Abstract
Kynurenines are the products of tryptophan metabolism. Among them, kynurenine and kynurenic acid are generally thought to have neuroprotective properties, while 3-hydroxykynurenine, 3-hydroxyanthranilic acid and quinolinic acid are considered neurotoxic. They participate in immunoregulation and inflammation and possess pro- or anti-excitotoxic properties, and their involvement in oxidative stress has also been suggested. Consequently, it is not surprising that kynurenines have been closely related to neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis and multiple sclerosis. More information about the less-known metabolites, picolinic and cinnabarinic acid, evaluation of new receptorial targets, such as aryl-hydrocarbon receptors, and intensive research on the field of the immunomodulatory function of kynurenines delineated the high importance of this pathway in general homeostasis. Emerging knowledge about the kynurenine pathway provides new target points for the development of therapeutical solutions against neurodegenerative diseases.
1. Introduction
Nowadays, the interpretation of neurotoxicity is not confined to the idea of external substances causing neuronal damage. Basically, every substance or phenomenon, whether it is internal or external, causing damage to the neurons is considered neurotoxic.
Neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), etc., all possess characteristics related to neurotoxic processes. Consequently, neurodegeneration in general could be attributable to various forms of neurotoxicity. Neurodegenerative diseases have different courses and distinct clinical symptoms; however, they share some common mechanisms, which eventually lead to neuronal death. These processes are the imbalance in intracellular energy homeostasis, excitotoxicity and inflammation [1,2,3]. A long line of evidence proves that the kynurenine pathway (KP) of tryptophan (TRP) metabolism and the pathomechanism of neurodegenerative diseases are associated at several points [4]. This review will focus on the properties of kynurenine metabolites related to neurotoxicity also occurring in neurodegenerative diseases.
2. Common Neurotoxic Mechanisms in Neurodegeneration
The neural tissue is the main energy consumer of the human body, and imbalance in energy homeostasis can lead to neuronal deficit and, eventually, to neuronal death. The energy, in the form of ATP, is provided by the mitochondria. These organelles are the scenes of the citric acid cycle, fatty acid oxidation, the urea cycle and oxidative phosphorylation. Impaired mitochondrial function leads to energy deficit (lack of ATP), which, in turn, leads to the disruption of Na+/K+-ATP-ase, Ca2+/H+-ATP-ase and the reversion of the Na+/Ca2+ transporter [5]. Under these circumstances, the cells are not able to maintain their normal membrane potential, resulting in depolarization. Cells with disrupted membrane potential are more prone to excitotoxic and oxidative damage [6].
Furthermore, impaired mitochondrial function causes the uncontrolled generation of reactive oxygen (ROS) and nitrogen species (RNS): superoxide anion (O2•−), hydroxyl radical (•OH), hydrogen peroxide (H2O2), nitric oxide (•NO), nitrogen dioxide (•NO2) and peroxynitrite anion (ONOO−). ROS and RNS can attack macromolecules, resulting in misfolded proteins, lipid peroxidation or nitrosylation and nucleic acid damage. However, mitochondria are not the only source of ROS in the cells; peroxisomes and the endoplasmic reticulum are also capable of ROS production. Besides, numerous enzymes are known to produce ROS, too, e.g., NADPH oxidases, cyclooxygenases, xanthine oxidase, cytochrome P450 enzymes and nitrogen oxide synthases [7]. The excess amount of reactive species, i.e., oxidative stress, is closely related to neurodegenerative diseases.
ROS can also originate from microglia, which are likely to contribute to the degenerative course after activation. Microglia are the resident immune cells of the central nervous system (CNS), providing defense against external pathogens and pollutants and clearing of cellular debris. Activated microglia are present in diseased brains [8,9,10], indicating the contribution of inflammatory processes to neurodegeneration [11,12]. Chronic neuroinflammation, prolonged activation of microglia and astrocytes and persistent exposure to inflammatory cytokines are considered neurotoxic.
Excitotoxicity is the neuronal death caused by excessive or prolonged activation of excitatory amino acid receptors. The main participant in this process is glutamate, acting on ionotropic N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainate receptors and on metabotropic glutamate receptors [3]. An excessive amount of glutamate in the synaptic cleft can result in the dysregulation of Ca2+ homeostasis, mitochondrial dysfunction and the generation of ROS and RNS. Under normal conditions, the presence and amount of glutamate is highly regulated, but in neurodegenerative diseases, this regulation is often disrupted, contributing to neuronal damage.
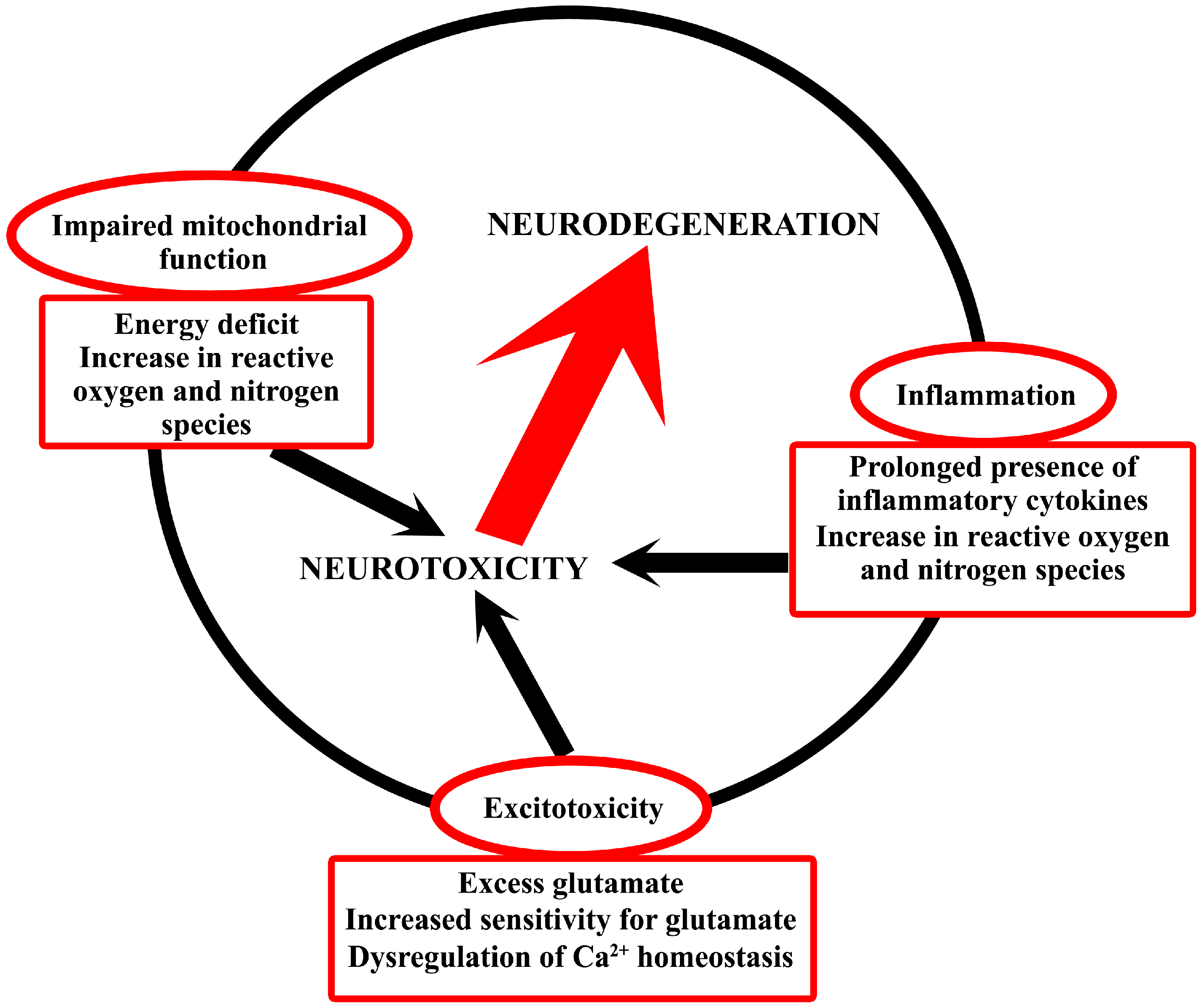
The mechanisms detailed above are in close relation in the development of neurodegenerative diseases [13,14,15] (Figure 1); however, their level of contribution to the pathological phenomena varies among the different disorders.
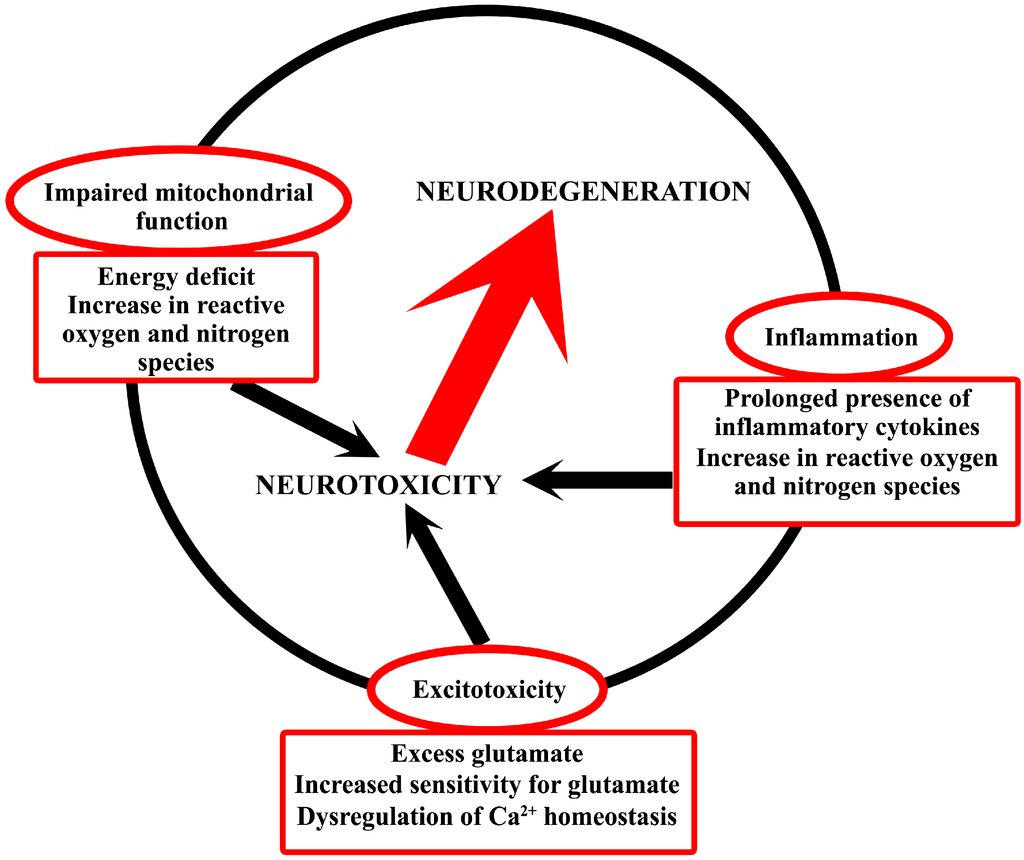
Figure 1.
Schematic drawing of the common neurotoxic mechanisms in neurodegeneration. The figure attempts to interpret that the involvement and interrelationship of metabolic disturbances, neuroinflammation and excitotoxicity causes neurotoxicity that eventually results in neurodegeneration.
Figure 1.
Schematic drawing of the common neurotoxic mechanisms in neurodegeneration. The figure attempts to interpret that the involvement and interrelationship of metabolic disturbances, neuroinflammation and excitotoxicity causes neurotoxicity that eventually results in neurodegeneration.
3. The Kynurenine Pathway
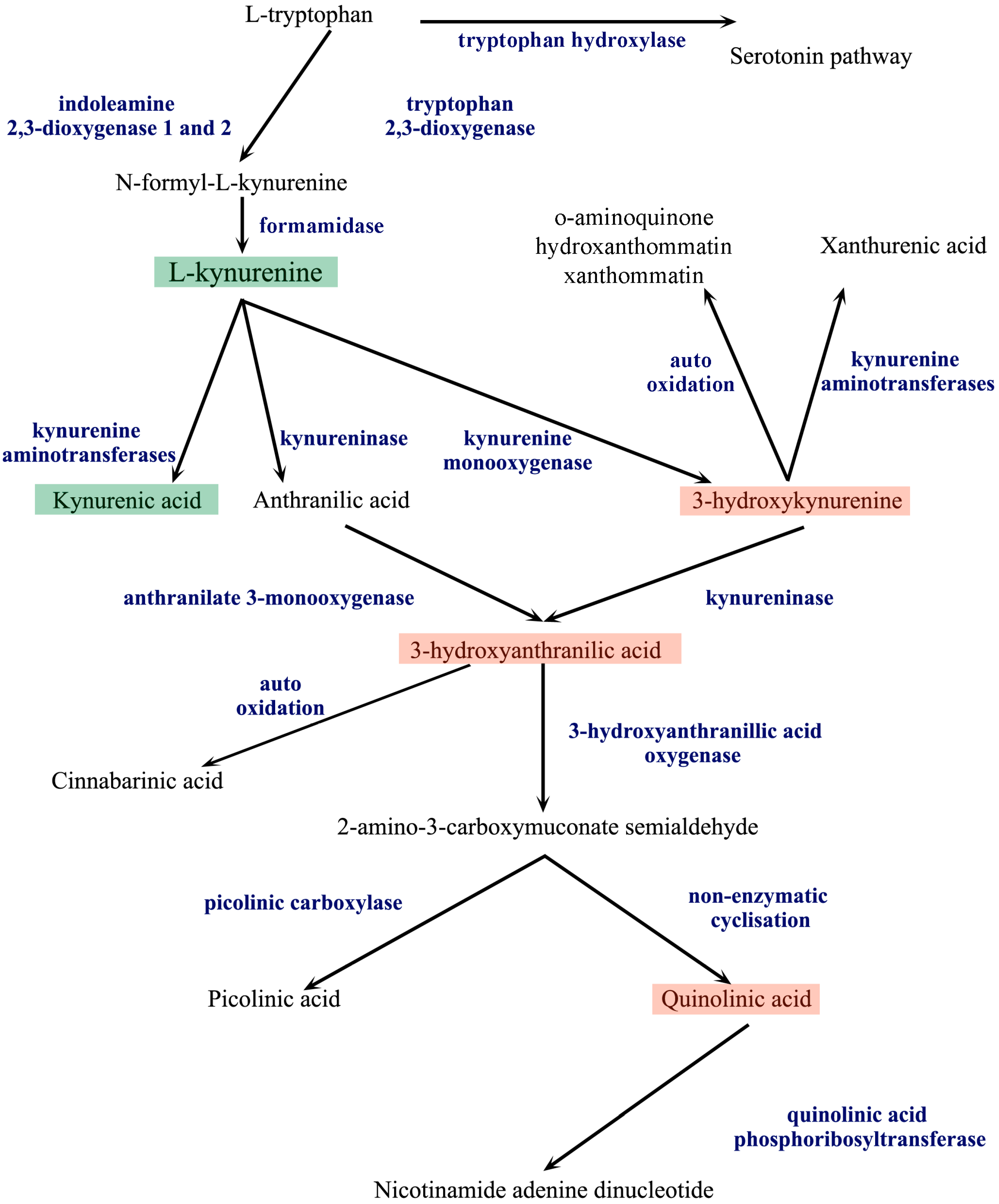
The main route of TRP metabolism is the kynurenine pathway (KP) (Figure 2), yielding neuroactive metabolites and nicotinamide adenine dinucleotide (NAD+). More than 95% of TRP is metabolized through the KP [16], while the remaining TRP is metabolized by the serotonin pathway.
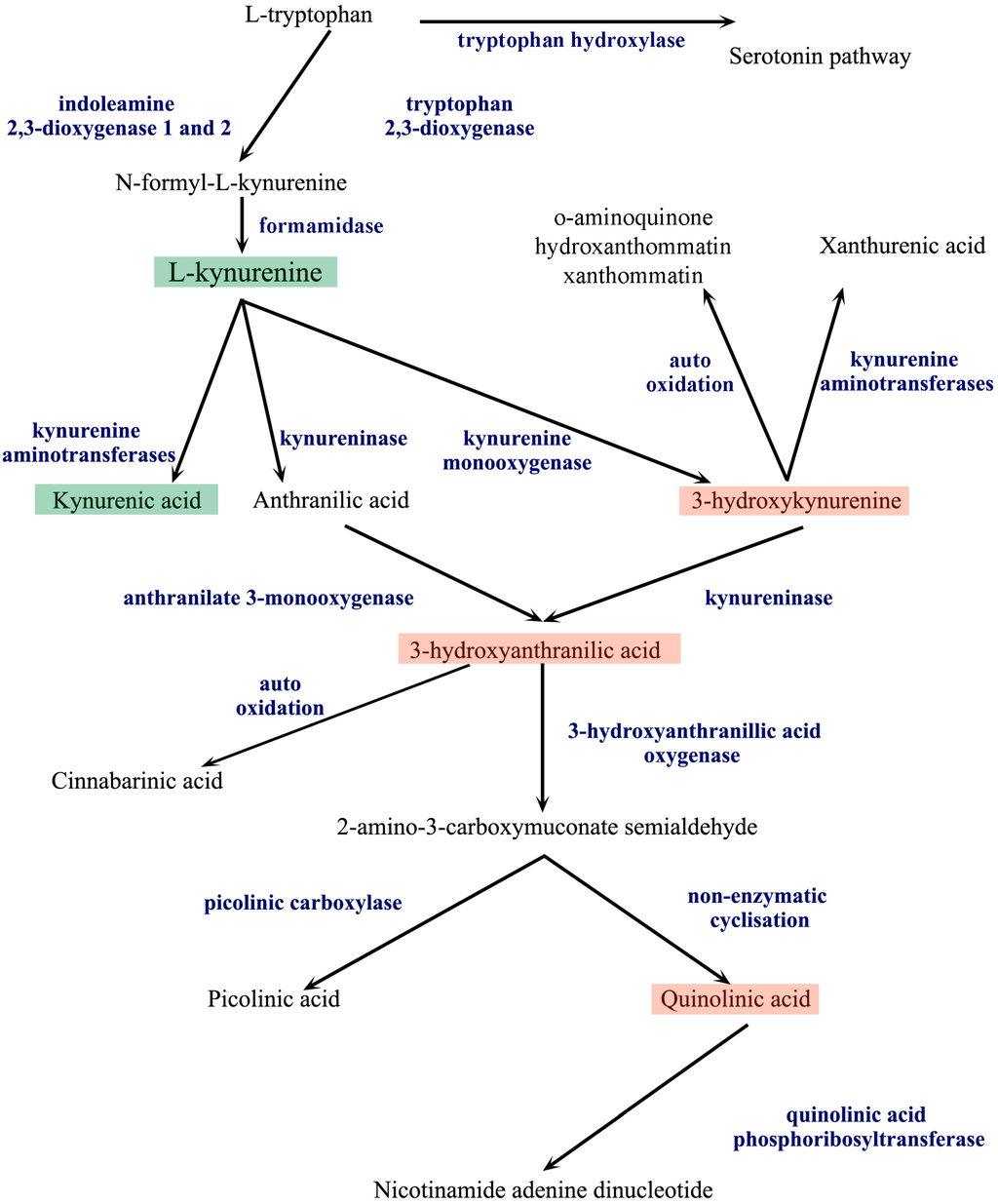
Figure 2.
The kynurenine pathway. The metabolism of l-tryptophan is divided into two distinct pathways, the serotonin and the kynurenine pathway (KP). Indoleamine 2,3-dioxygenase 1 and 2 and tryptophan 2,3-dioxygenase convert l-tryptophan to N-formyl-l-kynurenine in the first step of the KP. N-formyl-l-kynurenine is further processed by formamidase to l-kynurenine (l-KYN), the central metabolite of the KP. From l-KYN, three different enzymes produce the next metabolites, forming three branches of the metabolism. The first branch is the kynurenic acid branch, where kynurenine aminotransferases (KATs) produce kynurenic acid from l-KYN. On the second branch, kynureninase converts l-KYN to anthranilic acid, which is further metabolized by anthranilate 3-monoxygenase to 3-hydroxyanthranilic acid (3-HA). On the third branch, kynurenine monooxygenase produces 3-hydroxykynurenine (3-HK), which is further metabolized by kynureninase to 3-HA. 3-HK can be also metabolized by KATs to form xanthurenic acid or be auto-oxidized. 3-HA is converted by 3-hydroxyanthranilic acid oxygenase to 2-amino-3-carboxymuconate semialdehyde or suffers auto-oxidation to form cinnabarinic acid. 2-amino-3-carboxymuconate semialdehyde can be converted by picolinic carboxylase to picolinic acid or can be converted by non-enzymatic cyclisation to quinolinic acid, which, through conversion by quinolinic acid phosphoribosyltransferase, results in the formation of nicotinamide adenine dinucleotide.
Figure 2.
The kynurenine pathway. The metabolism of l-tryptophan is divided into two distinct pathways, the serotonin and the kynurenine pathway (KP). Indoleamine 2,3-dioxygenase 1 and 2 and tryptophan 2,3-dioxygenase convert l-tryptophan to N-formyl-l-kynurenine in the first step of the KP. N-formyl-l-kynurenine is further processed by formamidase to l-kynurenine (l-KYN), the central metabolite of the KP. From l-KYN, three different enzymes produce the next metabolites, forming three branches of the metabolism. The first branch is the kynurenic acid branch, where kynurenine aminotransferases (KATs) produce kynurenic acid from l-KYN. On the second branch, kynureninase converts l-KYN to anthranilic acid, which is further metabolized by anthranilate 3-monoxygenase to 3-hydroxyanthranilic acid (3-HA). On the third branch, kynurenine monooxygenase produces 3-hydroxykynurenine (3-HK), which is further metabolized by kynureninase to 3-HA. 3-HK can be also metabolized by KATs to form xanthurenic acid or be auto-oxidized. 3-HA is converted by 3-hydroxyanthranilic acid oxygenase to 2-amino-3-carboxymuconate semialdehyde or suffers auto-oxidation to form cinnabarinic acid. 2-amino-3-carboxymuconate semialdehyde can be converted by picolinic carboxylase to picolinic acid or can be converted by non-enzymatic cyclisation to quinolinic acid, which, through conversion by quinolinic acid phosphoribosyltransferase, results in the formation of nicotinamide adenine dinucleotide.
In the first step of the KP, TRP is converted to N-formyl-l-kynurenine, an instable compound, by the enzymes tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase 1 and 2 (IDO1, IDO2). These enzymes are the main rate limiting enzymes of the KP. Interferon-α, interferon-β (IFN-β) and interferon-γ (IFN-γ) are all able to induce IDO [17,18], but IFN-γ is considered as the main activator of IDO [19]. Furthermore, tumor necrosis factor-α and interleukin-6 are also able to activate IDO in an IFN-γ-independent way [20]. Previous investigations also showed that IDO has an important role in maternal-fetal tolerance, and both enzymes bear immunosuppressive properties [21,22].
N-formyl-l-kynurenine is further degraded by formamidase to l-kynurenine (l-KYN). l-KYN was long thought not to have neuroactive properties. l-KYN participation in oxidative processes has been proposed; pro-and also anti-oxidative properties have been documented [23].
Recently, evidence emerged that l-KYN is an endogenous ligand for the human aryl-hydrocarbon receptor (AHR) (Table 1) [24]. This receptor has an important role in cellular responses evoked by environmental toxins, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin [25] and polycyclic aromatic hydrocarbons [26,27,28]. The AHR is a ligand-activated transcription factor, a member of the family of basic helix-loop-helix transcription factors [29]. It is a cytosolic protein that is normally inactive, but after ligand binding, it translocates to the nucleus, where it binds to the regulatory regions of xenobiotic-responsive elements. DNA binding indicates diverse transcriptional responses, such as the activation of enzymes participating in xenobiotic metabolism. AHR also participates in immune response [30,31] and in tumor genesis [32]. Besides environmental pollutants, numerous endogenous AHR ligands have been identified, including kynurenines l-KYN and kynurenic acid (KYNA) [33], suggesting the involvement of the KP in the diverse transcriptional pathways, regulated by AHR.
In the CNS, 40% of l-KYN is generated locally, and 60% is taken up from the blood [34]. The KP continues in different branches deriving from l-KYN, the first one leading to the synthesis of KYNA by kynurenine aminotransferases (KATs). KYNA is present in the human and rat brain in nanomolar concentrations [35], and it is an endogenous broad-spectrum antagonist of NMDA receptors acting at low concentrations on the strychnine-insensitive glycine site [36], while at higher concentrations on the glutamate recognition site (Table 1) [37]. Furthermore, it is a weak antagonist of kainate and AMPA ionotropic glutamate receptors [38]. At low concentrations (nM–μM), KYNA has a facilitatory effect on AMPA receptors, while at higher concentrations, it acts as a competitive antagonist [39,40]. The broad spectrum of KYNA’s receptorial action is in favor of its important role in regulation of glutamatergic neurotransmission.
On α7-nicotinic acetylcholine receptors (α7nAch), KYNA exerts a non-competitive antagonistic effect [41], thus participating in both glutamatergic and nicotinergic neurotransmission. Modulation of α7nAch, thus presynaptic glutamate release by KYNA, seems to be an important site of action in protection against glutamate induced excitotoxicity. KYNA has been proposed as an endogenous agonist of the G-protein coupled receptor 35 (GPR35) [42], the function of which is yet poorly elucidated. GPR35 is expressed in the gastro-intestinal system and in immune cells [42], but it is functional in dorsal root ganglia and also in hippocampal neurons [43,44]. It also has been demonstrated, that KYNA, similarly to l-KYN, is a ligand to AHR [33]. The effects of KYNA on GPR35 and AHR suggest an important immunomodulatory role for this compound. Recently, it was shown that KYNA is able to scavenge ROS in an NMDA- and nicotinic receptor-independent way; thus, it can be a potential endogenous antioxidant [45]. Taken together, KYNA is involved in possible neurotoxic processes as a protective agent, underlining its importance in neurodegenerative mechanisms.
The second branch of the KP deriving from l-KYN proceeds to anthranilic acid (AA) with the aid of kynureninase. AA has been shown to inhibit citric acid cycle and the respiratory chain complexes I–III [46], interfering with mitochondrial function. It may have an anti-inflammatory effect by forming a complex with copper and acting as an •OH inactivating ligand [47,48]. Anthranilate 3-monooxygenase converts AA to 3-hydroxyanthranilic acid (3-HA), thus rallying to the third branch of l-KYN metabolism.
The first step of the third branch is the conversion of l-KYN by kynurenine monooxygenase (KMO) to 3-hydroxykynurenine (3-HK). KMO has the highest affinity for l-KYN among the enzymes of the three branches, suggesting that under physiological conditions, l-KYN is metabolized by this third branch [49]. 3-HK undergoes auto-oxidation, forming highly reactive o-aminoquinone and ROS—O2•− and H2O2 [50,51,52,53]—and dimerizes to hydroxanthommatin and xanthommatin under physiological conditions [51,53]. 3-HK is generally considered as a neurotoxic agent in vivo, causing convulsive attacks when administered intraventricularly [54] or leading to tissue damage when administered intrastriatally [55]. 3-HK is also present in eye lens, and it has been connected with cataract formation [56].
The toxicity of 3-HK can be attributable to its capability to produce free radicals during its auto-oxidation. However, the free radical scavenging effects of this compound have also been observed in vitro in rat cortex and in C6 glioma cells [57]. Pro- or anti-oxidative properties of 3-HK under different circumstances were thoroughly reviewed by Colín-Gonzalez and colleagues [58].
The next step in the metabolism of l-KYN is the generation of 3-HA from either 3-HK by kynureninase or from AA by anthranilate 3-monooxygenase. 3-HA is also prone to auto-oxidation, generating superoxide radicals, H2O2, and cinnabarinic acid [59]. Cinnabarinic acid is a ligand for the type 4 metabotropic glutamate receptor and also for AHR [60,61]. 3-HA can induce apoptosis in monocytes/macrophages [62], and it can inhibit the mitochondrial respiratory chain [46,63]. Furthermore, it has important immunoregulatory functions by interfering with T-cell survival [64].
3-HK also can be transaminated by KAT to xanthurenic acid, which, similarly to 3-HK and 3-HA, possesses both pro- and anti-oxidative properties [65,66,67].
3-HA is further processed by 3-hydroxyanthranillic acid oxygenase (3-HAO) to 2-amino-3-carboxymuconate semialdehyde. This intermediate can be metabolized by picolinic carboxylase to produce picolinic acid (PIC) or can be transformed by nonenzymatic cyclisation to quinolinic acid (QUIN).
PIC is a non-selective metal ion chelating agent [68], induces morphological changes in the rat hippocampus, substantia nigra and striatum when administered intraperitoneally [69] and has a macrophage induction activity [70]. PIC is able to prevent QUIN-induced neurotoxicity when injected into the nucleus basalis magnocellularis of the rat [71], and it is able to modulate kainate-induced glutamate release from the striatum [72].
QUIN under normal conditions is present in the brain in nanomolar concentrations and metabolized for the synthesis of NAD+. In vitro, QUIN is toxic for brain cells from above 150 nM [73]. QUIN is a weak endogenous agonist on NMDA receptors [74], the action of which is selective, involving the receptor subtypes containing the NR2A and NR2B subunits [75]. QUIN causes the greatest excitotoxic damage in brain areas rich in NMDA receptors containing NR2A and NR2B subunits, mainly in the striatum and in the hippocampus [76]. Furthermore, it can increase glutamate release by neurons and inhibit glutamate uptake by astrocytes, maintaining an elevated level to constantly stimulate NMDA receptors, resulting in excitotoxicity [77]. Lipid peroxidation also contributes to QUIN toxicity [78]; results suggest that QUIN forms a complex with iron, and this complex can contribute to the formation of ROS [79,80]. The toxicity of QUIN on brain cells is exerted mainly through NMDA-mediated excitotoxicity [73,81].

Table 1.
Short summary of the direct receptorial effects of kynurenine pathway metabolites.
| Kynurenine Pathway Metabolite | Receptorial Effect | References |
|---|---|---|
| l-Kynurenine | Aryl hydrocarbon receptor (AHR) agonist | [24] |
| Kynurenic acid | NMDA receptor antagonist | [36,37] |
| Dual effect on AMPA receptors: partial agonist at low nanomolar concentrations; antagonist at high micromolar-millimolar concentrations | [39,40] | |
| Kainate receptor antagonist | [38] | |
| α7-nicotinic acetylcholine receptor antagonist | [41] | |
| G-protein coupled receptor 35 agonist | [42] | |
| AHR agonist | [33] | |
| Cinnabarinic acid | Type 4 metabotropic glutamate receptor agonist | [60] |
| AHR agonist | [61] | |
| Quinolinic acid | NMDA agonist | [74] |
NMDA: N-methyl-d-aspartate; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid.
Quinolinic acid phosphoribosyltransferase converts QUIN to NAD+, finishing the metabolic process. NAD+ is thereafter utilized by different intracellular processes, serving as an electron transfer molecule.
The enzymes of the KP are differently distributed among the various cell types in the CNS, providing an important way of controlling the synthesis of different metabolites. Macrophages and microglia express the entire enzymatic machinery of the KP [82,83,84], and neurons are able to synthesize KYNA [85] and PIC [86], while astrocytes lack the enzyme KMO; therefore, they are not able to synthesize 3-HK under physiological conditions [83].
4. Kynurenines in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
Alzheimer’s disease is characterized by progressive cognitive decline and memory loss, mainly in the elderly population. Pathological protein aggregates in the form of amyloid β (Aβ)-formed plaques, and phosphorylated tau (p-tau)-constituted neurofibrillary tangles are the main hallmarks of the disorder. An increased l-KYN/TRP ratio in AD patients suggests an enhanced TRP metabolism and the increased activation of IDO, which could be connected to neuroinflammation being activated by IFN-γ [87]. 3-HK levels are increased in peripheral blood of AD patients compared to controls [88], further supporting a metabolic shift in diseased patients. Furthermore, results showed that QUIN accumulates in the brain of AD patients, and it is co-localized with p-tau and neurofibrillary tangles [89,90]. Levels of KYNA are also elevated in AD patients, mainly in the striatum and in the hippocampus [91]. These result suggest that the kynurenine pathway in general is upregulated in AD, but the origin of this phenomenon needs further clarification. Activated microglia may produce an increased amount of KP metabolites in response to Aβ and p-tau, and it is possible that the excess amount of the produced QUIN leads to the invigoration of tau phosphorylation and to the development of a vicious circle [90].
4.2. Parkinson’s Disease
Parkinson’s disease is the second most prevalent neurodegenerative disease, characterized by pathological presence of Lewy bodies and Lewy neurites, comprised mainly of α-synuclein, in the neuromelanin-containing dopaminergic cells. Selective death of dopaminergic neurons is most abundant in the substantia nigra pars compacta; however, expansive neurodegeneration is present in the CNS [92]. The selective degradation of the nigrostriatal pathway leads to the motor symptoms characteristic of PD. Oxidative stress, excitotoxicity and neuroinflammation can both be significant factors in the selective death of dopaminergic cells [93,94]. The involvement of the KP in PD has been investigated in several studies. Widner and colleagues found an increased l-KYN/TRP ratio in serum and cerebrospinal fluid (CSF) of PD patients [95], while Ogawa and co-workers demonstrated decreased l-KYN and KYNA and increased 3-HK levels in brain samples of PD patients [96]. These result suggest the upregulation of TRP metabolism in PD, further supported by the finding that 3-HK levels increased in CSF [97].
4.3. Huntington’s Disease
Huntington’s disease is an autosomal dominantly inherited disorder caused by the CAG expansion in the HD gene on chromosome 4. This gene encodes the protein huntingtin, the normal functions of which are still under intensive research. The disease presents with motor symptoms and progressive cognitive decline. Excitotoxicity is considered as one of the main factors in the disease.
Administration of QUIN can serve as an animal model of HD, because it causes lesions pathologically similar to that of human samples, as it spares the cholinergic and aspiny neurons of the striatum [98]. Based on these observations, the quinolinate hypothesis of HD emerged, suggesting that QUIN may have a causative role in this disease. Several studies measured the levels of the KP metabolites, especially QUIN, in brain and CSF in postmortem samples of HD patients. Brain tissue levels of QUIN were found to be reduced [99]; also, the levels of KYNA were found to be decreased both in CSF and brain in HD [100,101,102,103], while an increase in 3-HK levels was found [104]. Measuring the same compounds during the early stages of HD revealed that both 3-HK and QUIN are elevated, while at later stages, no change or a decrease can be observed [105]. These results suggest that 3-HK and QUIN may be participants in the degenerative processes early in the course of HD.
4.4. Amyotrophic Lateral Sclerosis
ALS is a progressive neurodegenerative disorder, affecting motor neurons at both spinal, brainstem and cortical levels. ALS is mainly sporadic, with a 5%–10% occurrence of familial cases, affecting the adult population. About 20% of the familial cases is attributable to the mutation of the superoxide dismutase 1 (SOD1) gene [106], coding an important free radical scavenging enzyme. The symptoms include muscle weakness, atrophy and paralysis, and in most cases, the involvement of breathing muscles results in death within 3–5 years from disease onset.
Perturbations of the kynurenines in ALS were studied by several groups, Chen and colleagues found increased l-KYN and QUIN levels in CSF and serum of ALS patients [107], while KYNA levels were elevated in CSF of bulbar onset patients, but decreased in the serum of patients with severe clinical status [108].
The exact pathomechanism of ALS is not understood, but the general view is that it is a multifactorial disease, involving glutamate excitotoxicity, mitochondrial dysfunction, oxidative stress and inflammation. The kynurenines are involved in all of the above-mentioned processes, thus providing a multitarget option for therapeutic intervention in ALS.
4.5. Multiple Sclerosis
Demyelinization and the forming of sclerotic plaques are the most well-known characteristic of MS. The loss of the myelin sheath and the inflammation at different sites of the CNS causes diverse symptoms and distinct disease courses in MS. MS can be categorized based on the disease course as clinically-isolated syndrome, relapsing-remitting MS, primary progressive MS, secondary progressive MS and progressive relapsing MS.
Alterations in the KP were noted in MS; levels of TRP were found to be reduced both in the CSF and plasma of MS patients compared with control subjects [109,110]. Furthermore, KYNA levels were shown to be significantly decreased in CSF of MS patients during remission, while a significant increase is present both in the plasma and CSF in the course of relapse [111,112,113]. These findings suggest that levels of KYNA may be directly involved in the alternation of relapsing-remitting phases of the disease.
IFN-β treatment, a first line therapy applied in MS, has been shown to significantly increase l-KYN levels and the l-KYN/TRP ration, which is indicative of IDO activity. These results suggest that IFN-β treatment leads to the induction of IDO [114,115]; thus, the important role of IDO and the KP in immune response and autoimmunity is further confirmed (thoroughly reviewed in [4,116]).
4.6. Acquired Immunodeficiency Syndrome Dementia Complex
Infection with human immunodeficiency virus type 1 (HIV-1) can lead to the development of acquired immunodeficiency syndrome dementia complex (ADC), also termed HIV-associated dementia. ADC is a subcortical type of dementia characterized by marked memory impairment and psychomotor slowing. It occurs in the severe forms of HIV-1 infection, affecting about 2% of the infected population receiving combined antiretroviral therapy, as demonstrated by the CHARTER study [117]. Heyes and colleagues proposed that QUIN could play a direct role in the development of ADC, as QUIN levels were elevated in HIV-infected patients and correlated with the severity of neurological deficits [118]. A more feasible hypothesis suggests that QUIN is elevated due to the inflammatory process generated by HIV-1, nevertheless emphasizing the importance of the KP in inflammatory processes [119].
5. Therapeutic Perspectives
Neurodegenerative diseases are a great socio-economic burden, and in most cases, the therapies available provide only symptomatic relief; they do not treat the cause of the disorders. Consequently, there is a constant need for new and effective therapeutic solutions. Manipulating the KP could result in beneficial effects, affecting each of the contributory mechanisms of neuroinflammation, oxidative stress and excitotoxicity.
One of the possible beneficiary therapeutic interventions is the shifting of KP metabolism towards the formation of protective agents, mainly KYNA (Table 2). This metabolic shift can be achieved by specific enzyme inhibitors of KMO, kynureninase and 3-HAO. Several compounds have been developed for the inhibition of these enzymes [120,121,122], but the inhibitors of KMO have been the most widely studied and may provide the most beneficial effects, because inhibition of KMO prevents the formation of the most neurotoxic kynurenines, 3-HK, 3-HAA and QUIN.
Table 2.
Therapeutic options for modulating the kynurenine pathway in neurodegenerative diseases, with some of the candidates developed and tested so far.
| Enzyme Inhibitors | Kynurenic Acid Prodrugs or Analogs |
|---|---|
| 3,4-dimethoxy-N-[4-(3-nitrophenyl)thiazol-2-yl]benzenesulfonamide (Ro-61-8048) | l-Kynurenine |
| 2-(3,4-dimethoxybenzenesulfonylamino)-4-(3-nitrophenyl)-5-(piperidin-1-yl)methylthiazole (JM6) | Combination of l-kynurenine and probenecid N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride |
| nicotinylalanine | 7-Chlorokynurenic acid |
| 4-Chlorokynurenine (AV-101) |
Inhibition of KMO leads to a decrease in the levels of 3-HK and QUIN in rats [123]. Blockade with 3,4-dimethoxy-N-[4-(3-nitrophenyl)thiazol-2-yl]benzenesulfonamide (Ro-61-8048), a potent KMO inhibitor, results in increased KYNA content in parkinsonian monkeys, and when combined with levodopa, it reduces the severity of dyskinesias, both acutely and after prolonged administration [124,125]. A prodrug of Ro-61-8048, 2-(3,4-dimethoxybenzenesulfonylamino)-4-(3-nitrophenyl)-5-(piperidin-1-yl)methylthiazole (JM6), was able to ameliorate anxiety-related behavior, spatial memory deficits and synaptic loss in a transgenic mouse model of AD and was able to decrease microglial activation and extend life span of R6/2 mice (a genetic mouse model of HD) [126]. However, the true prodrug properties of JM6 were questioned, and there is no doubt that Ro-61-8048 is an effective KMO inhibitor [127].
Another possibility for therapeutic intervention is the pharmacological increase of KYNA’s effect, either by administration of its prodrug or by synthetic analogs.
Combined administration of l-KYN and probenecid (PROB), an inhibitor of organic anion transport, causes elevation in the cortical levels of KYNA [128]. The same combination was shown to reduce the toxic effects of 6-hydroxydopamine, a widely-used model of PD in rats; joint administration was able to mitigate rotation behavior and neurodegeneration [129]. l-KYN administered together with PROB was able to attenuate histopathological changes and improve spatial memory in the Aβ model of AD [130]. Furthermore, l-KYN alone mitigated the neuronal cell loss and damage after ischemic insult in rats [131,132].
A combination of enzyme inhibition and pharmacological supplementation of KYNA also proved to be protective in a model of PD. l-KYN and PROB were combined with nicotinylalanine, an inhibitor of kynureninase and KMO, and were able to modulate QUIN-induced turning behavior [133].
During the past few years, several types of KYNA analogs have been synthesized (Scheme 1). A novel KYNA amide, N-(2-N,N-dimethylaminoethyl)-4-oxo-1H-quinoline-2-carboxamide hydrochloride, exerted beneficial effects in numerous paradigms. In micromolar concentrations, it reduced the amplitude of field excitatory postsynaptic potentials in the CA1 region of the hippocampus, while in nanomolar concentrations, it exerted a facilitatory effect, similar to KYNA [134]. In a transgenic mouse model of HD, the same analog increased survival time and prevented weight loss and striatal neuron loss of the animals [135]. The tests of the anti-inflammatory properties of this compound showed that it has a higher potency to inhibit tumor necrosis factor-α production than KYNA, suggesting a more potent immunoregulatory effect [136]. Similarly to KYNA, it also possesses protective characteristics against ischemia-induced neuronal loss [137]. The exact mechanism of action of this new compound is not known, but the lack of cognitive side effects makes it a promising candidate for further investigations [138].
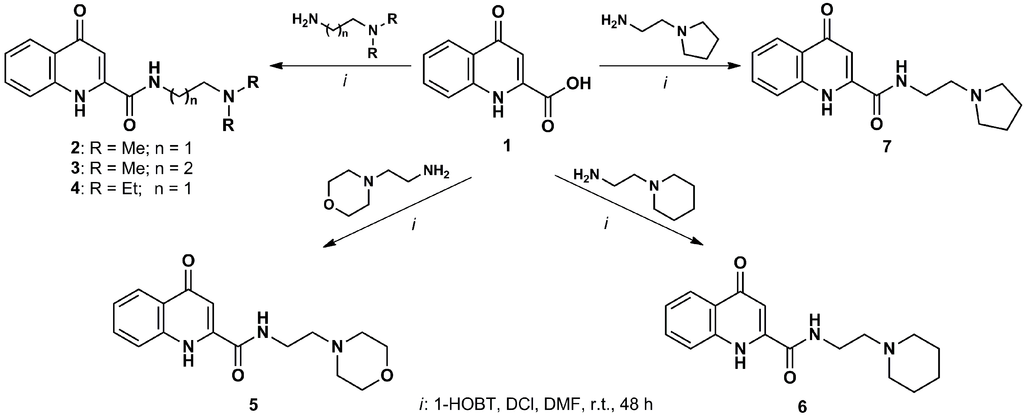
Scheme 1.
Transformational possibilities to develop kynurenic acid analogues. The transformations of kynurenic acid (KYNA) derivatives can be achieved through modification of the aromatic ring, the synthetically active 4-OH group, conversion of the 2-carboxylic function to pharmacologically interesting ester or amide derivatives of KYNA [139]. The amides of KYNA are pharmacologically and synthetically highly promising synthons in the patent literature. Coupling between KYNA and 2-dimethylaminoethylamine was achieved by using N,N'-diisopropylcarbodiimide (DCI) in the presence of 1-hydroxybenzotriazole hydrate (1-HOBT), yielding 2. Further transformations are also shown in Scheme 1 [140].
Scheme 1.
Transformational possibilities to develop kynurenic acid analogues. The transformations of kynurenic acid (KYNA) derivatives can be achieved through modification of the aromatic ring, the synthetically active 4-OH group, conversion of the 2-carboxylic function to pharmacologically interesting ester or amide derivatives of KYNA [139]. The amides of KYNA are pharmacologically and synthetically highly promising synthons in the patent literature. Coupling between KYNA and 2-dimethylaminoethylamine was achieved by using N,N'-diisopropylcarbodiimide (DCI) in the presence of 1-hydroxybenzotriazole hydrate (1-HOBT), yielding 2. Further transformations are also shown in Scheme 1 [140].
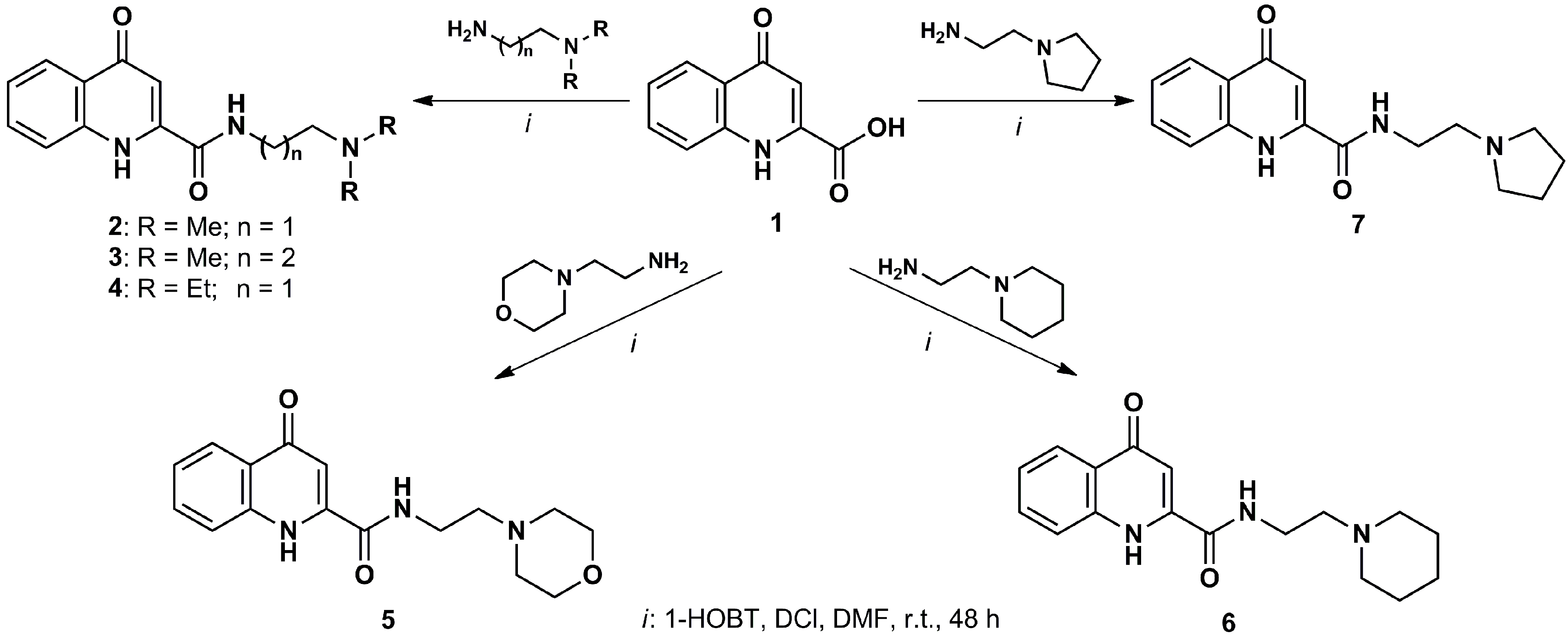
Halogenated KYNA analogues have also been tested; 7-chlorokynurenic acid is a selective antagonist of the glycine site of NMDA receptors [141], and it was able to modulate kainate-induced neurodegenerative changes [142,143]. However, the blood-brain barrier permeability of this drug is not optimal; therefore, its prodrug, 4-chlorokynurenine (also known as AV-101), was examined and has been shown to be protective against QUIN neurotoxicity [143]. AV-101 has successfully completed a phase I clinical trial for the evaluation of safety, tolerability and pharmacokinetic profile (ClinicalTrials.gov Identifier: NCT01483846) and hopefully will be further processed.
In summary, the interventions affecting the KP are promising targets in the development of neuroprotective strategies.
6. Conclusions
The KP is an important target for the development of new therapies against neurodegenerative disorders, as it is comprised of compounds influencing processes related to excitotoxicity, oxidative damage and inflammation. Compiling evidence suggests that the interplay between the immune and nervous system in neurodegeneration could be the main target of both diagnostic and therapeutic developments. Therefore, further characterization of the role of KP metabolites in the immune and nervous system could be the key to new pharmaceutical therapies. However, owing to the broad effects of certain KP metabolites, a more complex approach would be necessary to develop drugs with the fewest side effects.
Acknowledgments
This work was supported by the MTA-SZTE Neuroscience Research Group, Hungarian Scientific Research Fund-OTKA K105077 and the Hungarian Brain Research Program (Grant No. KTIA_13_NAP-A-III/9).
Abbreviations
| α7nAch | α7-nicotinic acetylcholine receptor |
| AA | anthranilic acid |
| Aβ | amyloid β |
| ADC | acquired immunodeficiency syndrome dementia complex |
| AD | Alzheimer’s disease |
| AHR | aryl-hydrocarbon receptor |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| ALS | amyotrophic lateral sclerosis |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| GPR35 | G-protein coupled receptor 35 |
| HIV-1 | human immunodeficiency virus type 1 |
| H2O2 | hydrogen peroxide |
| HD | Huntington’s disease |
| IDO1 | indoleamine 2,3-dioxygenase 1 |
| IDO2 | indoleamine 2,3-dioxygenase 2 |
| IFN-β | interferon-β |
| IFN-γ | interferon-γ |
| KAT | kynurenine aminotransferase |
| KMO | kynurenine monooxygenase |
| KP | kynurenine pathway |
| KYNA | kynurenic acid |
| l-KYN | l-kynurenine |
| MS | multiple sclerosis |
| NAD+ | nicotinamide adenine dinucleotide |
| NMDA | N-methyl-d-aspartate |
| •NO | nitric oxide |
| •NO2 | nitrogen dioxide |
| O2•− | superoxide anion |
| •OH | hydroxyl radical |
| ONOO− | peroxynitrite anion |
| PD | Parkinson’s disease |
| PIC | picolinic acid |
| p-tau | phosphorylated tau |
| QUIN | quinolinic acid |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| SOD1 | superoxide dismutase 1 |
| TDO | tryptophan 2,3-dioxygenase |
| TRP | tryptophan |
| 3-HA | 3-hydroxyanthranilic acid |
| 3-HK | 3-hydroxykynurenine |
| 3-HAO | 3-hydroxyanthranillic acid oxygenase |
Conflicts of Interest
The authors declare no conflict of interest.
References
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Robotka, H.; Toldi, J.; Vecsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Novelli, A.; Reilly, J.A.; Lysko, P.G.; Henneberry, R.C. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988, 451, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [PubMed]
- Ellrichmann, G.; Reick, C.; Saft, C.; Linker, R.A. The role of the immune system in Huntington’s disease. Clin. Dev. Immunol. 2013. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [PubMed]
- Wenk, G.L.; Parsons, C.G.; Danysz, W. Potential role of N-methyl-d-aspartate receptors as executors of neurodegeneration resulting from diverse insults: Focus on memantine. Behav. Pharmacol. 2006, 17, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H. The effect of hormones and vitamin B6 on urinary excretion of metabolites of the kynurenine pathway. Scand. J. Clin. Lab. Investig. Suppl. 1974, 136, 1–186. [Google Scholar]
- Guillemin, G.J.; Kerr, S.J.; Pemberton, L.A.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. IFN-β1b induces kynurenine pathway metabolism in human macrophages: Potential implications for multiple sclerosis treatment. J. Interferon Cytokine Res. 2001, 21, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, L.A.; Kerr, S.J.; Smythe, G.; Brew, B.J. Quinolinic acid production by macrophages stimulated with IFN-γ, TNF-α, and IFN-α. J. Interferon Cytokine Res. 1997, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Chen, C.Y.; Major, E.O.; Saito, K. Different kynurenine pathway enzymes limit quinolinic acid formation by various human cell types. Biochem. J. 1997, 326 Pt 2, 351–356. [Google Scholar] [PubMed]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: A role for IFN-γ? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.J.; Rendina, L.M. Targeting key dioxygenases in tryptophan-kynurenine metabolism for immunomodulation and cancer chemotherapy. Drug Discov. Today 2014. [Google Scholar] [CrossRef]
- Sedlmayr, P. Indoleamine 2,3-dioxygenase in materno–fetal interaction. Curr. Drug MeTable 2007, 8, 205–208. [Google Scholar] [CrossRef]
- Reyes Ocampo, J.; Lugo Huitron, R.; Gonzalez-Esquivel, D.; Ugalde-Muniz, P.; Jimenez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Rios, C.; Perez de la Cruz, V. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Denison, M.S.; Rogers, J.M.; Rushing, S.R.; Jones, C.L.; Tetangco, S.C.; Heath-Pagliuso, S. Analysis of the aryl hydrocarbon receptor (AhR) signal transduction pathway. Curr. Protoc. Toxicol. 2002. [Google Scholar] [CrossRef]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Kasai, A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007, 252, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Mason, G.G. Dioxin-receptor ligands in urban air and vehicle exhaust. Environ. Health Perspect. 1994, 102 (Suppl. 4), 111–116. [Google Scholar] [CrossRef] [PubMed]
- Bunger, M.K.; Glover, E.; Moran, S.M.; Walisser, J.A.; Lahvis, G.P.; Hsu, E.L.; Bradfield, C.A. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 2008, 106, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Julliard, W.; Fechner, J.H.; Mezrich, J.D. The aryl hydrocarbon receptor meets immunology: Friend or foe? A little of both. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Vogel, C.F.; Khan, E.M.; Leung, P.S.; Gershwin, M.E.; Chang, W.L.; Wu, D.; Haarmann-Stemmann, T.; Hoffmann, A.; Denison, M.S. Cross-talk between aryl hydrocarbon receptor and the inflammatory response: A role for nuclear factor-κB. J. Biol. Chem. 2014, 289, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gal, E.M.; Sherman, A.D. Synthesis and metabolism of l-kynurenine in rat brain. J. Neurochem. 1978, 30, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Russi, P.; Lombardi, G.; Beni, M.; Carla, V. Presence of kynurenic acid in the mammalian brain. J. Neurochem. 1988, 51, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-d-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Fadda, E.; Wroblewski, J.T.; Costa, E. Kynurenate and 2-amino-5-phosphonovalerate interact with multiple binding sites of the N-methyl-d-aspartate-sensitive glutamate receptor domain. Neurosci. Lett. 1989, 96, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.N.; Stone, T.W. Actions of kynurenic acid and quinolinic acid in the rat hippocampus in vivo. Exp. Neurol. 1985, 88, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rozsa, E.; Robotka, H.; Vecsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [PubMed]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed]
- Alkondon, M.; Pereira, E.F.; Todd, S.W.; Randall, W.R.; Lane, M.; Albuquerque, E.X. Functional G-protein coupled receptor 35 is expressed by neurons in the CA1 field of the hippocampus. Biochem. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Ohshiro, H.; Tonai-Kachi, H.; Ichikawa, K. GPR35 is a functional receptor in rat dorsal root ganglion neurons. Biochem. Biophys. Res. Commun. 2008, 365, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitron, R.; Blanco-Ayala, T.; Ugalde-Muniz, P.; Carrillo-Mora, P.; Pedraza-Chaverri, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzon, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.F.; Tonin, A.; da Costa Ferreira, G.; Viegas, C.M.; Latini, A.; Duval Wannmacher, C.M.; de Souza Wyse, A.T.; Dutra-Filho, C.S.; Wajner, M. Kynurenines impair energy metabolism in rat cerebral cortex. Cell. Mol. Neurobiol. 2007, 27, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Gaubert, S.; Bouchaut, M.; Brumas, V.; Berthon, G. Copper—Ligand interactions and the physiological free radical processes. Part 3. Influence of histidine, salicylic acid and anthranilic acid on copper-driven Fenton chemistry in vitro. Free Radic. Res. 2000, 32, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Miche, H.; Brumas, V.; Berthon, G. Copper(II) interactions with nonsteroidal antiinflammatory agents. II. Anthranilic acid as a potential. OH-inactivating ligand. J. Inorg. Biochem. 1997, 68, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Bender, D.A.; McCreanor, G.M. The preferred route of kynurenine metabolism in the rat. Biochim. Biophys. Acta 1982, 717, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Eastman, C.L.; Guilarte, T.R. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem. Res. 1990, 15, 1101–1107. [Google Scholar] [CrossRef]
- Ishii, T.; Iwahashi, H.; Sugata, R.; Kido, R. Formation of hydroxanthommatin-derived radical in the oxidation of 3-hydroxykynurenine. Arch. Biochem. Biophys. 1992, 294, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553–12558. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, S.; Garner, B.; Sheil, M.M.; Truscott, R.J. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Radic. Res. 2000, 32, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, A.; Ossi, C.; Colombo, R.; Tofanetti, O.; Spazzi, L. Experimental convulsions in rats induced by intraventricular administration of kynurenine and structurally related compounds. Neuropharmacology 1984, 23, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, Y.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine toxicity on the rat striatum in vivo. Jpn. J. Pharmacol. 1996, 71, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Mizdrak, J.; Hains, P.G.; Truscott, R.J.; Jamie, J.F.; Davies, M.J. Tryptophan-derived ultraviolet filter compounds covalently bound to lens proteins are photosensitizers of oxidative damage. Free Radic. Biol. Med. 2008, 44, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Schumacher, C.; Dalcin, K.B.; Scussiato, K.; Solano, A.; Funchal, C.; Dutra-Filho, C.S.; Wyse, A.T.; Wannmacher, C.M.; Latini, A.; et al. In vitro evidence for an antioxidant role of 3-hydroxykynurenine and 3-hydroxyanthranilic acid in the brain. Neurochem. Int. 2007, 50, 83–94. [Google Scholar]
- Colin-Gonzalez, A.L.; Maldonado, P.D.; Santamaria, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the central nervous system. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef] [PubMed]
- Dykens, J.A.; Sullivan, S.G.; Stern, A. Oxidative reactivity of the tryptophan metabolites 3-hydroxyanthranilate, cinnabarinate, quinolinate and picolinate. Biochem. Pharmacol. 1987, 36, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharmacol. 2012, 81, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.M.; Mold, J.E.; Kanwar, B.; Huang, Y.; Louie, A.; Pollastri, M.P.; Wang, C.; Patel, G.; Franks, D.G.; Schlezinger, J.; et al. Identification of cinnabarinic acid as a novel endogenous aryl hydrocarbon receptor ligand that drives IL-22 production. PLoS ONE 2014, 9, e87877. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Saito, K.; Takemura, M.; Maekawa, N.; Fujigaki, S.; Fujii, H.; Wada, H.; Takeuchi, S.; Noma, A.; Seishima, M.; et al. 3-Hydroxyanthranilic acid, an l-tryptophan metabolite, induces apoptosis in monocyte-derived cells stimulated by interferon-gamma. Ann. Clin. Biochem. 2001, 38, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, E.; Papa, S.; Saccone, C.; Alifano, A. Effect of 3-hydroxyanthranilic acid on the mitochondrial respiratory system. Biochem. J. 1964, 91, 137–146. [Google Scholar] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Christen, S.; Peterhans, E.; Stocker, R. Antioxidant activities of some tryptophan metabolites: Possible implication for inflammatory diseases. Proc. Natl. Acad. Sci. USA 1990, 87, 2506–2510. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Haneda, M.; Qiao, S.; Naruse, M.; Yoshino, M. Prooxidant action of rosmarinic acid: Transition metal-dependent generation of reactive oxygen species. Toxicol. In Vitro 2007, 21, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Ito, M.; Yoshino, M. Xanthurenic acid inhibits metal ion-induced lipid peroxidation and protects NADP-isocitrate dehydrogenase from oxidative inactivation. J. Nutr. Sci. Vitaminol. (Tokyo) 2001, 47, 306–310. [Google Scholar] [CrossRef]
- Aggett, P.J.; Fenwick, P.K.; Kirk, H. An in vitro study of the effect of picolinic acid on metal translocation across lipid bilayers. J. Nutr. 1989, 119, 1432–1437. [Google Scholar] [PubMed]
- Beskid, M.; Jachimowicz, J.; Taraszewska, A.; Kukulska, D. Histological and ultrastructural changes in the rat brain following systemic administration of picolinic acid. Exp. Toxicol. Pathol. 1995, 47, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bosco, M.C.; Rapisarda, A.; Massazza, S.; Melillo, G.; Young, H.; Varesio, L. The tryptophan catabolite picolinic acid selectively induces the chemokines macrophage inflammatory protein-1α and -1β in macrophages. J. Immunol. 2000, 164, 3283–3291. [Google Scholar] [CrossRef] [PubMed]
- Jhamandas, K.; Boegman, R.J.; Beninger, R.J.; Bialik, M. Quinolinate-induced cortical cholinergic damage: Modulation by tryptophan metabolites. Brain Res. 1990, 529, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Vrooman, L.; Jhamandas, K.; Boegman, R.J.; Beninger, R.J. Picolinic acid modulates kainic acid-evoked glutamate release from the striatum in vitro. Brain Res. 1993, 627, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Grant, R.; Adams, S.; Brew, B.J.; Guillemin, G.J. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox. Res. 2009, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, L.P.; Bochet, P.; Rossier, J. The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem. Int. 1996, 28, 445–452. [Google Scholar]
- Schwarcz, R.; Kohler, C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci. Lett. 1983, 38, 85–90. [Google Scholar] [CrossRef]
- Tavares, R.G.; Tasca, C.I.; Santos, C.E.; Alves, L.B.; Porciuncula, L.O.; Emanuelli, T.; Souza, D.O. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem. Int. 2002, 40, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Rios, C.; Santamaria, A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 1991, 16, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Goda, K.; Kishimoto, R.; Shimizu, S.; Hamane, Y.; Ueda, M. Quinolinic acid and active oxygens. Possible contribution of active oxygens during cell death in the brain. Adv. Exp. Med. Biol. 1996, 398, 247–254. [Google Scholar] [PubMed]
- Stipek, S.; Stastny, F.; Platenik, J.; Crkovska, J.; Zima, T. The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem. Int. 1997, 30, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brew, B.J.; Guillemin, G.J. Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2011, 118, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Chernyshev, O.N.; Reinhard, J.F., Jr.; Namboodiri, M.A.; Colton, C.A. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport 1997, 8, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320 Pt 2, 595–597. [Google Scholar] [PubMed]
- Rzeski, W.; Kocki, T.; Dybel, A.; Wejksza, K.; Zdzisinska, B.; Kandefer-Szerszen, M.; Turski, W.A.; Okuno, E.; Albrecht, J. Demonstration of kynurenine aminotransferases I and II and characterization of kynurenic acid synthesis in cultured cerebral cortical neurons. J. Neurosci. Res. 2005, 80, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Tryptophan degradation and immune activation in Alzheimer’s disease. J. Neural Transm. 2000, 107, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine metabolism in Alzheimer’s disease. J. Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zadori, D.; Szalardy, L.; Toldi, J.; Fulop, F.; Klivenyi, P.; Vecsei, L. Some molecular mechanisms of dopaminergic and glutamatergic dysfunctioning in Parkinson’s disease. J. Neural Transm. 2013, 120, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Leblhuber, F.; Fuchs, D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002, 109, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Kowall, N.W.; Ellison, D.W.; Mazurek, M.F.; Swartz, K.J.; Martin, J.B. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 1986, 321, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Swartz, K.J.; Markey, S.P.; Beal, M.F. Regional brain and cerebrospinal fluid quinolinic acid concentrations in Huntington’s disease. Neurosci. Lett. 1991, 122, 265–269. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matson, W.R.; Swartz, K.J.; Gamache, P.H.; Bird, E.D. Kynurenine pathway measurements in Huntington’s disease striatum: Evidence for reduced formation of kynurenic acid. J. Neurochem. 1990, 55, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115 Pt 5, 1249–1273. [Google Scholar] [CrossRef] [PubMed]
- Jauch, D.; Urbanska, E.M.; Guidetti, P.; Bird, E.D.; Vonsattel, J.P.; Whetsell, W.O., Jr.; Schwarcz, R. Dysfunction of brain kynurenic acid metabolism in Huntington’s disease: Focus on kynurenine aminotransferases. J. Neurol. Sci. 1995, 130, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.J.; Reynolds, G.P. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [PubMed]
- Chen, Y.; Stankovic, R.; Cullen, K.M.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Ilzecka, J.; Kocki, T.; Stelmasiak, Z.; Turski, W.A. Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Monaco, F.; Fumero, S.; Mondino, A.; Mutani, R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J. Neurol. Neurosurg. Psychiatry 1979, 42, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Demisch, L.; Engelhardt, W.; Fischer, P.A. Interleukin-2, soluble interleukin-2-receptor, neopterin, l-tryptophan and β2-microglobulin levels in CSF and serum of patients with relapsing-remitting or chronic-progressive multiple sclerosis. J. Neurol. 1993, 241, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Amirkhani, A.; Rajda, C.; Arvidsson, B.; Bencsik, K.; Boda, K.; Seres, E.; Markides, K.E.; Vecsei, L.; Bergquist, J. Interferon-β affects the tryptophan metabolism in multiple sclerosis patients. Eur. J. Neurol. 2005, 12, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Durastanti, V.; Lugaresi, A.; Bramanti, P.; Amato, M.; Bellantonio, P.; de Luca, G.; Picconi, O.; Fantozzi, R.; Locatelli, L.; Solda, A.; et al. Neopterin production and tryptophan degradation during 24-months therapy with interferon β-1a in multiple sclerosis patients. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: Charter Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Brew, B.J.; Martin, A.; Price, R.W.; Salazar, A.M.; Sidtis, J.J.; Yergey, J.A.; Mouradian, M.M.; Sadler, A.E.; Keilp, J.; et al. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: Relationship to clinical and neurological status. Ann. Neurol. 1991, 29, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Brew, B.J. Involvement of quinolinic acid in AIDS dementia complex. Neurotox. Res. 2005, 7, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Carpenedo, R.; Molina, M.T.; Mattoli, L.; Pellicciari, R.; Moroni, F. Comparison of the neurochemical and behavioral effects resulting from the inhibition of kynurenine hydroxylase and/or kynureninase. J. Neurochem. 1995, 65, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Colabroy, K.L.; Zhai, H.; Li, T.; Ge, Y.; Zhang, Y.; Liu, A.; Ealick, S.E.; McLafferty, F.W.; Begley, T.P. The mechanism of inactivation of 3-hydroxyanthranilate-3,4-dioxygenase by 4-chloro-3-hydroxyanthranilate. Biochemistry 2005, 44, 7623–7631. [Google Scholar] [CrossRef] [PubMed]
- Walsh, H.A.; O’Shea, K.C.; Botting, N.P. Comparative inhibition by substrate analogues 3-methoxy- and 3-hydroxydesaminokynurenine and an improved 3 step purification of recombinant human kynureninase. BMC Biochem. 2003, 4. [Google Scholar] [CrossRef]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the relationship between the two branches of the kynurenine pathway in the rat brain in vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, L.; Rassoulpour, A.; Guidetti, P.; Samadi, P.; Bedard, P.J.; Izzo, E.; Schwarcz, R.; di Paolo, T. Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav. Brain Res. 2008, 186, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Gregoire, L.; Rassoulpour, A.; Guidetti, P.; Izzo, E.; Schwarcz, R.; Bedard, P.J. Effect of kynurenine 3-hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in Parkinsonian monkeys. Mov. Disord. 2005, 20, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Beconi, M.G.; Yates, D.; Lyons, K.; Matthews, K.; Clifton, S.; Mead, T.; Prime, M.; Winkler, D.; O’Connell, C.; Walter, D.; et al. Metabolism and pharmacokinetics of JM6 in mice: JM6 is not a prodrug for Ro-61-8048. Drug Metab. Dispos. 2012, 40, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Chauvel, V.; Vamos, E.; Pardutz, A.; Vecsei, L.; Schoenen, J.; Multon, S. Effect of systemic kynurenine on cortical spreading depression and its modulation by sex hormones in rat. Exp. Neurol. 2012, 236, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Silva-Adaya, D.; Perez-de la Cruz, V.; Villeda-Hernandez, J.; Carrillo-Mora, P.; Gonzalez-Herrera, I.G.; Garcia, E.; Colin-Barenque, L.; Pedraza-Chaverri, J.; Santamaria, A. Protective effect of l-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: Implications of modulating kynurenate as a protective strategy. Neurotoxicol. Teratol. 2011, 33, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Mendez-Cuesta, L.A.; Perez-de la Cruz, V.; Fortoul-van der Goes, T.I.; Santamaria, A. Protective effect of systemic l-kynurenine and probenecid administration on behavioural and morphological alterations induced by toxic soluble amyloid β(25–35) in rat hippocampus. Behav. Brain Res. 2010, 210, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Robotka, H.; Sas, K.; Agoston, M.; Rozsa, E.; Szenasi, G.; Gigler, G.; Vecsei, L.; Toldi, J. Neuroprotection achieved in the ischaemic rat cortex with l-kynurenine sulphate. Life Sci. 2008, 82, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Robotka, H.; Rozsa, E.; Agoston, M.; Szenasi, G.; Gigler, G.; Marosi, M.; Kis, Z.; Farkas, T.; Vecsei, L.; et al. Kynurenine diminishes the ischemia-induced histological and electrophysiological deficits in the rat hippocampus. Neurobiol. Dis. 2008, 32, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.F.; Sutton, M.A.; Beninger, R.J.; Jhamandas, K.; Boegman, R.J. Quinolinic acid lesion of the nigrostriatal pathway: Effect on turning behaviour and protection by elevation of endogenous kynurenic acid in Rattus norvegicus. Neurosci. Lett. 1999, 262, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Marosi, M.; Nagy, D.; Farkas, T.; Kis, Z.; Rozsa, E.; Robotka, H.; Fulop, F.; Vecsei, L.; Toldi, J. A novel kynurenic acid analogue: A comparison with kynurenic acid. An in vitro electrophysiological study. J. Neural Transm. 2010, 117, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Zadori, D.; Nyiri, G.; Szonyi, A.; Szatmari, I.; Fulop, F.; Toldi, J.; Freund, T.F.; Vecsei, L.; Klivenyi, P. Neuroprotective effects of a novel kynurenic acid analogue in a transgenic mouse model of Huntington’s disease. J. Neural Transm. 2011, 118, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Tiszlavicz, Z.; Nemeth, B.; Fulop, F.; Vecsei, L.; Tapai, K.; Ocsovszky, I.; Mandi, Y. Different inhibitory effects of kynurenic acid and a novel kynurenic acid analogue on tumour necrosis factor-alpha (TNF-α) production by mononuclear cells, HMGB1 production by monocytes and HNP1-3 secretion by neutrophils. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383, 447–455. [Google Scholar] [CrossRef]
- Gellert, L.; Fuzik, J.; Goblos, A.; Sarkozi, K.; Marosi, M.; Kis, Z.; Farkas, T.; Szatmari, I.; Fulop, F.; Vecsei, L.; et al. Neuroprotection with a new kynurenic acid analog in the four-vessel occlusion model of ischemia. Eur. J. Pharmacol. 2011, 667, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Gellert, L.; Varga, D.; Ruszka, M.; Toldi, J.; Farkas, T.; Szatmari, I.; Fulop, F.; Vecsei, L.; Kis, Z. Behavioural studies with a newly developed neuroprotective KYNA-amide. J. Neural Transm. 2012, 119, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Fulop, F.; Szatmari, I.; Vamos, E.; Zadori, D.; Toldi, J.; Vecsei, L. Syntheses, transformations and pharmaceutical applications of kynurenic acid derivatives. Curr. Med. Chem. 2009, 16, 4828–4842. [Google Scholar] [CrossRef] [PubMed]
- Fulop, F.; Szatmari, I.; Toldi, J.; Vecsei, L. Modifications on the carboxylic function of kynurenic acid. J. Neural Transm. 2012, 119, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; Foster, A.C.; Leeson, P.D.; Priestley, T.; Tridgett, R.; Iversen, L.L.; Woodruff, G.N. 7-Chlorokynurenic acid is a selective antagonist at the glycine modulatory site of the N-methyl-d-aspartate receptor complex. Proc. Natl. Acad. Sci. USA 1988, 85, 6547–6550. [Google Scholar] [CrossRef] [PubMed]
- Domenici, M.R.; Longo, R.; Sagratella, S. 7-Chlorokynurenic acid prevents in vitro epileptiform and neurotoxic effects due to kainic acid. Gen. Pharmacol. 1996, 27, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Lee, S.C.; Schwarcz, R. Systemic administration of 4-chlorokynurenine prevents quinolinate neurotoxicity in the rat hippocampus. Eur. J. Pharmacol. 2000, 390, 267–274. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).