
Pathogenesis of Renal Disease in Systemic Lupus Erythematosus—The Role of Autoantibodies and Lymphocytes Subset Abnormalities

{kind=link}
Abstract
:1. Introduction
2. The Role of Autoantibodies
2.1. Anti-dsDNA Antibody
2.2. Anti-C1q Autoantibody and Other Autoantibodies
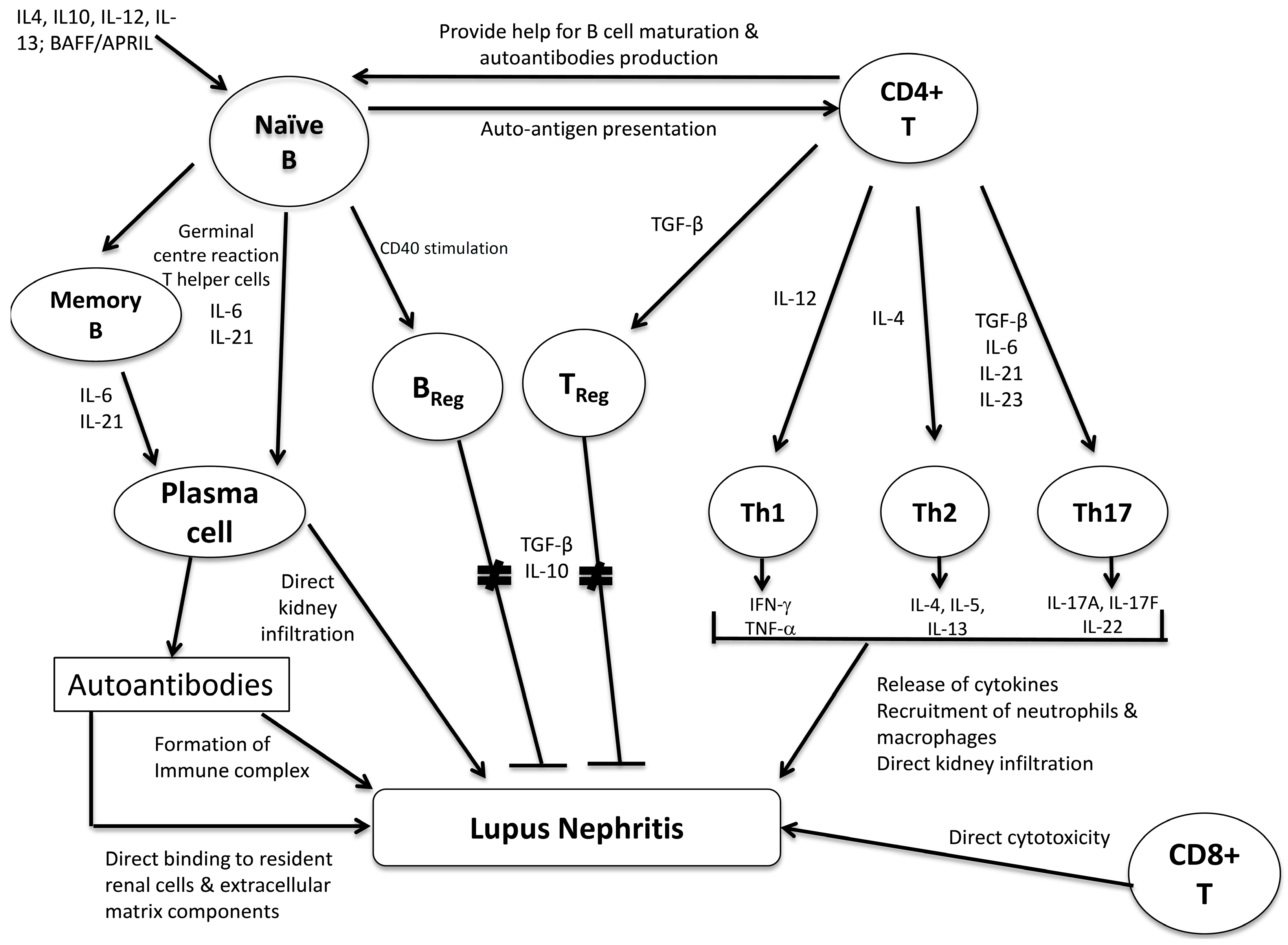
3. The Role of B Lymphocytes and Its Subsets
4. The Role of T lymphocytes and Its Subsets
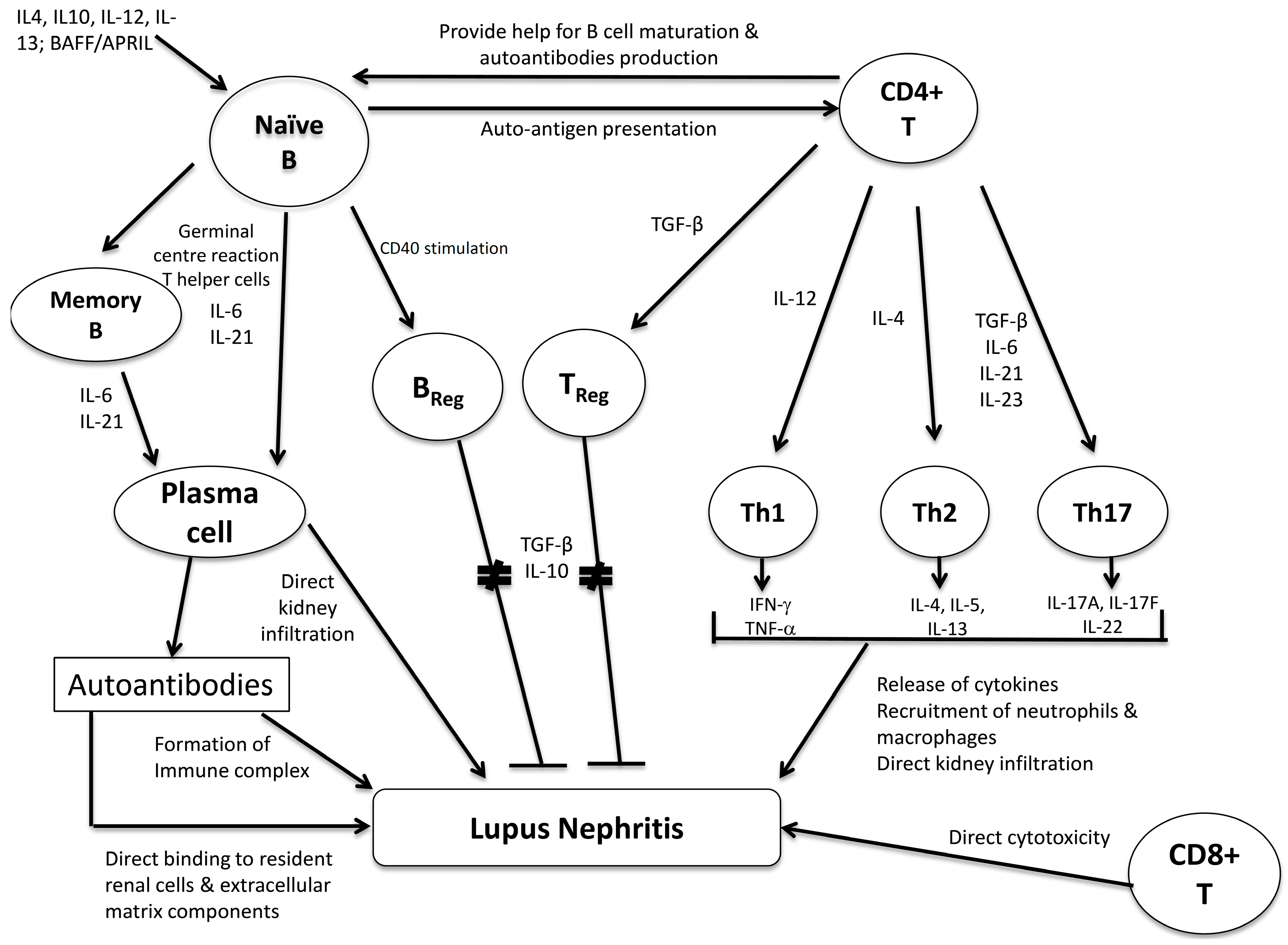
4.1. T-Helper Cells
4.1.1. Th1/Th2 Subsets
4.1.2. Th17 and TReg
4.1.3. Other T Helper Subsets (Follicular T Helper Cells, Th9 and Th22 Cells)
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Danchenko, N.; Satia, J.A.; Anthony, M.S. Epidemiology of systemic lupus erythematosus: A comparison of worldwide disease burden. Lupus 2006, 15, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Osio-Salido, E.; Manapat-Reyes, H. Epidemiology of systemic lupus erythematosus in Asia. Lupus 2010, 19, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Korbet, S.M.; Schwartz, M.M.; Evans, J.; Lewis, E.J. Severe lupus nephritis: Racial differences in presentation and outcome. J. Am. Soc. Nephrol. 2007, 18, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Zhang, X. Biomarkers for lupus nephritis: The quest continues. Clin. J. Am. Soc. Nephrol. 2009, 4, 1858–1865. [Google Scholar] [CrossRef] [PubMed]
- Winfield, J.B.; Faiferman, I.; Koffler, D. Avidity of anti-DNA antibodies in serum and IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity antinative DNA antibody with glomerulonephritis. J. Clin. Investig. 1977, 59, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kalunian, K.C.; Panosian-Sahakian, N.; Ebling, F.M.; Cohen, A.H.; Louie, J.S.; Kaine, J.; Hahn, B.H. Idiotypic characteristics of immunoglobulins associated with systemic lupus erythematosus. Studies of antibodies deposited in glomeruli of humans. Arthritis Rheumatol. 1989, 32, 513–522. [Google Scholar] [CrossRef]
- Termaat, R.M.; Assmann, K.J.; van Son, J.P.; Dijkman, H.B.; Koene, R.A.; Berden, J.H. Antigen-specificity of antibodies bound to glomeruli of mice with systemic lupus erythematosus-like syndromes. Lab. Investig. 1993, 68, 164–173. [Google Scholar] [PubMed]
- Vlahakos, D.V.; Foster, M.H.; Adams, S.; Katz, M.; Ucci, A.A.; Barrett, K.J.; Datta, S.K.; Madaio, M.P. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992, 41, 1690–1700. [Google Scholar] [CrossRef]
- Yung, S.; Cheung, K.F.; Zhang, Q.; Chan, T.M. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J. Am. Soc. Nephrol. 2010, 21, 1912–1927. [Google Scholar] [CrossRef] [PubMed]
- Fenton, K.A.; Tommeras, B.; Marion, T.N.; Rekvig, O.P. Pure anti-dsDNA mAbs need chromatin structures to promote glomerular mesangial deposits in BALB/c mice. Autoimmunity 2010, 43, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M.; Leung, J.K.; Ho, S.K.; Yung, S. Mesangial cell-binding anti-DNA antibodies in patients with systemic lupus erythematosus. J. Am. Soc. Nephrol. 2002, 13, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Croquefer, S.; Renaudineau, Y.; Jousse, S.; Gueguen, P.; Ansart, S.; Saraux, A.; Youinou, P. The ananti-α-actinin test completes ananti-DNA determination in systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2005, 1050, 170–175. [Google Scholar] [CrossRef]
- Zhao, Z.; Weinstein, E.; Tuzova, M.; Davidson, A.; Mundel, P.; Marambio, P.; Putterman, C. Cross-reactivity of human lupus anti-DNA antibodies with α-actinin and nephritogenic potential. Arthritis Rheumatol. 2005, 52, 522–530. [Google Scholar] [CrossRef]
- Zhao, Z.; Deocharan, B.; Scherer, P.E.; Ozelius, L.J.; Putterman, C. Differential binding of cross-reactive anti-DNA antibodies to mesangial cells: The role of α-actinin. J. Immunol. 2006, 176, 7704–7714. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, E.S.; Rekvig, O.P. Nephritogenic potential of anti-DNA antibodies against necrotic nucleosomes. J. Am. Soc. Nephrol. 2009, 20, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Mjelle, J.E.; Rekvig, O.P.; van der Vlag, J.; Fenton, K.A. Nephritogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity 2011, 44, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, A.; Fismen, S.; Fenton, K.A.; Fenton, C.; Osterud, B.; Mortensen, E.S.; Rekvig, O.P. Heparin exerts a dual effect on murine lupus nephritis by enhancing enzymatic chromatin degradation and preventing chromatin binding in glomerular membranes. Arthritis Rheumatol. 2011, 63, 1065–1075. [Google Scholar] [CrossRef]
- Yung, S.; Tsang, R.C.; Sun, Y.; Leung, J.K.; Chan, T.M. Effect of human anti-DNA antibodies on proximal renal tubular epithelial cell cytokine expression: Implications on tubulointerstitial inflammation in lupus nephritis. J. Am. Soc. Nephrol. 2005, 16, 3281–3294. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Koscec, M.; Wolfson-Reichlin, M.; Ebling, F.M.; Tsao, B.; Hahn, B.H.; Reichlin, M. Murine and human antibodies to native DNA that cross-react with the A and D SnRNP polypeptides cause direct injury of cultured kidney cells. J. Immunol. 1995, 154, 4857–4864. [Google Scholar] [PubMed]
- Zack, D.J.; Stempniak, M.; Wong, A.L.; Taylor, C.; Weisbart, R.H. Mechanisms of cellular penetration and nuclear localization of an anti-double strand DNA autoantibody. J. Immunol. 1996, 157, 2082–2088. [Google Scholar] [PubMed]
- Chan, T.M.; Frampton, G.; Jayne, D.R.; Perry, G.J.; Lockwood, C.M.; Cameron, J.S. Clinical significance of anti-endothelial cell antibodies in systemic vasculitis: A longitudinal study comparing anti-endothelial cell antibodies and anti-neutrophil cytoplasm antibodies. Am. J. Kidney Dis. 1993, 22, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M.; Cheng, I.K. A prospective study on anti-endothelial cell antibodies in patients with systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1996, 78, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M.; Frampton, G.; Cameron, J.S. Identification of DNA-binding proteins on human umbilical vein endothelial cell plasma membrane. Clin. Exp. Immunol. 1993, 91, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.M.; Frampton, G.; Staines, N.A.; Hobby, P.; Perry, G.J.; Cameron, J.S. Different mechanisms by which anti-DNA MoAbs bind to human endothelial cells and glomerular mesangial cells. Clin. Exp. Immunol. 1992, 88, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Leung, J.C.; Lai, B.K.; Li, P.K.; Lai, C.K. Anti-DNA autoantibodies stimulate the release of interleukin-1 and interleukin-6 from endothelial cells. J. Pathol. 1996, 178, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Leung, J.C.; Lai, K.B.; Lai, F.M.; Wong, K.C. Increased release of von Willebrand factor antigen from endothelial cells by anti-DNA autoantibodies. Ann. Rheum. Dis. 1996, 55, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Leung, J.C.; Lai, K.B.; Lai, C.K. Effect of anti-DNA autoantibodies on the gene expression of interleukin 8, transforming growth factor-β, and nitric oxide synthase in cultured endothelial cells. Scand. J. Rheumatol. 1997, 26, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.R.; Wang, C.; Marion, T.N. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. 2012, 82, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.P.; Rubin, K.; Laurell, A.B.; Loos, M. Common epitopes in Clq and collagen type II. Mol. Immunol. 1989, 26, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Wener, M.H.; Mannik, M.; Schwartz, M.M.; Lewis, E.J. Relationship between renal pathology and the size of circulating immune complexes in patients with systemic lupus erythematosus. Medicine (Baltimore) 1987, 66, 85–97. [Google Scholar] [CrossRef]
- Uwatoko, S.; Mannik, M. Low-molecular weight C1q-binding immunoglobulin G in patients with systemic lupus erythematosus consists of autoantibodies to the collagen-like region of C1q. J. Clin. Investig. 1988, 82, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Yang, X.W.; Song, Y.; Yu, F.; Zhao, M.H. Anti-C1q autoantibodies from active lupus nephritis patients could inhibit the clearance of apoptotic cells and complement classical pathway activation mediated by C1q in vitro. Immunobiology 2014, 219, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Moroni, G.; Trendelenburg, M.; del Papa, N.; Quaglini, S.; Raschi, E.; Panzeri, P.; Testoni, C.; Tincani, A.; Banfi, G.; Balestrieri, G.; et al. Anti-C1q antibodies may help in diagnosing a renal flare in lupus nephritis. Am. J. Kidney Dis. 2001, 37, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Siegert, C.E.; Daha, M.R.; Tseng, C.M.; Coremans, I.E.; van Es, L.A.; Breedveld, F.C. Predictive value of IgG autoantibodies against C1q for nephritis in systemic lupus erythematosus. Ann. Rheum. Dis. 1993, 52, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Coremans, I.E.; Spronk, P.E.; Bootsma, H.; Daha, M.R.; van der Voort, E.A.; Kater, L.; Breedveld, F.C.; Kallenberg, C.G. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am. J. Kidney Dis. 1995, 26, 595–601. [Google Scholar] [CrossRef]
- Yap, D.Y.; Yung, S.; Zhang, Q.; Tang, C.; Chan, T.M. Mesangial cell-binding activity of serum immunoglobulin g in patients with lupus nephritis. PLoS ONE 2014, 9, e101987. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Manson, J.J.; Mills, K.; Jury, E.; Mason, L.; D’Cruz, D.P.; Ni, L.; Saleem, M.; Mathieson, P.; Isenberg, D.; Rahman, A. Pathogenic autoantibodies from patients with lupus nephritis cause reduced tyrosine phosphorylation of podocyte proteins, including tubulin. Lupus Sci. Med. 2014, 1, e000013. [Google Scholar] [CrossRef]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Gatto, M. Nephritogenic-antinephritogenic antibody network in lupus glomerulonephritis. Lupus 2012, 21, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Sekine, H.; Watanabe, H.; Gilkeson, G.S. Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J. Immunol. 2004, 172, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Espeli, M.; Bokers, S.; Giannico, G.; Dickinson, H.A.; Bards;ey, V.; Fogo, A.B.; Smith, K.G.C. Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol. 2011, 22, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ishikawa, S.; Sato, T.; Akadegawa, K.; Yurino, H.; Kitabatake, M.; Hontsu, S.; Ezaki, T.; Kimura, H.; Matsushima, K. Defective B1 cell homing to the peritoneal cavity and preferential recruitment of B1 cells in the target organs in a murine model for systemic lupus erythematosus. J. Immunol. 2004, 172, 3628–3634. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999, 189, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.T.; Madaio, M.P.; Shlomchik, M.J. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immunol. 1999, 163, 3592–3596. [Google Scholar] [PubMed]
- Harris, D.P.; Haynes, L.; Sayles, P.C.; Duso, D.K.; Eaton, S.M.; Lepak, N.M.; Johnson, L.L.; Swain, S.L.; Lund, F.E. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 2000, 1, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Anolik, J.H.; Barnard, J.; Cappione, A.; Pugh-Bernard, A.E.; Felgar, R.E.; Looney, R.J.; Sanz, I. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheumatol. 2004, 50, 3580–3590. [Google Scholar] [CrossRef]
- Dorner, T.; Jacobi, A.M.; Lee, J.; Lipsky, P.E. Abnormalities of B cell subsets in patients with systemic lupus erythematosus. J. Immunol. Methods 2011, 363, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Tiller, T.; Tsuiji, M.; Yurasov, S.; Velinzon, K.; Nussenzweig, M.C.; Wardemann, H. Autoreactivity in human IgG+ memory B cells. Immunity 2007, 26, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Odendahl, M.; Jacobi, A.; Hansen, A.; Feist, E.; Hiepe, F.; Burmester, G.R.; Lipsky, P.E.; Radbruch, A.; Dorner, T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J. Immunol. 2000, 165, 5970–5979. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.M.; Odendahl, M.; Reiter, K.; Bruns, A.; Burmester, G.R.; Radbruch, A.; Valet, G.; Lipsky, P.E.; Dorner, T. Correlation between circulating CD27 high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2003, 48, 1332–1342. [Google Scholar] [CrossRef]
- Sun, C.Y.; Shen, Y.; Chen, X.W.; Yan, Y.C.; Wu, F.X.; Dai, M.; Li, T.; Yang, C.D. The characteristics and significance of locally infiltrating B cells in lupus nephritis and their association with local BAFF expression. Int. J. Rheumatol. 2013, 2013, 954292. [Google Scholar] [PubMed]
- Renaudineau, Y.; Pers, J.O.; Bendaoud, B.; Jamin, C.; Youinou, P. Dysfunctional B cells in systemic lupus erythematosus. Autoimmun. Rev. 2004, 3, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rangel-Moreno, J.; Owen, T.; Barnard, J.; Nevarez, S.; Ichikawa, H.T.; Anolik, J.H. Long-term B cell depletion in murine lupus eliminates autoantibody-secreting cells and is associated with alterations in the kidney plasma cell niche. J. Immunol. 2014, 192, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheumatol. 2010, 62, 222–233. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheumatol. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Gunnarsson, I.; Jonsdottir, T. Rituximab treatment in lupus nephritis—Where do we stand? Lupus 2013, 22, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.; Houssiau, F.; Aranow, C.; D’Cruz, D.P.; Askanase, A.; Roth, D.A.; Zhong, Z.J.; Cooper, S.; Freimuth, W.W.; Ginzler, E.M.; et al. Effect of belimumab treatment on renal outcomes: Results from the phase 3 belimumab clinical trials in patients with SLE. Lupus 2013, 22, 63–72. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Appel, G.B.; Estes, D.; Knowles, D.M., 2nd; Pirani, C.L. Monoclonal antibody identification of infiltrating mononuclear leukocytes in lupus nephritis. Kidney Int. 1986, 30, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Mozes, E.; Lovchik, J.; Zinger, H.; Singer, D.S. MHC class I expression regulates susceptibility to spontaneous autoimmune disease in (NZBxNZW)F1 mice. Lupus 2005, 14, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Merino, R.; Fossati, L.; Iwamoto, M.; Takahashi, S.; Lemoine, R.; Ibnou-Zekri, N.; Pugliatti, L.; Merino, J.; Izui, S. Effect of long-term anti-CD4 or anti-CD8 treatment on the development of lpr CD4−CD8− double negative T cells and of the autoimmune syndrome in MRL-lpr/lpr mice. J. Autoimmun. 1995, 8, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Inaba, M.; Sugihara, A.; Koshiji, M.; Sugiura, K.; Amoh, Y.; Mori, S.; Kamiya, T.; Genba, H.; Ikehara, S. Effects of administration of monoclonal antibodies (anti-CD4 or anti-CD8) on the development of autoimmune diseases in (NZW × BXSB)F1 mice. Immunobiology 1998, 198, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Masutani, K.; Akahoshi, M.; Tsuruya, K.; Tokumoto, M.; Ninomiya, T.; Kohsaka, T.; Fukuda, K.; Kanai, H.; Nakashima, H.; Otsuka, T.; et al. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheumatol. 2001, 44, 2097–2106. [Google Scholar] [CrossRef]
- Okamoto, A.; Fujio, K.; Tsuno, N.H.; Takahashi, K.; Yamamoto, K. Kidney-infiltrating CD4+ T-cell clones promote nephritis in lupus-prone mice. Kidney Int. 2012, 82, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Lombardi, L.; Richards, H.B.; Dammacco, F.; Silvestris, F. Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clin. Exp. Immunol. 2008, 154, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Akahoshi, M.; Nakashima, H.; Tanaka, Y.; Kohsaka, T.; Nagano, S.; Ohgami, E.; Arinobu, Y.; Yamaoka, K.; Niiro, H.; Shinozaki, M.; et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheumatol. 1999, 42, 1644–1648. [Google Scholar]
- Masutani, K.; Taniguchi, M.; Nakashima, H.; Yotsueda, H.; Kudoh, Y.; Tsuruya, K.; Tokumoto, M.; Fukuda, K.; Kanai, H.; Hirakata, H.; Iida, M. Up-regulated interleukin-4 production by peripheral T-helper cells in idiopathic membranous nephropathy. Nephrol. Dial. Transplant. 2004, 19, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, A.; Wada, T.; Kinoshita, K.; Tesch, G.; Kelley, V.R. IFN-γ receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J. Immunol. 1998, 161, 494–503. [Google Scholar] [PubMed]
- Haas, C.; Ryffel, B.; Le Hir, M. IFN-γ is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J. Immunol. 1997, 158, 5484–5491. [Google Scholar] [PubMed]
- Kikawada, E.; Lenda, D.M.; Kelley, V.R. IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-γ expression, and diminishes systemic pathology. J. Immunol. 2003, 170, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Sugiyama, N.; Masutani, K.; Sadanaga, A.; Miyazaki, Y.; Inoue, Y.; Akahoshi, M.; Katafuchi, R.; Hirakata, H.; Harada, M.; et al. Membranous glomerulonephritis development with Th2-type immune deviations in MRL/lpr mice deficient for IL-27 receptor (WSX-1). J. Immunol. 2005, 175, 7185–7192. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.D.; Monestier, M.; Tsokos, G.C. Accelerated ischemia/reperfusion-induced injury in autoimmunity-prone mice. J. Immunol. 2004, 173, 4230–4235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kyttaris, V.C.; Tsokos, G.C. The role of IL-23/IL-17 axis in lupus nephritis. J. Immunol. 2009, 183, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Kyttaris, V.C.; Zhang, Z.; Kuchroo, V.K.; Oukka, M.; Tsokos, G.C. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J. Immunol. 2010, 184, 4605–4609. [Google Scholar] [CrossRef] [PubMed]
- Kyttaris, V.C.; Kampagianni, O.; Tsokos, G.C. Treatment with anti-interleukin 23 antibody ameliorates disease in lupus-prone mice. Biomed. Res. Int. 2013, 2013, 861028. [Google Scholar] [CrossRef] [PubMed]
- Crispin, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Dolff, S.; Quandt, D.; Wilde, B.; Feldkamp, T.; Hua, F.; Cai, X.; Specker, C.; Kribben, A.; Kallenberg, C.G.; Witzke, O. Increased expression of costimulatory markers CD134 and CD80 on interleukin-17 producing T cells in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R150. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Y.; Chen, Y.M.; Wen, M.C.; Hsieh, T.Y.; Hung, W.T.; Lan, J.L. The potential role of Th17 cells and Th17-related cytokines in the pathogenesis of lupus nephritis. Lupus 2012, 21, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Zickert, A.; Amouddruz, P.; Sundstrom, Y.; Ronnelid, J.; Malmstrom, V.; Gunnarsson, I. Il-17 and IL-23 in lupus nephritis—Association to histopathology and response to treatment. BMC Immunol. 2015, 16, 7. [Google Scholar] [CrossRef]
- Schmidt, T.; Paust, H.J.; Krebs, C.F.; Turner, J.E.; Kaffke, A.; Bennstein, S.B.; Koyro, T.; Peters, A.; Velden, J.; Hünemörder, S.; et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol. 2015, 67, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Woodworth, L.; Smith, K.; Coco, J.; Vitsky, A.; McPherson, J. Therapeutic benefit of treatment with anti-thymocyte globulin and latent TGF-β1 in the MRL/lpr lupus mouse model. Lupus 2008, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.; Su, H.; Cui, J.; Wang, B. Role of Treg cells and TGF-β1 in patients with systemic lupus erythematosus: A possible relation with lupus nephritis. Immunol. Investig. 2012, 41, 15–27. [Google Scholar] [CrossRef]
- Xing, Q.; Wang, B.; Su, H.; Cui, J.; Li, J. Elevated Th17 cells are accompanied by FoxP3+ Treg cells decrease in patients with lupus nephritis. Rheumatol. Int. 2012, 32, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, D.; Chen, J.; Lu, L.; Hua, B.; Li, X.; Tsao, B.P.; Sun, L. Inhibition of aberrant circulating Tfh cell proportions by corticosteroids in patients with systemic lupus erythematosus. PLoS ONE 2012, 7, e51982. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Wong, P.T.; Tam, L.S.; Li, E.K.; Chen, D.P.; Lam, C.W. Elevated production of B cell chemokine CXCL13 is correlated with systemic lupus erythematosus disease activity. J. Clin. Immunol. 2010, 30, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Henderson, S.G.; Brandt, D.; Liu, N.; Guttikonda, R.; Hsieh, C.; Kaverina, N.; Utset, T.O.; Meehan, S.M.; Quigg, R.J.; et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J. Immunol. 2011, 186, 1849–1860. [Google Scholar]
- Zhao, L.; Jiang, Z.; Jiang, Y.; Ma, N.; Wang, K.; Zhang, Y.; Feng, L. IL-22+CD4+ T-cells in patients with active systemic lupus erythematosus. Exp. Biol. Med. (Maywood) 2013, 238, 193–199. [Google Scholar] [CrossRef]
- Yang, X.Y.; Wang, H.Y.; Zhao, X.Y.; Wang, L.J.; Lv, Q.H.; Wang, Q.Q. Th22, but not Th17 might be a good index to predict the tissue involvement of systemic lupus erythematosus. J. Clin. Immunol. 2013, 33, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.X.; Pan, H.F.; Ye, D.Q.; Xu, Y. Potential roles of IL-9 in the pathogenesis of systemic lupus erythematosus. Am. J. Clin. Exp. Immunol. 2012, 1, 28–32. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, D.Y.H.; Lai, K.N. Pathogenesis of Renal Disease in Systemic Lupus Erythematosus—The Role of Autoantibodies and Lymphocytes Subset Abnormalities. Int. J. Mol. Sci. 2015, 16, 7917-7931. https://doi.org/10.3390/ijms16047917
Yap DYH, Lai KN. Pathogenesis of Renal Disease in Systemic Lupus Erythematosus—The Role of Autoantibodies and Lymphocytes Subset Abnormalities. International Journal of Molecular Sciences. 2015; 16(4):7917-7931. https://doi.org/10.3390/ijms16047917
Chicago/Turabian StyleYap, Desmond Y. H., and Kar N. Lai. 2015. "Pathogenesis of Renal Disease in Systemic Lupus Erythematosus—The Role of Autoantibodies and Lymphocytes Subset Abnormalities" International Journal of Molecular Sciences 16, no. 4: 7917-7931. https://doi.org/10.3390/ijms16047917
APA StyleYap, D. Y. H., & Lai, K. N. (2015). Pathogenesis of Renal Disease in Systemic Lupus Erythematosus—The Role of Autoantibodies and Lymphocytes Subset Abnormalities. International Journal of Molecular Sciences, 16(4), 7917-7931. https://doi.org/10.3390/ijms16047917