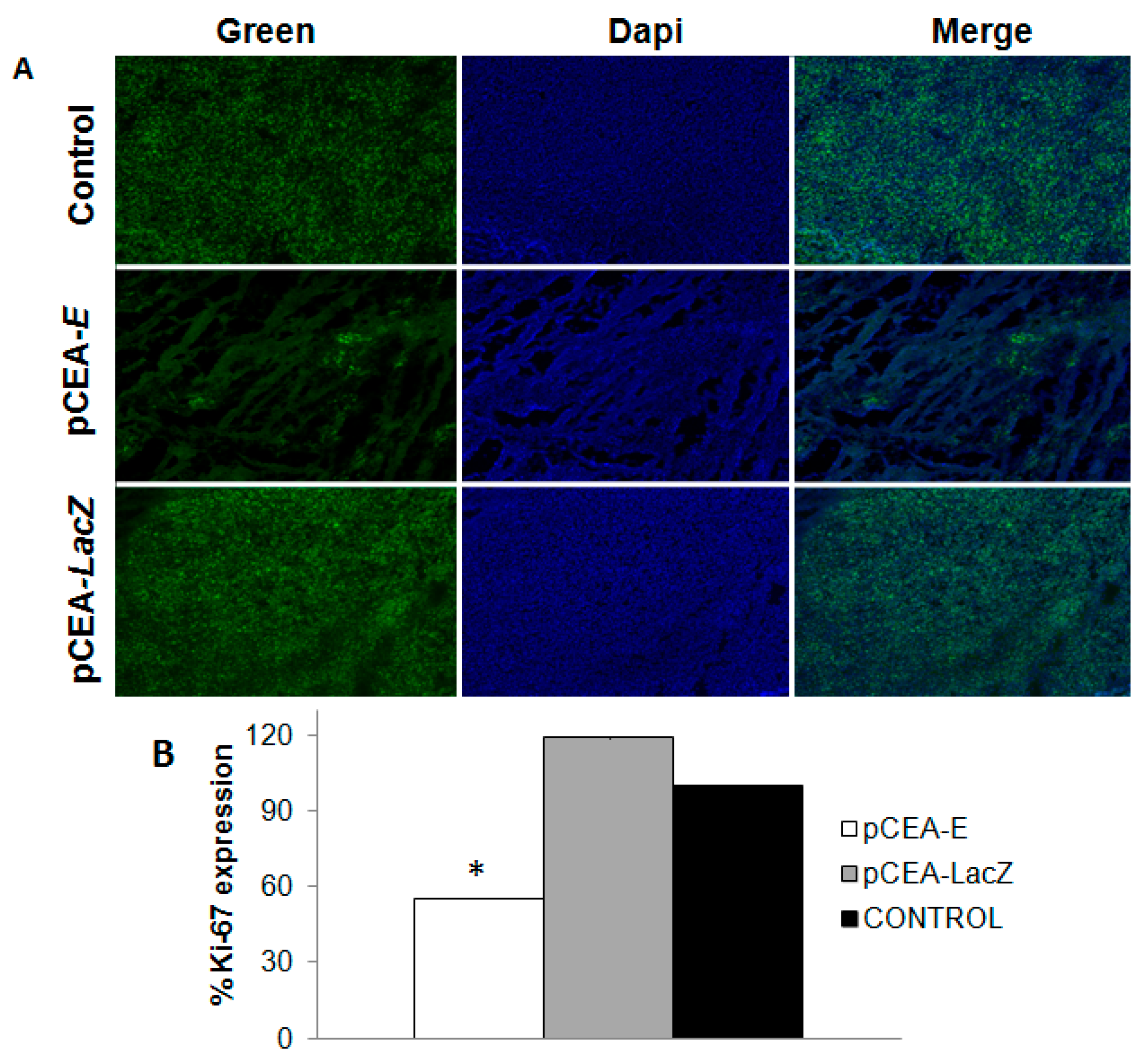
2.5. Effect of pCEA-E on Cell Proliferation and Apoptosis
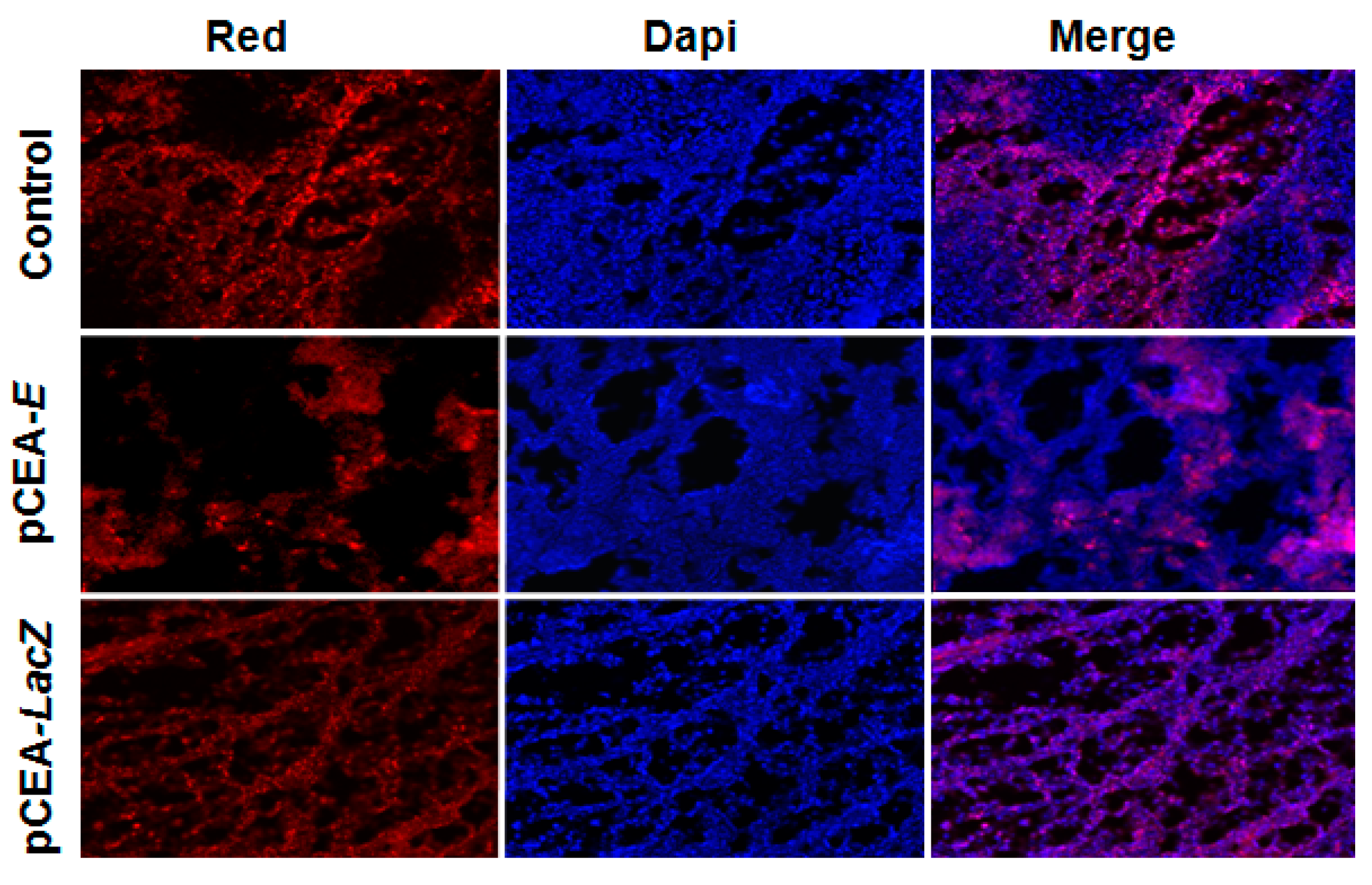
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. Construction of Luciferase and E Expression Vectors
4.3. Luciferase Assay
4.4. In Vitro Proliferation Assay
4.5. Microscopic Analysis
4.6. In Vivo Study
4.7. Histological Studies
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Labianca, R.; Beretta, G.D.; Kildani, B.; Milesi, L.; Merlin, F.; Mosconi, S.; Pessi, M.A.; Prochilo, T.; Quadri, A.; Gatta, G.; et al. Colon cancer. Crit. Rev. Oncol. Hematol. 2010, 74, 106–133. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Cripps, A.; Wei, M.Q. New strategies for cancer gene therapy: Progress and opportunities. Clin. Exp. Pharmacol. Physiol. 2010, 37, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Prados, J.; Melguizo, C.; Rama, A.R.; Ortiz, R.; Segura, A.; Boulaiz, H.; Velez, C.; Caba, O.; Ramos, J.L.; Aranega, A. Gef gene therapy enhances the therapeutic efficacy of doxorubicin to combat growth of MCF-7 breast cancer cells. Cancer Chemother. Pharmacol. 2010, 66, 69–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ortiz, R.; Prados, J.; Melguizo, C.; Arias, J.L.; Ruiz, M.A.; Alvarez, P.J.; Caba, O.; Luque, R.; Segura, A.; Aranega, A. 5-Fluorouracil-loaded poly(ε-caprolactone) nanoparticles combined with phage E gene therapy as a new strategy against colon cancer. Int. J. Nanomed. 2012, 7, 95–107. [Google Scholar]
- Rama, A.R.; Prados, J.; Melguizo, C.; Alvarez, P.J.; Ortiz, R.; Madeddu, R.; Aranega, A. E phage gene transfection associated to chemotherapeutic agents increases apoptosis in lung and colon cancer cells. Bioeng. Bugs 2011, 2, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Tian, J.; An, L.; Yang, K. Prognostic utility of gene therapy with herpes simplex virus thymidine kinase for patients with high-grade malignant gliomas: A systematic review and meta analysis. J. Neuro Oncol. 2014, 118, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Nasu, Y.; Saika, T.; Ebara, S.; Kusaka, N.; Kaku, H.; Abarzua, F.; Manabe, D.; Thompson, T.C.; Kumon, H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol. Ther. 2007, 15, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Mazzolini, G.; Ruiz, M.; Ruiz, J.; Quiroga, J.; Herrero, I.; Qian, C.; Benito, A.; Olague, C.; Larrache, J.; et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010, 17, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Amit, D.; Hochberg, A. Development of targeted therapy for a broad spectrum of cancers (pancreatic cancer, ovarian cancer, glioblastoma and HCC) mediated by a double promoter plasmid expressing diphtheria toxin under the control of H19 and IGF2-P4 regulatory sequences. Int. J. Clin. Exp. Med. 2012, 5, 296–305. [Google Scholar] [PubMed]
- Thakur, M.; Mergel, K.; Weng, A.; von Mallinckrodt, B.; Gilabert-Oriol, R.; Durkop, H.; Fuchs, H.; Melzig, M.F. Targeted tumor therapy by epidermal growth factor appended toxin and purified saponin: An evaluation of toxicity and therapeutic potential in syngeneic tumor bearing mice. Mol. Oncol. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y. Apoptin gene transfer via modified wheat histone H4 facilitates apoptosis of human ovarian cancer cells. Cancer Biother. Radiopharm. 2011, 26, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Rama, A.R.; Prados, J.; Melguizo, C.; Ortiz, R.; Alvarez, P.J.; Rodriguez-Serrano, F.; Hita, F.; Ramos, J.L.; Burgos, M.; Aranega, A. E phage gene transfection enhances sensitivity of lung and colon cancer cells to chemotherapeutic agents. Int. J. Oncol. 2010, 37, 1503–1514. [Google Scholar] [PubMed]
- Ortiz, R.; Prados, J.; Melguizo, C.; Rama, A.R.; Segura, A.; Rodriguez-Serrano, F.; Boulaiz, H.; Hita, F.; Martinez-Amat, A.; Madeddu, R.; et al. The cytotoxic activity of the phage E protein suppress the growth of murine B16 melanomas in vitro and in vivo. J. Mol. Med. Berl. 2009, 87, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Wanner, G.; Blasi, U.; Halfmann, G.; Szostak, M.; Lubitz, W. Endogenous transmembrane tunnel formation mediated by phi X174 lysis protein E. J. Bacteriol. 1990, 172, 4109–4114. [Google Scholar] [PubMed]
- Qiu, Y.; Peng, G.L.; Liu, Q.C.; Li, F.L.; Zou, X.S.; He, J.X. Selective killing of lung cancer cells using carcinoembryonic antigen promoter and double suicide genes, thymidine kinase and cytosine deaminase (pCEA-TK/CD). Cancer Lett. 2012, 316, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Hazama, S.; Araki, A.; Yoshimura, K.; Iizuka, N.; Yoshino, S.; Noma, T.; Oka, M. A novel cancer vaccine strategy with combined IL-18 and HSV-TK gene therapy driven by the hTERT promoter in a murine colorectal cancer model. Int. J. Oncol. 2014, 45, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Danda, R.; Krishnan, G.; Ganapathy, K.; Krishnan, U.M.; Vikas, K.; Elchuri, S.; Chatterjee, N.; Krishnakumar, S. Targeted expression of suicide gene by tissue-specific promoter and microRNA regulation for cancer gene therapy. PLoS ONE 2013, 8, e83398. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Li, Q.; Wang, Y.; Liu, T.; Peng, J. Effective combination gene therapy using CEACAM6-shRNA and the fusion suicide gene yCDglyTK for pancreatic carcinoma in vitro. Exp. Ther. Med. 2013, 5, 155–161. [Google Scholar] [PubMed]
- Zhou, X.; Xie, G.; Wang, S.; Wang, Y.; Zhang, K.; Zheng, S.; Chu, L.; Xiao, L.; Yu, Y.; Zhang, Y.; et al. Potent and specific antitumor effect for colorectal cancer by CEA and Rb double regulated oncolytic adenovirus harboring ST13 gene. PLoS ONE 2012, 7, e47566. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, Y.; Wang, Y.; Yan, Y.; Shi, Z.; Chen, L.; Lin, H.; Lu, S.; Zhu, M.; Su, C.; et al. CEA promoter-regulated oncolytic adenovirus-mediated Hsp70 expression in immune gene therapy for pancreatic cancer. Cancer Lett. 2012, 319, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, M.; Maeda, K.; Nagahara, H.; Ohtani, H.; Sakurai, K.; Toyokawa, T.; Kubo, N.; Tanaka, H.; Muguruma, K.; Ohira, M.; et al. Significance of CEA and CA19-9 combination as a prognostic indicator and for recurrence monitoring in patients with stage II colorectal cancer. Anticancer Res. 2014, 34, 3753–3758. [Google Scholar] [PubMed]
- Vukobrat-Bijedic, Z.; Husic-Selimovic, A.; Sofic, A.; Bijedic, N.; Bjelogrlic, I.; Gogov, B.; Mehmedovic, A. Cancer antigens (CEA and CA 19-9) as markers of advanced stage of colorectal carcinoma. Med. Arch. 2013, 67, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Michl, M.; Koch, J.; Laubender, R.P.; Modest, D.P.; Giessen, C.; Schulz, C.; Heinemann, V. Tumor markers CEA and CA 19-9 correlate with radiological imaging in metastatic colorectal cancer patients receiving first-line chemotherapy. Tumour Biol. 2014, 35, 10121–10127. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Zhang, X.; Jing, J.; Zhao, X.; Wang, Y.; Han, C. Evaluating the significance of expression of CEA mRNA and levels of CEA and its related proteins in colorectal cancer patients. J. Surg. Oncol. 2014, 109, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M. Getting into the colon: Approaches to target colorectal cancer. Expert Opin. Drug Deliv. 2014, 11, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, T.; Chen, Y.H.; Chen, Y.; Xu, M.; Peng, J.; Yu, S.; Yuan, J.; Zhang, X. Tissue specific cytotoxicity of colon cancer cells mediated by nanoparticle-delivered suicide gene in vitro and in vivo. Clin. Cancer Res. 2009, 15, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.Y.; Wang, J.P.; Gui, Z.F.; Shen, L.Z. Antitumor activity of mutant bacterial cytosine deaminase gene for colon cancer. World J. Gastroenterol. 2011, 17, 2958–2964. [Google Scholar] [CrossRef] [PubMed]
- Yawata, T.; Maeda, Y.; Okiku, M.; Ishida, E.; Ikenaka, K.; Shimizu, K. Identification and functional characterization of glioma-specific promoters and their application in suicide gene therapy. J. Neuro Oncol. 2011, 104, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Kagiava, A.; Sargiannidou, I.; Bashiardes, S.; Richter, J.; Schiza, N.; Christodoulou, C.; Gritti, A.; Kleopa, K.A. Gene delivery targeted to oligodendrocytes using a lentiviral vector. J. Gene Med. 2014, 16, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Evans, T.R.; Somanath, S.; Armesilla, A.L.; Darling, J.L.; Schatzlein, A.; Cassidy, J.; Wang, W. In vitro evaluation of cancer-specific NF-κB-CEA enhancer-promoter system for 5-fluorouracil prodrug gene therapy in colon cancer cell lines. Br. J. Cancer 2007, 97, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, A.; Szary, J.; Kowalczuk, M.; Szala, S.; Ugorski, M. CEA-negative glioblastoma and melanoma cells are sensitive to cytosine deaminase/5-fluorocytosine therapy directed by the carcinoembryonic antigen promoter. Acta Biochim. Pol. 2004, 51, 723–732. [Google Scholar] [PubMed]
- Liu, T.; Zhang, G.; Chen, Y.H.; Chen, Y.; Liu, X.; Peng, J.; Xu, M.H.; Yuan, J.W. Tissue specific expression of suicide genes delivered by nanoparticles inhibits gastric carcinoma growth. Cancer Biol. Ther. 2006, 5, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Okabe, S.; Arai, T.; Yamashita, H.; Sugihara, K. Adenovirus-mediated prodrug-enzyme therapy for CEA-producing colorectal cancer cells. J. Cancer Res. Clin. Oncol. 2003, 129, 367–373. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rama, A.R.; Hernandez, R.; Perazzoli, G.; Burgos, M.; Melguizo, C.; Vélez, C.; Prados, J. Specific Colon Cancer Cell Cytotoxicity Induced by Bacteriophage E Gene Expression under Transcriptional Control of Carcinoembryonic Antigen Promoter. Int. J. Mol. Sci. 2015, 16, 12601-12615. https://doi.org/10.3390/ijms160612601
Rama AR, Hernandez R, Perazzoli G, Burgos M, Melguizo C, Vélez C, Prados J. Specific Colon Cancer Cell Cytotoxicity Induced by Bacteriophage E Gene Expression under Transcriptional Control of Carcinoembryonic Antigen Promoter. International Journal of Molecular Sciences. 2015; 16(6):12601-12615. https://doi.org/10.3390/ijms160612601
Chicago/Turabian StyleRama, Ana R., Rosa Hernandez, Gloria Perazzoli, Miguel Burgos, Consolación Melguizo, Celia Vélez, and Jose Prados. 2015. "Specific Colon Cancer Cell Cytotoxicity Induced by Bacteriophage E Gene Expression under Transcriptional Control of Carcinoembryonic Antigen Promoter" International Journal of Molecular Sciences 16, no. 6: 12601-12615. https://doi.org/10.3390/ijms160612601
APA StyleRama, A. R., Hernandez, R., Perazzoli, G., Burgos, M., Melguizo, C., Vélez, C., & Prados, J. (2015). Specific Colon Cancer Cell Cytotoxicity Induced by Bacteriophage E Gene Expression under Transcriptional Control of Carcinoembryonic Antigen Promoter. International Journal of Molecular Sciences, 16(6), 12601-12615. https://doi.org/10.3390/ijms160612601