2.2. Phase-Selective Gelation (PSG)
A preliminary screening of the peptide library synthesized in our previous work [
38] showed a significant ability to selectively gel organic solvents from their mixtures with water after heating-cooling treatment. In these experiments the water phase remained in the fluid state. Due to very similar physical properties of the gels observed within each group [
38,
39], PSG of oil/water mixtures (1:2
v/
v) was studied using only some representative compounds,
i.e.,
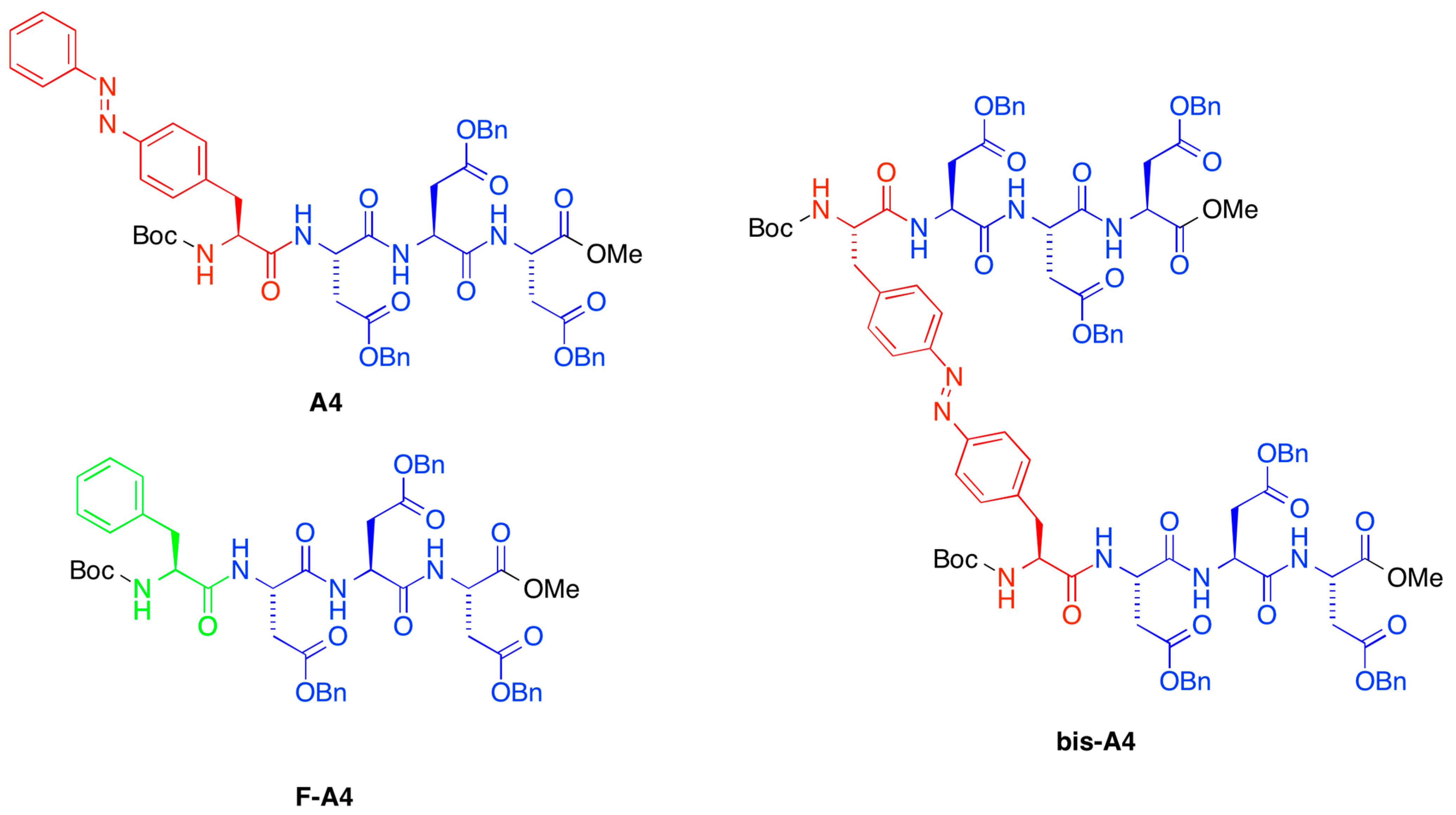
A4,
bis-A4,
F-A4, as shown in
Figure 1. Very interestingly, we found that the presence of side-chain azobenzene moieties in the gelator structure (
A4,
bis-A4) allowed PSG of the oil phase within minutes at the same concentration as the described minimum gelation concentration (MGC) for pure organic phases (POP) [
38] (
Table 1, entries 1–14). In contrast, the use of Phe-based gelators (
F-A4) required
ca. two times higher concentration than the corresponding MGC in POP (
Table 1, entries 15–21). Therefore, we decided to continue further studies with
A4 and
bis-A4 as model systems, which also showed the ability to gel ionic liquids (
i.e., 1-butyl-3-methylimidazolium hexafluorophosphate, BMIM·PF
6) either as pure solvents or in aqueous mixtures.
Table 1.
Comparison of the gelation properties of pure organic phases (POP) and phase-selective gelation (PSG) for selected organic phases.
Table 1.
Comparison of the gelation properties of pure organic phases (POP) and phase-selective gelation (PSG) for selected organic phases.
| Entry | Organic Phase | Gelator | Minimum Gelation Concentration, MGC (%
w/v) | Gel-to-Sol Transition Temperature,
Tgel (°C) |
|---|
| POP | PSG | POP | PSG |
|---|
| 1 | 2-BuOH | A4 | 2.8 ± 0.3 | 3.0 ± 0.4 | 55 ± 1 | 54 ± 2 |
| 2 | BMIM·PF6 | A4 | 4.0 ± 0.3 | 4.0 ± 0.3 | 78 ± 2 | 76 ± 1 |
| 3 | Toluene | A4 | 1.9 ± 0.2 | 2.0 ± 0.2 | 37 ± 2 | 39 ± 1 |
| 4 | Xylene | A4 | 7.0 ± 0.5 | 7.0 ± 0.5 | 59 ± 1 | 62 ± 2 |
| 5 | Olive Oil | A4 | 1.3 ± 0.2 | 1.5 ± 0.2 | 93 ± 2 | 89 ± 1 |
| 6 | Gasoline | A4 | 2.2 ± 0.2 | 2.4 ± 0.2 | 52 ± 2 | 53 ± 2 |
| 7 | Diesel | A4 | 2.0 ± 0.2 | 2.0 ± 0.2 | 62 ± 1 | 61 ± 1 |
| 8 | 2-BuOH | Bis-A4 | 0.8 ± 0.1 | 1.0 ± 0.1 | 62 ± 2 | 63 ± 1 |
| 9 | BMIM·PF6 | Bis-A4 | 2.2 ± 0.2 | 2.2 ± 0.2 | 69 ± 1 | 68 ± 2 |
| 10 | Toluene | Bis-A4 | 0.6 ± 0.1 | 0.5 ± 0.1 | 70 ± 1 | 70 ± 2 |
| 11 | Xylene | Bis-A4 | 3.3 ± 0.2 | 3.5 ± 0.3 | 76 ± 2 | 74 ± 2 |
| 12 | Olive Oil | Bis-A4 | 0.5 ± 0.1 | 0.5 ± 0.1 | 104 ± 2 | 101 ± 2 |
| 13 | Gasoline | Bis-A4 | 2.5 ± 0.2 | 2.5 ± 0.2 | 56 ± 1 | 55 ± 2 |
| 14 | Diesel | Bis-A4 | 1.8 ± 0.2 | 2.0 ± 0.2 | 59 ± 2 | 61 ± 1 |
| 15 | 2-BuOH | F-A4 | 10.0 ± 1.0 | 13.0 ± 1.5 | 57 ± 1 | 52 ± 2 |
| 16 | BMIM·PF6 | F-A4 | - | - | - | - |
| 17 | Toluene | F-A4 | 2.5 ± 0.3 | 4.4 ± 0.3 | 35 ± 1 | 44 ± 1 |
| 18 | Xylene | F-A4 | 2.0 ± 0.2 | 3.2 ± 0.3 | 37 ± 1 | 36 ± 2 |
| 19 | Olive Oil | F-A4 | 2.0 ± 0.2 | 2.8 ± 0.3 | 72 ± 2 | 71 ± 1 |
| 20 | Gasoline | F-A4 | 3.3 ± 0.3 | 4.5 ± 0.5 | 54 ± 2 | 57 ± 2 |
| 21 | Diesel | F-A4 | 1.8 ± 0.2 | 2.4 ± 0.2 | 56 ± 2 | 54 ± 1 |
It should be emphasized that many other solvents previously reported to be gelled by the different peptides could also be selectively gelled from their mixtures with water [
38,
39]. Remarkably, the presence of the water phase during PSG did not cause a significant impact on the thermal, mechanical, and morphological properties of the corresponding organogels bodies as determined by
Tgel measurements (
Table 1), dynamic rheological measurements (
Figure 2) and electron microscopy (
Figure 3), respectively. Thus, the presence of water during PSG does not disrupt (e.g., by partial diffusion to the organic layer) the pattern of non-covalent interactions responsible for the gelation of the oil phase [
38]. In water, the peptidic gelator excludes water due a hydrophobic effect caused by the presence of lipophilic moieties (an effect which is larger in
A4 than in
F-A4 due to the presence of side-chain azobenzene units, which could explain the differences observed on the MGC). This gives room for the self-assembly of the gelator in the oil phase driven by non-covalent interactions such as hydrogen-bonding, π–π stacking and/or van der Waals [
38]. However, further studies are still necessary to determine the exact conformation of these peptides in hydrophobic environments, and quantify the interactions that are responsible for the gelation process.
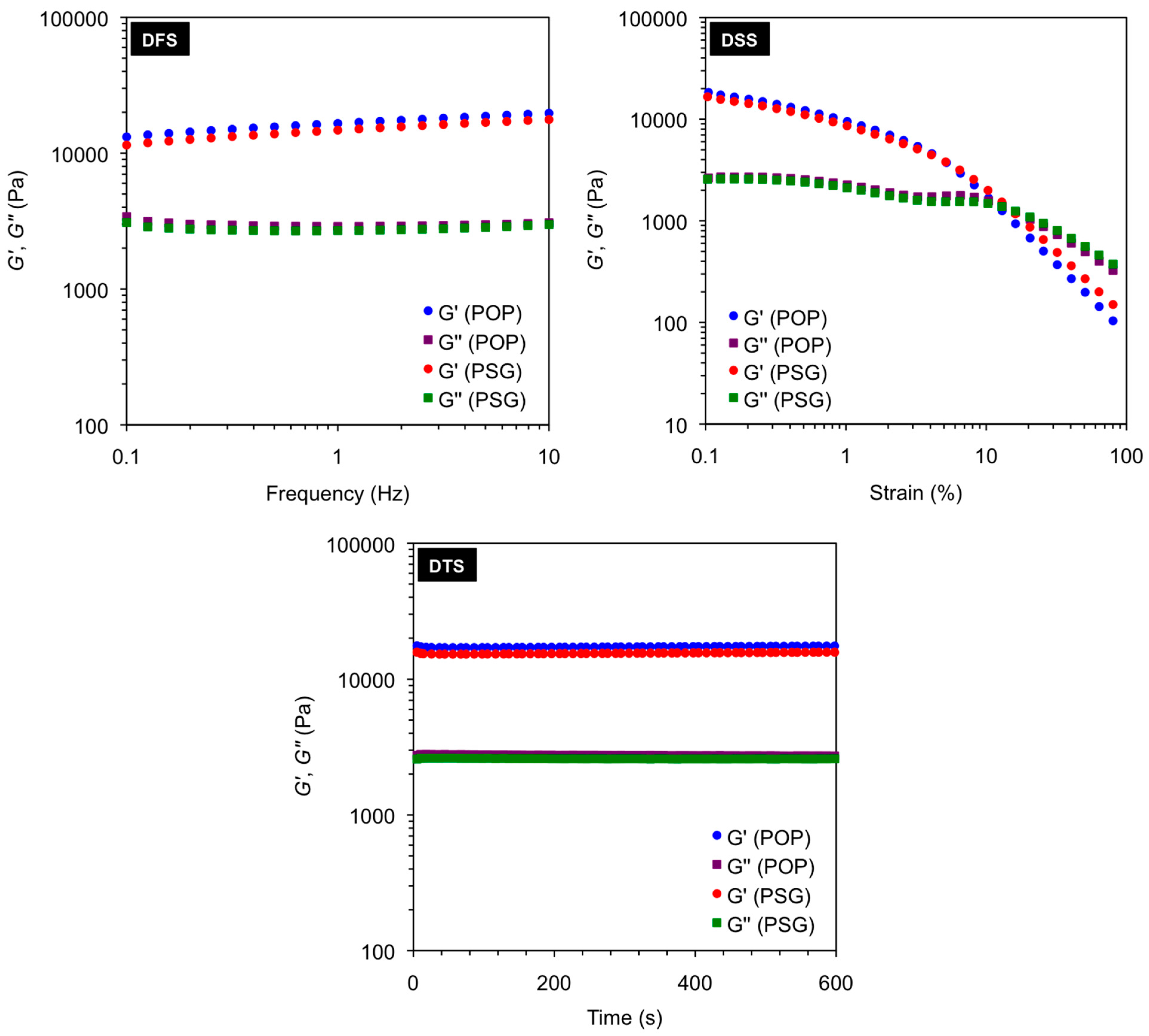
Figure 2.
Dynamic oscillatory rheological measurements (i.e., DFS, DSS, DTS) of the gels in toluene obtained after gelation of pure organic phase (POP) and phase selective gelation (PSG) of toluene/water mixture (1:2 v/v) using A4 as gelator (2.0% w/v). Heating-cooling treatment was used in these examples to induce gelation. Average storage modulus G' (16.5 ± 1.97 kPa for POP; 14.7 ± 1.89 kPa for PSG), average loss modulus G'' (3.0 ± 0.12 kPa for POP; 2.8 ± 0.11 kPa for PSG), average tan δ (0.18 ± 0.0028 for POP; 0.19 ± 0.0019 for PSG), and maximum strain at break (16% ± 2.0% for POP; 13% ± 1.5% for PSG) remained constant within the limits of the experimental error regardless the material. As observed in POP, the gels obtained from PSG displayed G' at least one order of magnitude higher than G'' and relatively low dependence with frequency, indicating the viscoelastic nature of the samples as well as a good tolerance to external forces.
Figure 2.
Dynamic oscillatory rheological measurements (i.e., DFS, DSS, DTS) of the gels in toluene obtained after gelation of pure organic phase (POP) and phase selective gelation (PSG) of toluene/water mixture (1:2 v/v) using A4 as gelator (2.0% w/v). Heating-cooling treatment was used in these examples to induce gelation. Average storage modulus G' (16.5 ± 1.97 kPa for POP; 14.7 ± 1.89 kPa for PSG), average loss modulus G'' (3.0 ± 0.12 kPa for POP; 2.8 ± 0.11 kPa for PSG), average tan δ (0.18 ± 0.0028 for POP; 0.19 ± 0.0019 for PSG), and maximum strain at break (16% ± 2.0% for POP; 13% ± 1.5% for PSG) remained constant within the limits of the experimental error regardless the material. As observed in POP, the gels obtained from PSG displayed G' at least one order of magnitude higher than G'' and relatively low dependence with frequency, indicating the viscoelastic nature of the samples as well as a good tolerance to external forces.
Figure 3.
(
A–
F) Representative field emission scanning electron microscope (FESEM) images of xerogels prepared by freeze-drying the corresponding gel materials obtained at the minimum gelation concentration (MGC) as indicated in
Table 1: (
A)
A4 in toluene (POP) [
38]; (
B)
A4 in toluene (PSG); (
C)
F-A4 in toluene (POP) [
38]; (
D)
F-A4 in toluene (PSG); (
E)
Bis-A4 in toluene (POP) [
38]; (
F)
Bis-A4 in toluene (PSG); and (
G,
H) Representative transmission electron microscope (TEM) images of xerogels prepared by freeze-drying the corresponding gel materials obtained at the MGC as indicated in
Table 1; (
G)
A4 in toluene (POP) [
38]; (
H)
A4 in toluene (PSG).
Figure 3.
(
A–
F) Representative field emission scanning electron microscope (FESEM) images of xerogels prepared by freeze-drying the corresponding gel materials obtained at the minimum gelation concentration (MGC) as indicated in
Table 1: (
A)
A4 in toluene (POP) [
38]; (
B)
A4 in toluene (PSG); (
C)
F-A4 in toluene (POP) [
38]; (
D)
F-A4 in toluene (PSG); (
E)
Bis-A4 in toluene (POP) [
38]; (
F)
Bis-A4 in toluene (PSG); and (
G,
H) Representative transmission electron microscope (TEM) images of xerogels prepared by freeze-drying the corresponding gel materials obtained at the MGC as indicated in
Table 1; (
G)
A4 in toluene (POP) [
38]; (
H)
A4 in toluene (PSG).
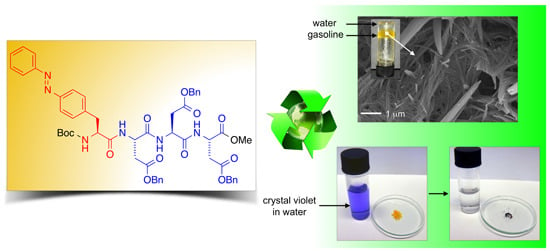
Additionally, our LMW peptidic gelators also allowed PSG of environmental hazardous materials including gasoline and diesel fuel (
Figure 4A,B), affording gel phases that were characterized by fibrillar networks as shown in
Figure 4C.
Figure 4.
(
A) Typical appearance of an inverted vial containing PSG of a mixture xylene/water prepared with
A4 as shown in
Table 1; (
B) PSG of gasoline/water mixture; and (
C) FESEM image of the xerogel prepared by freeze-drying the gel obtained by PSG of a mixture of gasoline/water (1:2
v/
v) using
bis-A4 as shown in
Table 1.
Figure 4.
(
A) Typical appearance of an inverted vial containing PSG of a mixture xylene/water prepared with
A4 as shown in
Table 1; (
B) PSG of gasoline/water mixture; and (
C) FESEM image of the xerogel prepared by freeze-drying the gel obtained by PSG of a mixture of gasoline/water (1:2
v/
v) using
bis-A4 as shown in
Table 1.
Encouraged by these results, we also investigated the more challenging PSG of water-miscible organic solvents (
i.e., CH
3CN, EtOH,
i-PrOH) where gelation of the POP was previously observed [
38]. Surprisingly, we found gelation of the entire organic solvent/water mixture (1:2
v/
v) resulting in the formation of quasi-hydrogels with a total content of organic phase of 33% (
v/
v) and different morphological features in comparison to the organogels prepared in pure organic solvents. For instance, the organogel obtained in
i-PrOH was characterized by spherical structures with a fibrillar porous inner part [
38], whereas the material obtained from
i-PrOH/water mixture revealed a high-density fibrillar network without globular microstructures (
Figure 5). Interestingly enough, a much lower concentration of peptide was necessary to gel the mixture of solvent in comparison to the pure organic phase (
i.e., 0.7%
w/
v for
i-PrOH/water (1:2
v/
v)
versus 2.0%
w/
v for pure
i-PrOH). This opens the door for future research to minimize the oil phase content and/or the use of biocompatible oil phases, which would expand the potential of these gels for the encapsulation and controlled release of hydrophobic drugs.
Figure 5.
(
A) FESEM image of the xerogel obtained from the corresponding organogel made in
i-PrOH using
A4 (2.0%
w/
v) (inset: photograph of the gel) [
38]; and (
B) FESEM image of the xerogel obtained from the corresponding organogel made in
i-PrOH/water mixture (1:2
v/
v) using
A4 (0.7%
w/
v) (inset: photograph of the gel).
Figure 5.
(
A) FESEM image of the xerogel obtained from the corresponding organogel made in
i-PrOH using
A4 (2.0%
w/
v) (inset: photograph of the gel) [
38]; and (
B) FESEM image of the xerogel obtained from the corresponding organogel made in
i-PrOH/water mixture (1:2
v/
v) using
A4 (0.7%
w/
v) (inset: photograph of the gel).
One of the major drawbacks of some PSG described in the literature is the heating-cooling protocol necessary to induce gelation, which makes it impractical for the removal of oil spills due to the high flammability of most oil-phases (e.g., gasoline, diesel). Thus, taking advantage of our previous observation of ultrasound-induced gelation [
38] we were able to successfully use this triggering mechanism for PSG at room temperature. Ultrasound-induced gelation, mainly by solvent cavitation and mismatched intra-/intermolecular interactions, has been recognized as a paradigm shift within the field of supramolecular gels [
40]. With the aim of improving the viability of the process, we were delighted to observe that the addition of slightly warmed or ultrasound-aided concentrated solutions (20%–40%
w/
v) of the suitable gelator in an oil-phase to a mixture of the same oil and water resulted in very effective PSG. Importantly, no discrepancies in MGC values were observed for the different methods used to induce PSG (
Table 2). The latter strategy avoids the use of other water-miscible cosolvents in order to predissolve the gelator, which could be potentially toxic and exhibit a negative impact on foreseeable practical applications.
Table 2.
Comparison of the MGC necessary to induce PSG in selected oil/water mixtures either by the heating-cooling method (HC), ultrasound treatment of the mixture (US) or by adding an ultrasound-aided concentrated solution of the gelator in an oil-phase to the corresponding oil/water mixture (CS).
Table 2.
Comparison of the MGC necessary to induce PSG in selected oil/water mixtures either by the heating-cooling method (HC), ultrasound treatment of the mixture (US) or by adding an ultrasound-aided concentrated solution of the gelator in an oil-phase to the corresponding oil/water mixture (CS).
| Entry | Organic Phase | Gelator | Minimum Gelation Concentration, MGC (%
w/v) |
|---|
| POP | PSG (HC) | PSG (US) | PSG (CS) |
|---|
| 1 | Toluene | A4 | 1.9 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 |
| 2 | Gasoline | A4 | 2.2 ± 0.2 | 2.4 ± 0.2 | 2.4 ± 0.2 | 2.4 ± 0.2 |
| 3 | Diesel | A4 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 |
| 4 | Toluene | Bis-A4 | 0.6 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| 5 | Gasoline | Bis-A4 | 2.5 ± 0.2 | 2.5 ± 0.2 | 2.5 ± 0.2 | 2.5 ± 0.2 |
| 6 | Diesel | Bis-A4 | 1.8 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ±0.2 |
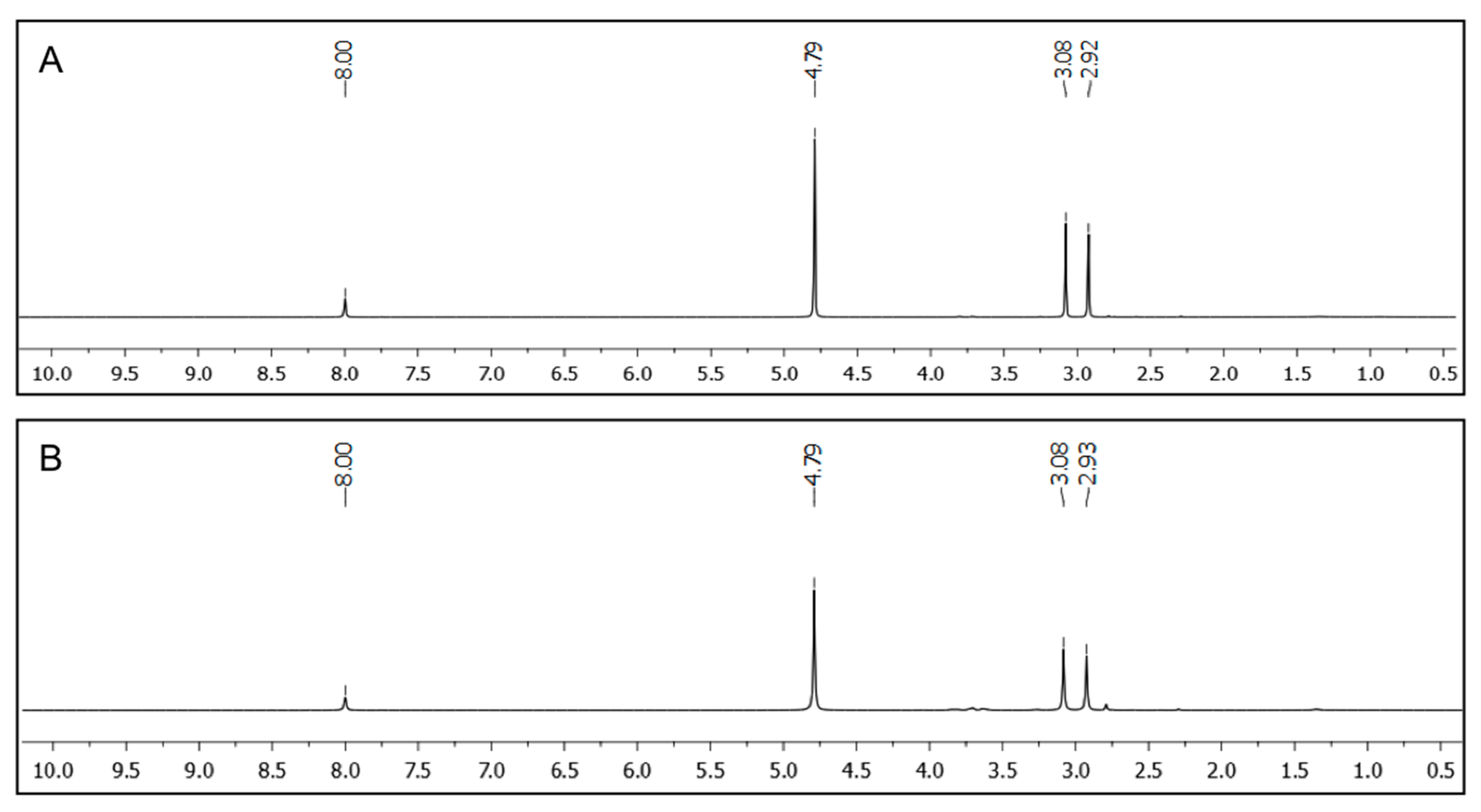
The complete removal of the oil-phase from oil/water model mixtures was confirmed by
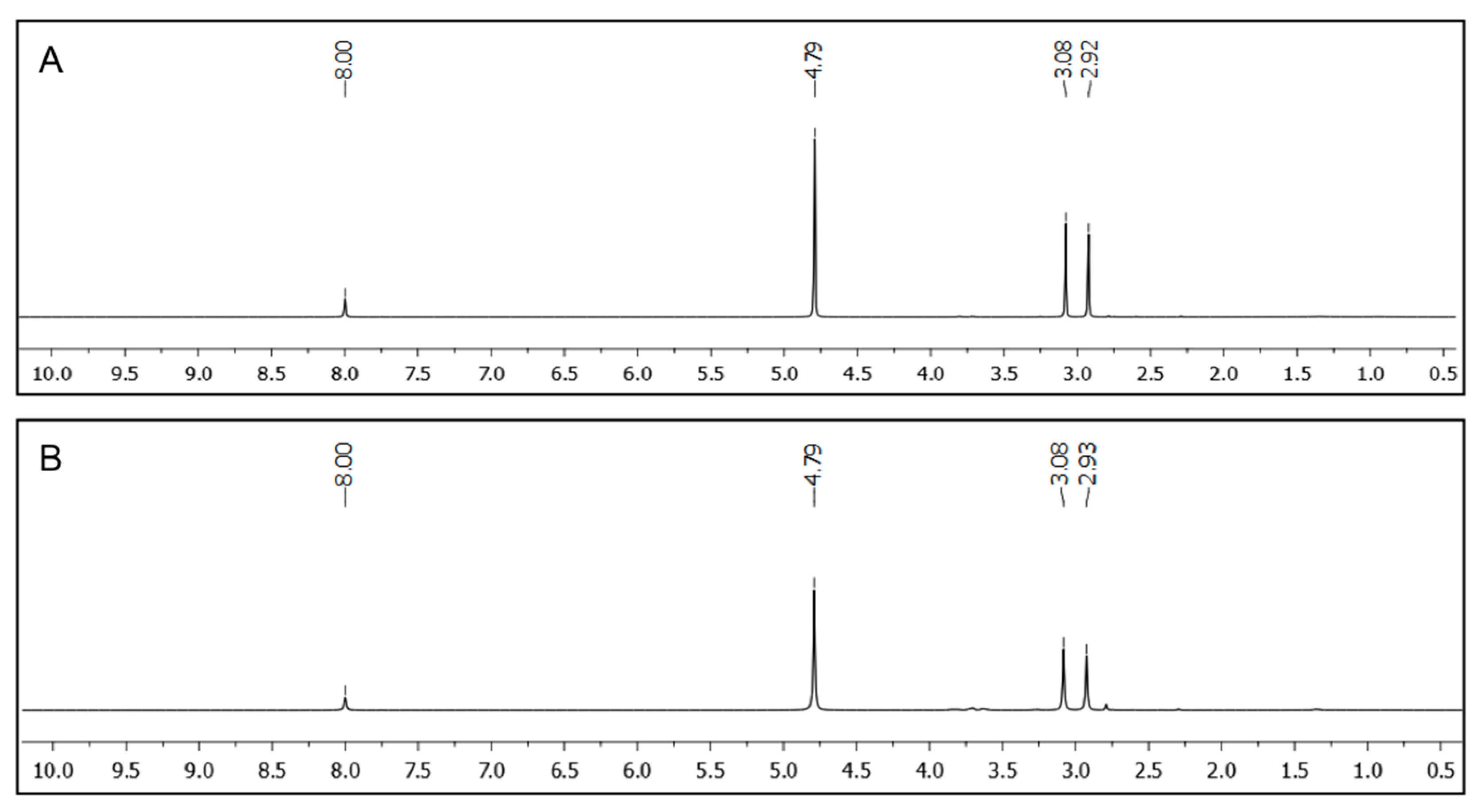
1H NMR spectroscopy using D
2O as aqueous phase containing DMF as internal standard (
Figure 6). No indication of signals referring to toluene or xylene could be found in the corresponding spectra when peptidic gelator
A4 (2.0%
w/
v and 7.0%
w/
v, respectively) was used for the PSG induced by the heating-cooling method. Additionally, no gelator could be detected by NMR analysis of the aqueous phase. Therefore, the term PSG is not strictly correct in these situations [
18] because there is not a significant partition of the gelator into the aqueous phase. However, the term has been widely adopted to describe similar cases [
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37]. Furthermore, it should be considered that only a minor fraction of known non-water soluble organogelators has shown PSG ability, which makes the described tetrapeptides very interesting for both fundamental and applied sciences.
In order to evaluate the potential application of PSG triggered by these peptides for natural oil spills, we made comparative experiments using biphasic systems containing pure water, salty water (NaCl 3.3%
w/
v), and natural river water (
Table 3). The results showed no differences between the mixtures in terms of gelation efficiency and MGC that could be caused by the modification of the ionic strength or the presence of other impurities in natural water. These results demonstrate the robustness of the process.
Figure 6.
1H NMR spectra of the aqueous phases separated from D2O/toluene mixtures after PSG using A4 as gelator. PSG was induced by the heating-cooling method and DMF (0.1 mmol) was used as internal standard. (A) Solvent mixture: D2O/toluene (2:1 v/v); [A4] = 2.0% w/v; (B) Solvent mixture: D2O/toluene (2:1 v/v); [A4] = 7.0% w/v.
Figure 6.
1H NMR spectra of the aqueous phases separated from D2O/toluene mixtures after PSG using A4 as gelator. PSG was induced by the heating-cooling method and DMF (0.1 mmol) was used as internal standard. (A) Solvent mixture: D2O/toluene (2:1 v/v); [A4] = 2.0% w/v; (B) Solvent mixture: D2O/toluene (2:1 v/v); [A4] = 7.0% w/v.
Table 3.
Comparison of the MGC necessary to induce PSG in selected oil/aqueous mixtures using different aqueous phases.
Table 3.
Comparison of the MGC necessary to induce PSG in selected oil/aqueous mixtures using different aqueous phases.
| Entry | Organic Phase | Gelator | Minimum Gelation Concentration, MGC (%
w/v) |
|---|
| Pure Water | NaCl 3.3%
w/v | River Water |
|---|
| 1 | Toluene | A4 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 |
| 2 | Gasoline | A4 | 2.4 ± 0.2 | 2.4 ± 0.2 | 2.4 ± 0.2 |
| 3 | Diesel | A4 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 |
| 4 | Toluene | Bis-A4 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| 5 | Gasoline | Bis-A4 | 2.5 ± 0.2 | 2.5 ± 0.2 | 2.5 ± 0.2 |
| 6 | Diesel | Bis-A4 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 |
The relatively high mechanical strength of the gel-materials as well as their high temporal stability (
i.e., inverted vials containing the gelled oil phase could keep the weight of the aqueous phase for at least two months without any visible gravitational flow) make these LMW peptidic gelators suitable for application in oil spill recovery if the process can be scaled-up. In order to demonstrate this potential, a model scenario was set up as shown in
Figure 7. Herein, toluene (5 mL) was poured into a much larger volume of salty water (NaCl 3.3%
w/
v, 40 mL). The aqueous layer was stained with CuSO
4 (
Figure 7A) for better visualization of the two-phase nature of the mixture (as with NaCl, the presence of the metal salt did not affect the gelation). The entire toluene-layer could be efficiently gelled by adding an ultrasound-aided concentrated solution of gelator
A4 (27.5%
w/
v) in a small volume of toluene (0.4 mL). This resulted in a gelator concentration of 2.0%
w/
v with respect to the toluene-phase (
Figure 7B). Taking advantage of the thermoreversibility of the resulting supramolecular gel, which melts at
ca. 40 °C, the gel-phase (
Figure 7C) could be easily separated by filtration and subsequently distilled (
Figure 7D–G) to recover both the gelator (yellowish residue that stays in the round bottom flask) and the toluene phase in almost quantitative yield (87% ± 7% in volume) (
Figure 7H). The recovered gelator was reused in a new cycle with a fresh oil/water mixture at the same original concentration (2.0%
w/
v) affording the same yield of distilled oil. It is important to mention that gelation could be also achieved even under mechanical agitation (mimicking a real emulsion-like oil spill), albeit the gel body suffered partial fragmentation into floating gel pieces. The same experiment was also repeated using other oil-phases such as gasoline and diesel fuel with practically identical results for the oil recovery. Such efficient recovery of the oil from the gel phase and recycling of the gelator have been only reported recently on a few occasions [
24,
25,
26].
Figure 7.
Model lab bench set up for oil spill recovery using the PSG ability of azobenzene-containing peptide
A4 (2.0%
w/
v with respect to the volume of the oil-phase). (
A) Mixture of salty water (NaCl 3.3%
w/
v, 40 mL) and toluene (5 mL). The aqueous layer was stained with CuSO
4 (
Figure 7A) for better visualization of the two-phases indicated by red arrows; (
B) Addition ultrasound-aided concentrated solution of gelator
A4 (27.5%
w/
v) in a small volume of toluene (0.4 mL); (
C) Formation of a yellowish gel-phase containing the toluene layer; (
D,
E) Separation of the gel-phase by filtration; (
F,
G) Distillation of the gel-phase to recover both the yellowish gelator and the oil phase; and (
H) Isolated toluene phase after distillation.
Figure 7.
Model lab bench set up for oil spill recovery using the PSG ability of azobenzene-containing peptide
A4 (2.0%
w/
v with respect to the volume of the oil-phase). (
A) Mixture of salty water (NaCl 3.3%
w/
v, 40 mL) and toluene (5 mL). The aqueous layer was stained with CuSO
4 (
Figure 7A) for better visualization of the two-phases indicated by red arrows; (
B) Addition ultrasound-aided concentrated solution of gelator
A4 (27.5%
w/
v) in a small volume of toluene (0.4 mL); (
C) Formation of a yellowish gel-phase containing the toluene layer; (
D,
E) Separation of the gel-phase by filtration; (
F,
G) Distillation of the gel-phase to recover both the yellowish gelator and the oil phase; and (
H) Isolated toluene phase after distillation.
2.3. Dye Removal
To demonstrate another practical application of our system, we took advantage of the PSG ability of the LMW peptidic gelators to quantitatively remove water-soluble dyes [
41] from aqueous mixtures within minutes (
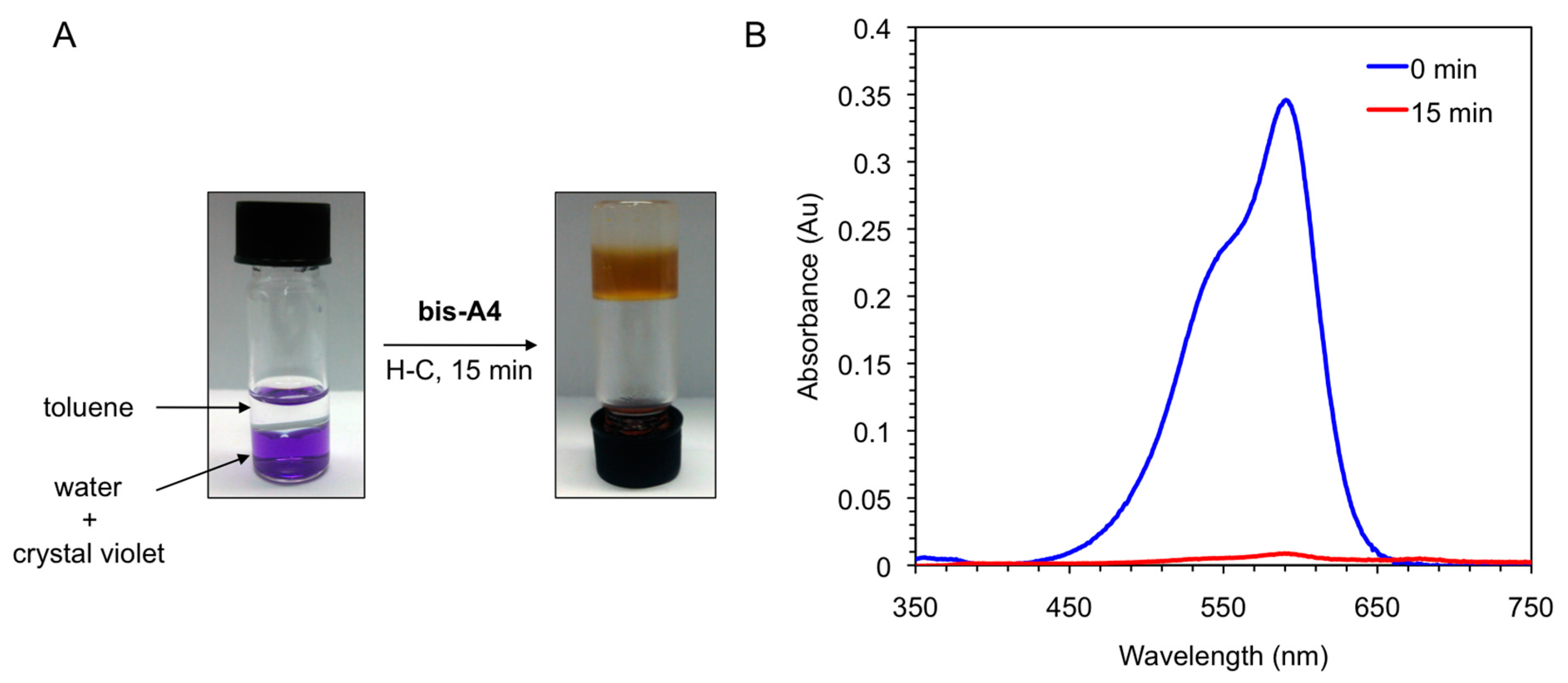
Figure 8). In two model examples, gelator
bis-A4 was used to remove crystal violet (CV) from aqueous phases. The addition of a toluene phase and the peptidic gelator at a concentration that should guarantee the complete gelation of the organic phase, caused a rapid migration of the dye from the aqueous to the organic layer with simultaneous entrapment of the dye after heating-cooling treatment of the mixture. The dye was almost quantitatively (>95%) removed from the aqueous phase within minutes as determined by UV-vis spectroscopy (
Figure 8). Preliminary experiments [
39] showed that other dyes such as ruthenium red could also be removed from aqueous solutions using other peptidic gelators bearing a side-chain azobenzene moiety (data not shown) [
38].
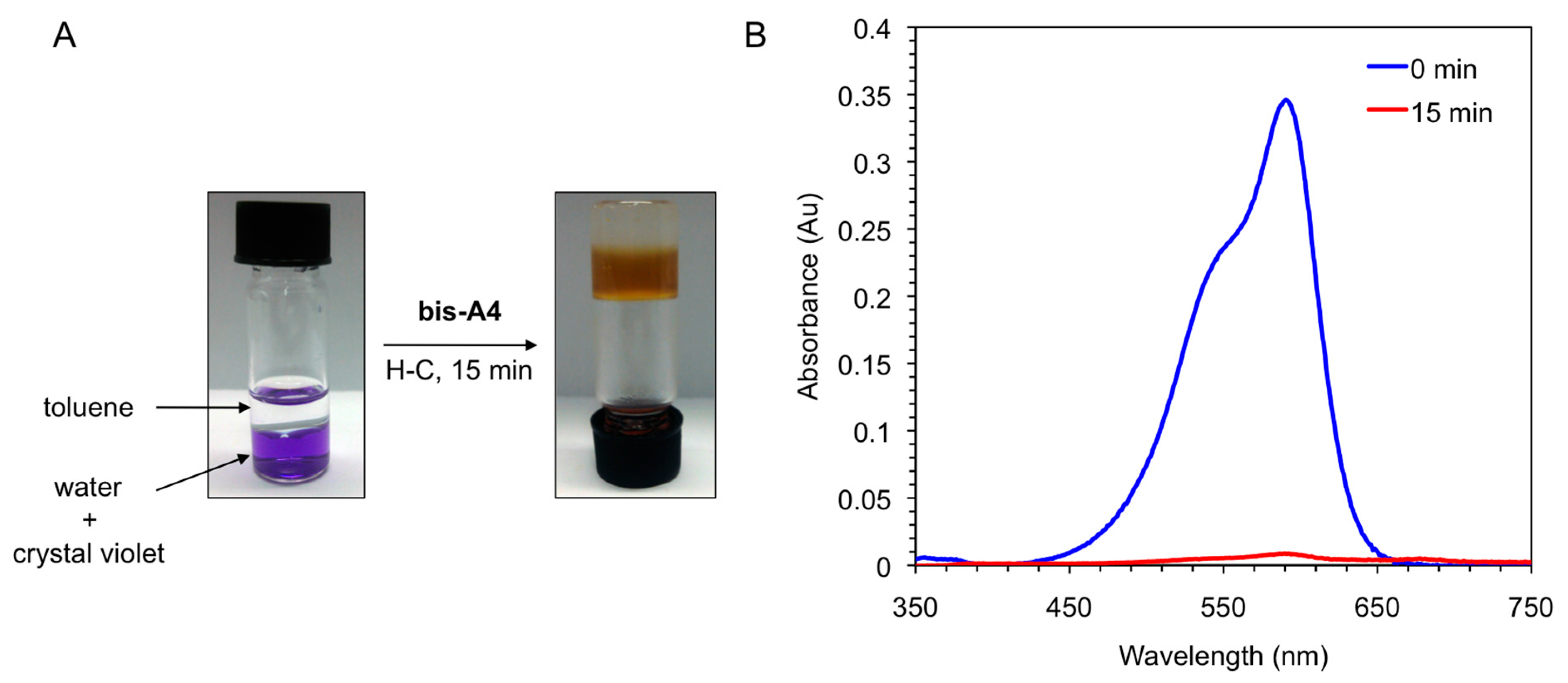
Figure 8.
(A) Photographs illustrating the removal of crystal violet from an aqueous solution (c = 1 × 10−5 mol·L−1) by addition of toluene (left vial, water/toluene 1:1 v/v) and gelator bis-A4 (1.0% w/v). PSG was induced by heating-cooling (H-C) treatment (right vial); and (B) UV-vis absorption spectra of the initial aqueous solution of crystal violet (t = 0 min; blue line) and of the aqueous phase after PSG (t = 15 min; red line).
Figure 8.
(A) Photographs illustrating the removal of crystal violet from an aqueous solution (c = 1 × 10−5 mol·L−1) by addition of toluene (left vial, water/toluene 1:1 v/v) and gelator bis-A4 (1.0% w/v). PSG was induced by heating-cooling (H-C) treatment (right vial); and (B) UV-vis absorption spectra of the initial aqueous solution of crystal violet (t = 0 min; blue line) and of the aqueous phase after PSG (t = 15 min; red line).
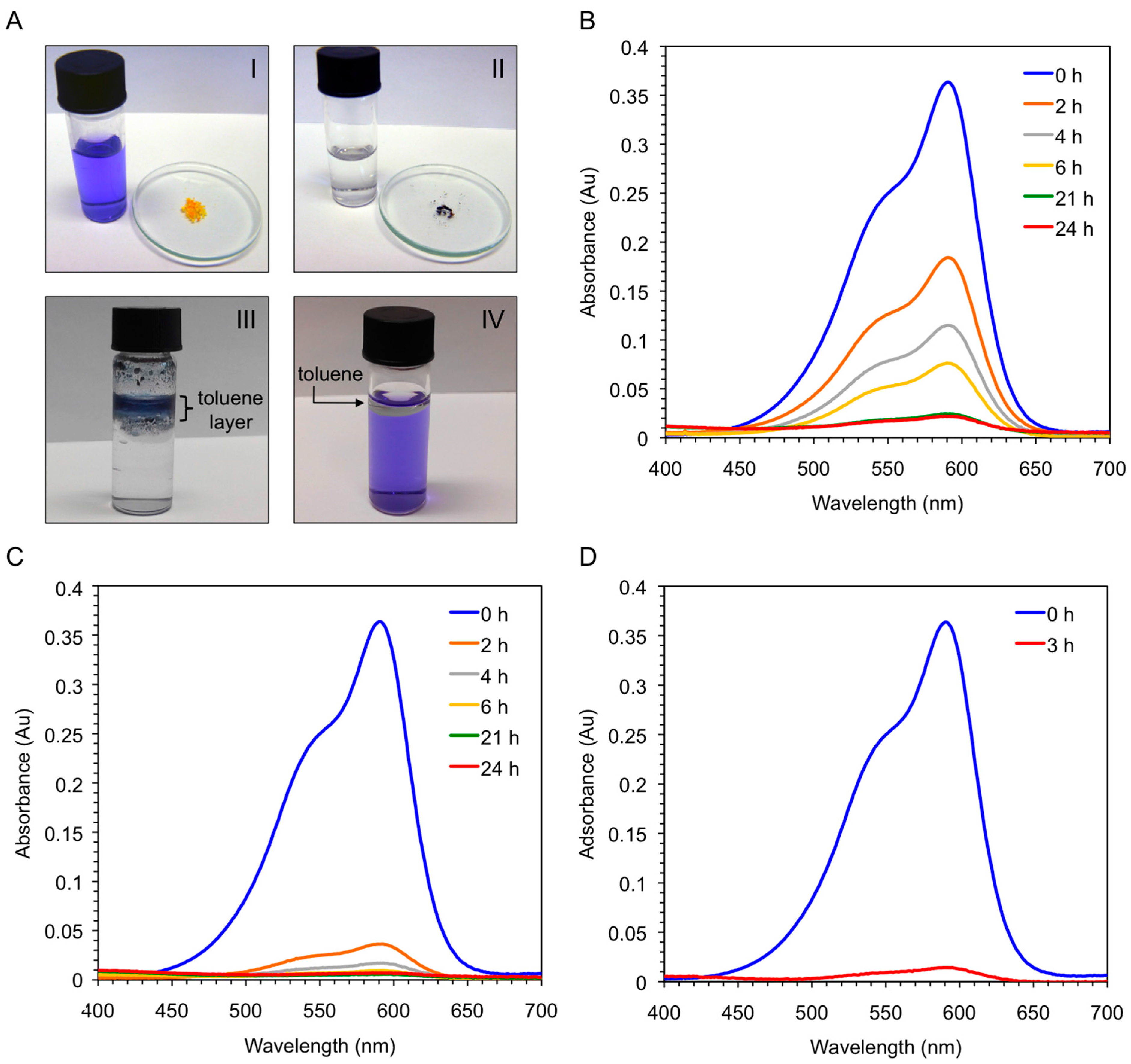
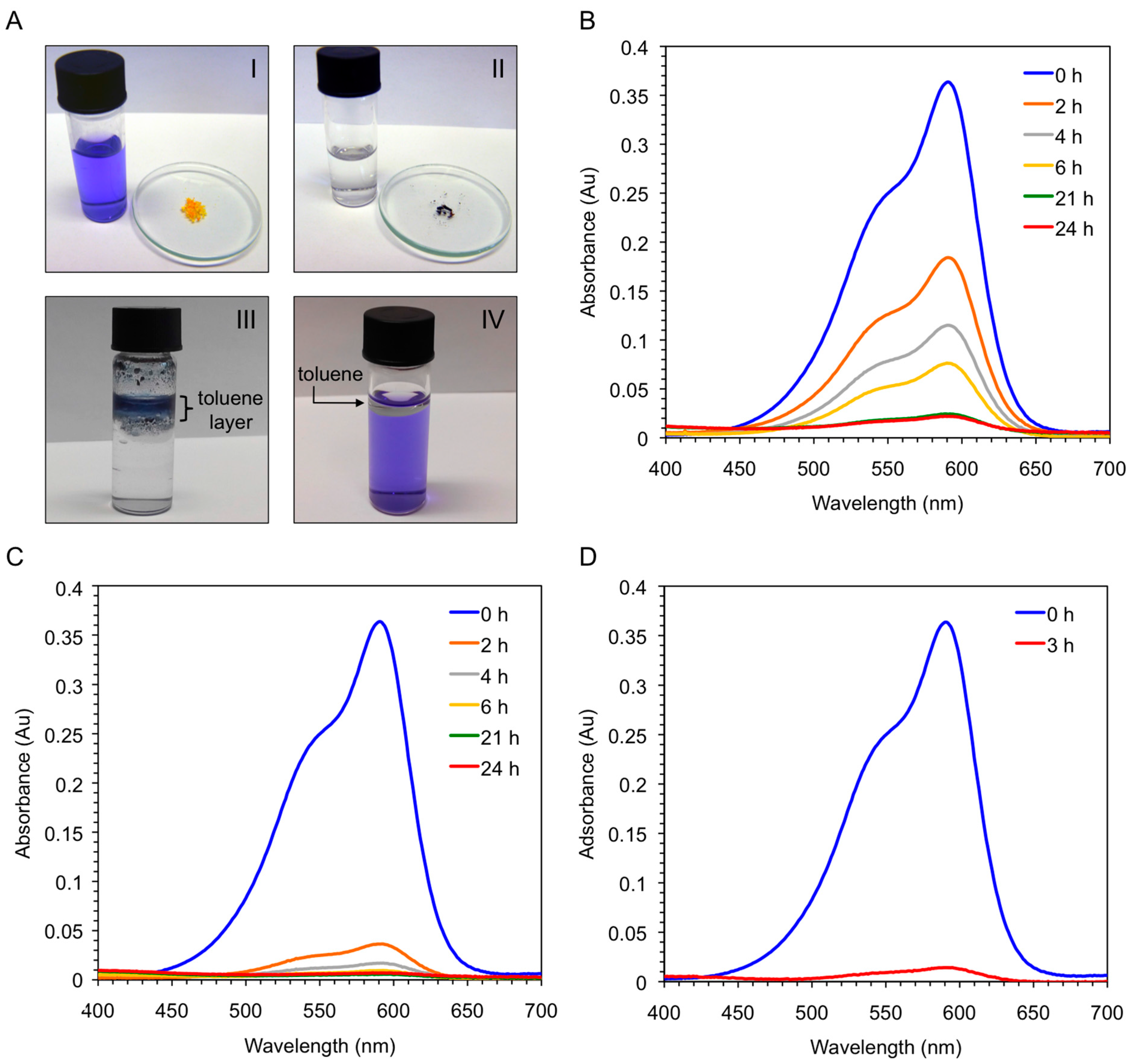
Vey interestingly, the xerogel material obtained by freeze-drying the corresponding organogel was also able to uptake CV (>95%) within 6 h directly from the aqueous solution, whereas the freshly synthesized peptide needed one day to achieve the same result (
Figure 9B,C). In principle, this result could be explained because the ability of the peptide to form supramolecular aggregates is considerably hampered in pure water, whereas the xerogel constitutes already a hydrophobic self-assembled network. Therefore, we decided to run a new experiment using the peptide and adding a small amount of toluene to the aqueous phase. Very interestingly, a quick migration of the peptide and the dye to the organic phase was observed at room temperature without the necessity of heating or sonicating the mixture. The kinetics of the dye removal was comparable to that using the xerogel material (
Figure 9C,D), and the dye-containing organic layer could be separated from the clean aqueous phase by simple decantation. Notably, unless the organic phase is heated or sonicated the peptide remains insoluble in it without gel formation (
Figure 9A-III). Moreover, simple addition of toluene to the dye-containing aqueous phase did not cause visible partition of the dye into the organic phase (
Figure 9A-IV). Therefore, the peptide and/or peptide-dye complex is likely to adopt an energetically more favorable aggregate in the organic phase. However, further detailed experiments are still necessary in order to confirm this hypothesis and unequivocally determine the substrate specificity of the process and the exact nature of the intermolecular interactions between peptide and dye.
Figure 9.
(A-I) Aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1) before addition of either bis-A4 (1.0% w/v; yellow powder in watch glass) or the corresponding xerogel obtained by freeze-drying the gel prepared from bis-A4 in toluene (1.0% w/v); (A-II) Previous solution after dye removal by the xerogel (dark-blue powder in watch glass); (A-III) Mixture obtained after addition of toluene and bis-A4 (1.0% w/v) to an aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1). Ratio water:toluene 1:0.1 v/v. The picture was taken 3 h after shaking the mixture at room temperature; (A-IV) Mixture containing an aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1) and toluene. Ratio water:toluene 1:0.1 v/v. The picture was taken after shaking the mixture at room temperature; (B) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent adsorption of the dye by freshly synthesized peptide bis-A4; (C) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent adsorption of the dye by the corresponding xerogel; and (D) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent removal of the dye after addition of toluene (ratio water:toluene 1:0.1 v/v) and peptide bis-A4 at room temperature.
Figure 9.
(A-I) Aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1) before addition of either bis-A4 (1.0% w/v; yellow powder in watch glass) or the corresponding xerogel obtained by freeze-drying the gel prepared from bis-A4 in toluene (1.0% w/v); (A-II) Previous solution after dye removal by the xerogel (dark-blue powder in watch glass); (A-III) Mixture obtained after addition of toluene and bis-A4 (1.0% w/v) to an aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1). Ratio water:toluene 1:0.1 v/v. The picture was taken 3 h after shaking the mixture at room temperature; (A-IV) Mixture containing an aqueous solution of crystal violet (c = 1 × 10−5 mol·L−1) and toluene. Ratio water:toluene 1:0.1 v/v. The picture was taken after shaking the mixture at room temperature; (B) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent adsorption of the dye by freshly synthesized peptide bis-A4; (C) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent adsorption of the dye by the corresponding xerogel; and (D) UV-vis absorption spectra of the aqueous solution of crystal violet showing the time-dependent removal of the dye after addition of toluene (ratio water:toluene 1:0.1 v/v) and peptide bis-A4 at room temperature.
![Ijms 16 11766 g009]()




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}