
Identification of Inhibitors of Biological Interactions Involving Intrinsically Disordered Proteins


Abstract
:1. Structural Features and Models of PPI Hubs in Networks
2. IDPs and Diseases

{kind=link}
{kind=link}
| Protein | Diseases |
|---|---|
| Aβ | Alzheimer’s disease, Dutch hereditary cerebral hemorrhage with amyloidosis, Congophilic angiopathy |
| Tau | Tauopathies, Alzheimer’s disease, Corticobasal degeneration, Pick’s disease, Progressive supranuclear palsy |
| Prion protein | Prion diseases, Creutzfeld-Jacob disease, Gerstmann-Strӓussler-Schneiker syndrome, Fatal familial insomnia, Kuru, Bovine spongiform encephalopathy, Scrapie, Chronic wasting disease |
| α-Synuclein | Synucleinopathies, Parkinson’s disease, Lowy body variant of Alzheimer’s disease, Diffuse Lowy body disease, Dementia with Lowy bodies, Multiple system atrophy, Neurodegeneration with brain iro accumulation type I |
| β-Synuclein | Parkinson’s disease, Diffuse Lowy body disease |
| γ-Synuclein | Parkinson’s disease, Diffuse Lowy body disease |
| Huntingtin’s protein | Huntington’s disease |
| DRPLA protein | Hereditary dentatorubral-pallidoluysian atrophy |
| Androgen receptor | Kennedy’s disease or X-link spinal and bulbar muscular atrophy |
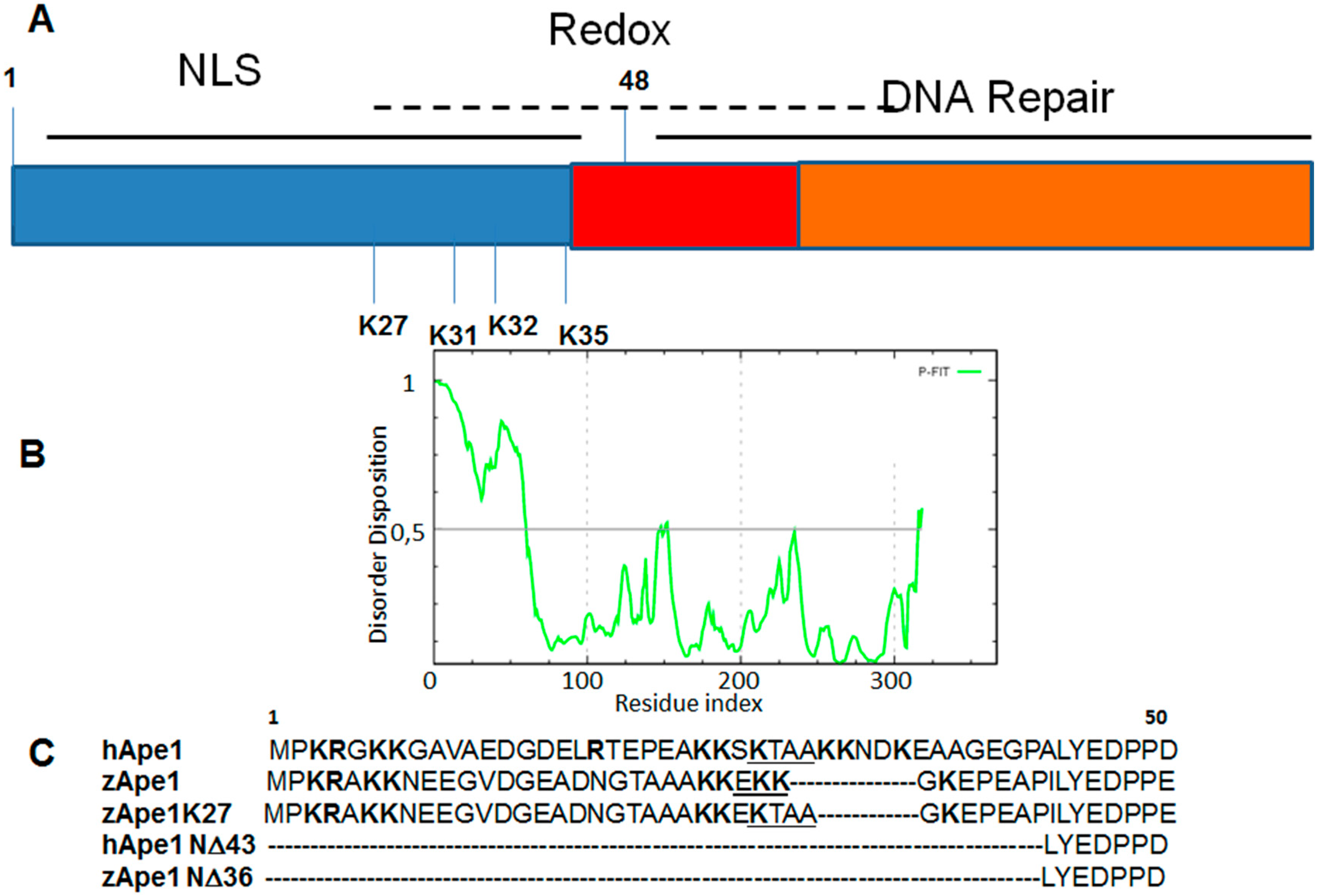
3. DNA Binding Proteins
| APE1 Mutants | Tm (°C) |
|---|---|
| hAPE1 | 41.5 |
| zAPE1 | 46.5 |
| zAPE1 K27 | 39.5 |
| hAPE1 N∆43 | 44.0 |
| zAPE1 N∆36 | 50.5 |
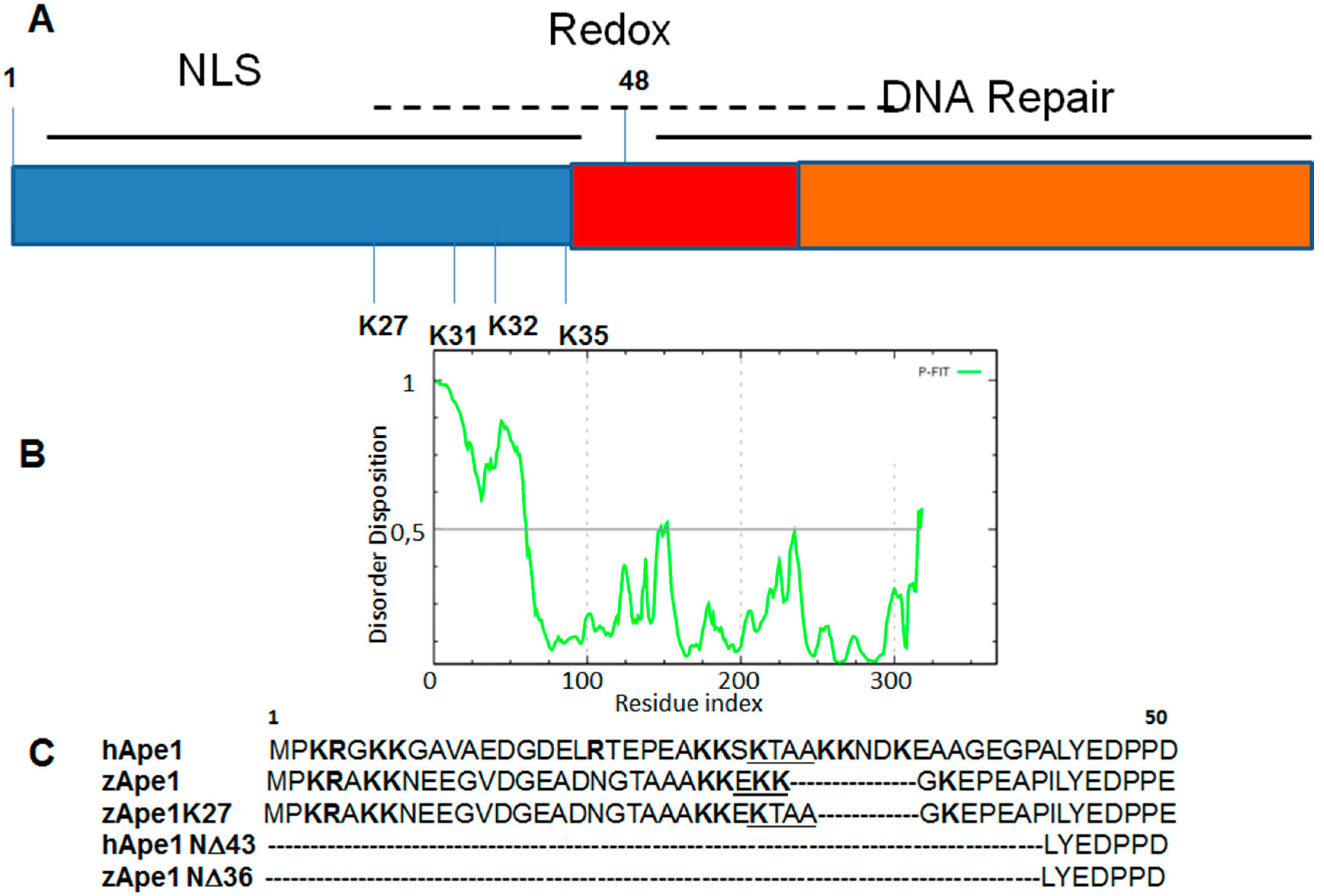
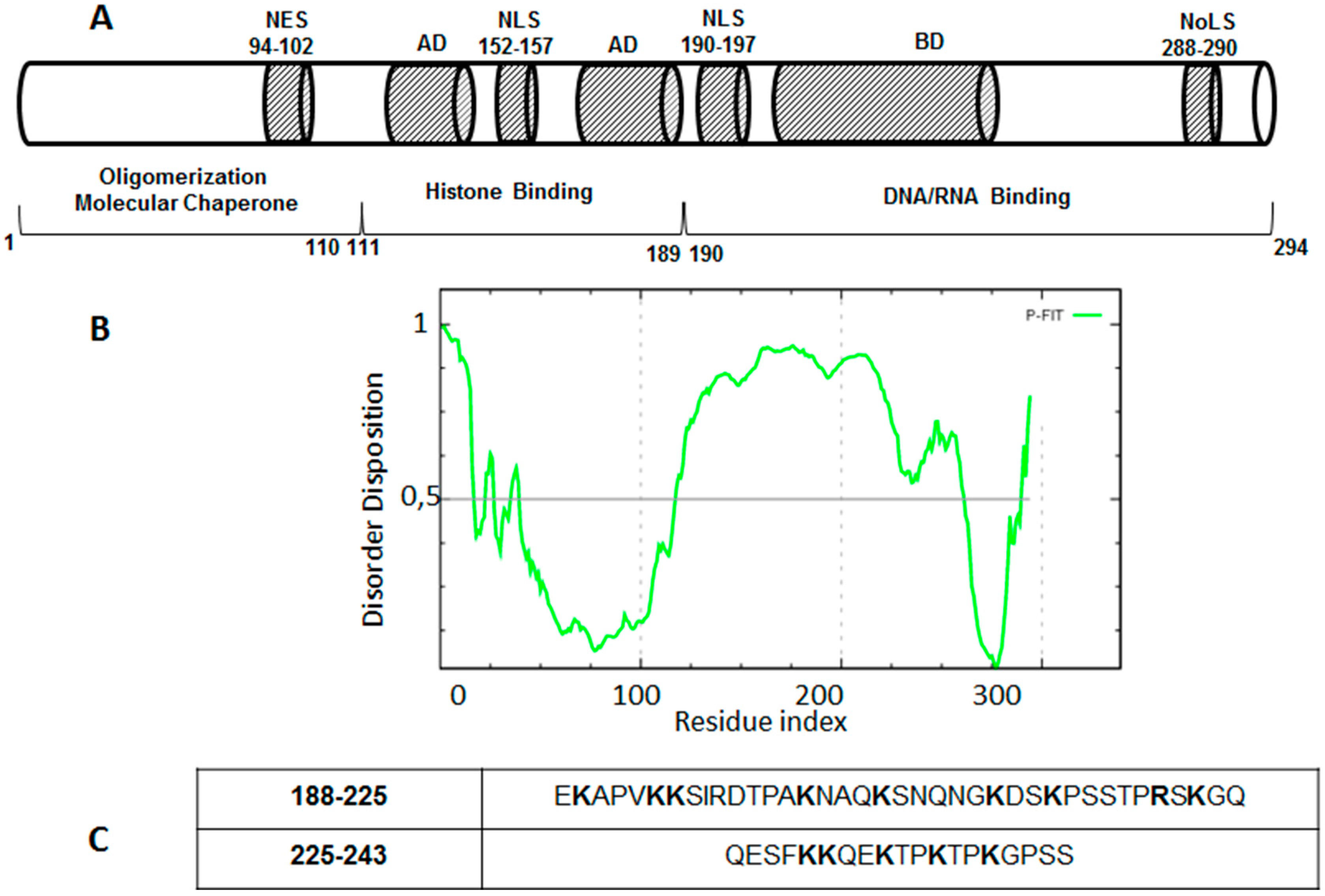
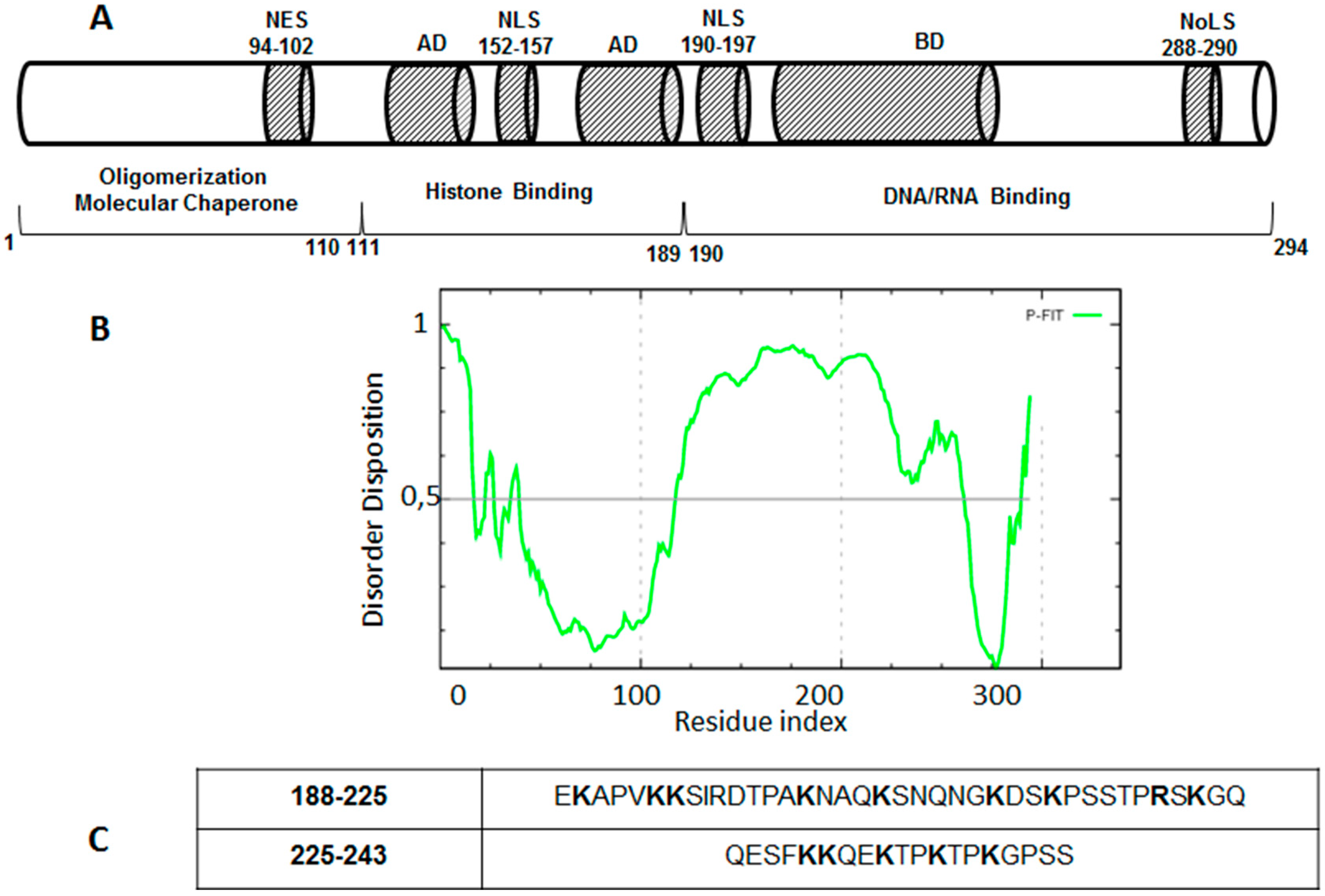
4. Drug Design for the Disordered Proteins
| Protein/Complex | Inhibitor | Structural Technique | Reference |
|---|---|---|---|
| Aβ42 | Curcumin, Congo red | In silico FBDD | [86] |
| Aβ42, αsynuclein, IAPP | EGCG | CD, NMR | [87] |
| Aβ42 | carnosine | NMR | [90] |
| τ-protein | PcTS | SAXS, NMR, EPR | [88] |
| β2 microglobulin | rifamycin SV | ESI-IMS-MS | [89] |
| p53/MDM2 | peptidomimetics, small molecules | Virtual screening | [93] |
| KSHV Pr | small molecule | NMR | [96] |
| c-Myc/Max | peptidomimetic, small molecule | Virtual screening, FRET, NMR, FP | [97,98,99,100] |
| c-Fos/c-Jun | peptidomimetic, small molecule | MD, FP | [101,102] |
| androgen receptor | peptidomimetic | X-ray | [103] |
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining α-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Under-folded proteins: Conformational ensembles and their roles in protein folding, function, and pathogenesis. Biopolymers 2013, 99, 870–887. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 473–484. [Google Scholar]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 2006, 5, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed]
- Weatheritt, R.J.; Jehl, P.; Dinkel, H.; Gibson, T.J. iELM—A web server to explore short linear motif-mediated interactions. Nucleic Acids Res. 2012, 40, W364–W369. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J. Cell regulation: Determined to signal discrete cooperation. Trends Biochem. Sci. 2009, 34, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets: The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- De Laureto, P.P.; Frare, E.; Battaglia, F.; Mossuto, M.F.; Uversky, V.N.; Fontana, A. Protein dissection enhances the amyloidogenic properties of α-lactalbumin. FEBS J. 2005, 272, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.Z.; Wan, Y.M.; He, B.F.; Ying, H.J.; Ouyang, P.K. New gene cluster from the thermophile Bacillus fordii MH602 in the conversion of dl-5-substituted hydantoins to l-amino acids. J. Microbiol. Biotechnol. 2009, 19, 1497–1505. [Google Scholar] [CrossRef]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Dosztanyi, Z.; Simon, I. Disordered binding regions and linear motifs—Bridging the gap between two models of molecular recognition. PLoS ONE 2012, 7, e46829. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Protein disorder in the human diseasome: Unfoldomics of human genetic diseases. BMC Genomics 2009, 10, S12. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Rochet, J.C.; Lansbury, P.T. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol. 2000, 10, 60–68. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Moritsugu, K.; Terada, T.; Kidera, A. Disorder-to-order transition of an intrinsically disordered region of sortase revealed by multiscale enhanced sampling. J. Am. Chem. Soc. 2012, 134, 7094–7101. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Mark, W.Y.; Liao, J.C.; Lu, Y.; Ayed, A.; Laister, R.; Szymczyna, B.; Chakrabartty, A.; Arrowsmith, C.H. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein–protein and protein–DNA interactions. J. Mol. Biol. 2005, 345, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Potikyan, G.; Savene, R.O.; Denny, C.T.; Uversky, V.N.; Lee, K.A. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Roman, A.; Oldfield, C.J.; Dunker, A.K. Protein intrinsic disorder and human papillomaviruses: Increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J. Proteome Res. 2006, 5, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Pathak, R.R.; Xue, B.; Uversky, V.N.; Dave, V. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci. Rep. 2013, 3, 2035. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of α-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Midic, U.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Unfoldomics of human genetic diseases: Illustrative examples of ordered and intrinsically disordered members of the human diseasome. Protein Pept. Lett. 2009, 16, 1533–1547. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pal, U.; Bagga, K.; Roy, A.; Mrigwani, A.; Maiti, N.C. Sequence complexity of amyloidogenic regions in intrinsically disordered human proteins. PLoS ONE 2014, 9, e89781. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Esteras-Chopo, A.; Serrano, L.; Lopez de la Paz, M. The amyloid stretch hypothesis: Recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. USA 2005, 102, 16672–16677. [Google Scholar] [CrossRef] [PubMed]
- Vuzman, D.; Levy, Y. Intrinsically disordered regions as affinity tuners in protein–DNA interactions. Mol. Biosyst. 2012, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Toth-Petroczy, A.; Simon, I.; Fuxreiter, M.; Levy, Y. Disordered tails of homeodomains facilitate DNA recognition by providing a trade-off between folding and specific binding. J. Am. Chem. Soc. 2009, 131, 15084–15085. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Vascotto, C.; Scognamiglio, P.L.; Lirussi, L.; Marasco, D.; Tell, G. Role of the unstructured N-terminal domain of the hAPE1 (human apurinic/apyrimidinic endonuclease 1) in the modulation of its interaction with nucleic acids and NPM1 (nucleophosmin). Biochem. J. 2013, 452, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Tell, G.; Damante, G.; Caldwell, D.; Kelley, M.R. The intracellular localization of APE1/Ref-1: More than a passive phenomenon. Antioxid. Redox Signal. 2005, 7, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Fantini, D.; Vascotto, C.; Marasco, D.; D’Ambrosio, C.; Romanello, M.; Vitagliano, L.; Pedone, C.; Poletto, M.; Cesaratto, L.; Quadrifoglio, F.; et al. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010, 38, 8239–8256. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.N.; Roviello, G.; Musumeci, D.; Bucci, E.M.; Pedone, C. Dakin-West reaction on 1-thyminyl acetic acid for the synthesis of 1,3-bis(1-thyminyl)-2-propanone, a heteroaromatic compound with nucleopeptide-binding properties. Amino Acids 2012, 43, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.N.; Roviello, G.; Musumeci, D.; Bucci, E.M.; Pedone, C. Evidences for supramolecular organization of nucleopeptides: Synthesis, spectroscopic and biological studies of a novel dithymine l-serine tetrapeptide. Mol. Biosyst. 2011, 7, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Lirussi, L.; Antoniali, G.; Vascotto, C.; D’Ambrosio, C.; Poletto, M.; Romanello, M.; Marasco, D.; Leone, M.; Quadrifoglio, F.; Bhakat, K.K.; et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell 2012, 23, 4079–4096. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Dorjsuren, D.; Scognamiglio, P.L.; Marasco, D.; Vascotto, C.; Jadhav, A.; Malon, D.J.; Wilson, D.M.; Simeonov, A.; Tell, G. Inhibitors of the apurinic/apyrimidinic endonuclease 1 (APE1)/nucleophosmin (NPM1) interaction that display anti-tumor properties. Mol. Carcinog. 2015, in press. [Google Scholar]
- Dunker, A.K.; Uversky, V.N. Drugs for “protein clouds”: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Gruschus, J.M.; Tsao, D.H.; Wang, L.H.; Nirenberg, M.; Ferretti, J.A. Interactions of the vnd/NK-2 homeodomain with DNA by nuclear magnetic resonance spectroscopy: Basis of binding specificity. Biochemistry 1997, 36, 5372–5380. [Google Scholar] [CrossRef] [PubMed]
- Khazanov, N.; Levy, Y. Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol. 2011, 408, 335–355. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Kar, R.K.; Sa, G. Structural and sequential context of p53: A review of experimental and theoretical evidence. Prog. Biophys. Mol. Biol. 2014. [Google Scholar] [CrossRef]
- Zhou, H.X. The affinity-enhancing roles of flexible linkers in two-domain DNA-binding proteins. Biochemistry 2001, 40, 15069–15073. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Batey, S.; Nickson, A.A.; Teichmann, S.A.; Clarke, J. The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 2007, 8, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Kim, J.; Truong, K.; Sherman, M.; Yuan, T.; Ikura, M. Calmodulin target database. J. Struct. Funct. Genomics 2000, 1, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Babu, Y.S.; Bugg, C.E.; Cook, W.J. Structure of calmodulin refined at 2.2 A resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Maestre-Martinez, M.; Wolff, S.; Becker, S.; Griesinger, C. Interdomain dynamics explored by paramagnetic NMR. J. Am. Chem. Soc. 2013, 135, 17111–17120. [Google Scholar] [CrossRef] [PubMed]
- Nagulapalli, M.; Parigi, G.; Yuan, J.; Gsponer, J.; Deraos, G.; Bamm, V.V.; Harauz, G.; Matsoukas, J.; de Planque, M.R.; Gerothanassis, I.P.; et al. Recognition pliability is coupled to structural heterogeneity: A calmodulin intrinsically disordered binding region complex. Structure 2012, 20, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Olson, M.O.; Jones, C.; Busch, H. Nucleolar phosphoproteins of normal rat liver and Novikoff hepatoma ascites cells. Cancer Res. 1975, 35, 1470–1475. [Google Scholar] [PubMed]
- Okuwaki, M.; Matsumoto, K.; Tsujimoto, M.; Nagata, K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001, 506, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Borer, R.A.; Lehner, C.F.; Eppenberger, H.M.; Nigg, E.A. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989, 56, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Okuwaki, M.; Iwamatsu, A.; Tsujimoto, M.; Nagata, K. Identification of nucleophosmin/B23, an acidic nucleolar protein, as a stimulatory factor for in vitro replication of adenovirus DNA complexed with viral basic core proteins. J. Mol. Biol. 2001, 311, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S. NPM1/B23: A Multifunctional chaperone in ribosome biogenesis and chromatin remodeling. Biochem. Res. Int. 2011. [Google Scholar] [CrossRef]
- Balusu, R.; Fiskus, W.; Rao, R.; Chong, D.G.; Nalluri, S.; Mudunuru, U.; Ma, H.; Chen, L.; Venkannagari, S.; Ha, K.; et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood 2011, 118, 3096–3106. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Bolli, N.; Martelli, M.P.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Martelli, M.F. Translocations and mutations involving the nucleophosmin (NPM1) gene in lymphomas and leukemias. Haematologica 2007, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Namboodiri, V.M.; Akey, I.V.; Schmidt-Zachmann, M.S.; Head, J.F.; Akey, C.W. The structure and function of Xenopus NO38-core, a histone chaperone in the nucleolus. Structure 2004, 12, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Kim, H.S.; Kang, J.Y.; Lee, B.I.; Ha, J.Y.; Yoon, H.J.; Lim, S.O.; Jung, G.; Suh, S.W. Crystal structure of human nucleophosmin-core reveals plasticity of the pentamer-pentamer interface. Proteins 2007, 69, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, M.; Nagata, K.; Okuwaki, M. Intrinsically disordered regions of nucleophosmin/B23 regulate its RNA binding activity through their inter- and intra-molecular association. Nucleic Acids Res. 2014, 42, 1180–1195. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; di Natale, C.; Leone, M.; Poletto, M.; Vitagliano, L.; Tell, G.; Marasco, D. G-quadruplex DNA recognition by nucleophosmin: new insights from protein dissection. Biochim. Biophys. Acta 2014, 1840, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Grummitt, C.G.; Townsley, F.M.; Johnson, C.M.; Warren, A.J.; Bycroft, M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 2008, 283, 23326–23332. [Google Scholar] [CrossRef] [PubMed]
- Marasco, D.; Ruggiero, A.; Vascotto, C.; Poletto, M.; Scognamiglio, P.L.; Tell, G.; Vitagliano, L. Role of mutual interactions in the chemical and thermal stability of nucleophosmin NPM1 domains. Biochem. Biophys. Res. Commun. 2013, 430, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, S.; Federici, L.; di Matteo, A.; Brunori, M.; Gianni, S. The folding pathway of a functionally competent C-terminal domain of nucleophosmin: Protein stability and denatured state residual structure. Biochem. Biophys. Res. Commun. 2013, 435, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Scaloni, F.; Federici, L.; Brunori, M.; Gianni, S. Deciphering the folding transition state structure and denatured state properties of nucleophosmin C-terminal domain. Proc. Natl. Acad. Sci. USA 2010, 107, 5447–5452. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Lo Sterzo, C.; Mori, M.; Di Matteo, A.; Bertini, I.; Banci, L.; Brunori, M.; Federici, L. Structure of nucleophosmin DNA-binding domain and analysis of its complex with a G-quadruplex sequence from the c-MYC promoter. J. Biol. Chem. 2012, 287, 26539–26548. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Arcovito, A.; Scaglione, G.L.; Scaloni, F.; Lo Sterzo, C.; di Matteo, A.; Falini, B.; Giardina, B.; Brunori, M. Nucleophosmin C-terminal leukemia-associated domain interacts with G-rich quadruplex forming DNA. J. Biol. Chem. 2010, 285, 37138–37149. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, S.; de Cola, A.; Scaglione, G.L.; Carletti, E.; Graziano, V.; Barcaroli, D.; Lo Sterzo, C.; di Matteo, A.; di Ilio, C.; Falini, B.; et al. Nucleophosmin mutations alter its nucleolar localization by impairing G-quadruplex binding at ribosomal DNA. Nucleic Acids Res. 2013, 41, 3228–3239. [Google Scholar] [CrossRef] [PubMed]
- Arcovito, A.; Chiarella, S.; della Longa, S.; di Matteo, A.; Lo Sterzo, C.; Scaglione, G.L.; Federici, L. Synergic role of nucleophosmin three-helix bundle and a flanking unstructured tail in the interaction with G-quadruplex DNA. J. Biol. Chem. 2014, 289, 21230–21241. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Falini, B. Nucleophosmin mutations in acute myeloid leukemia: A tale of protein unfolding and mislocalization. Protein Sci. 2013, 22, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Tou, W.I. How to design a drug for the disordered proteins. Drug Discov. Today 2013, 18, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Dey, S.; Bhattacharyya, N.P.; Mukhopadhyay, D. The role of intrinsically unstructured proteins in neurodegenerative diseases. PLoS ONE 2009, 4, e5566. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Uceda, D.; Santoro, J. Direct correlation of consecutive C'–N groups in proteins: A method for the assignment of intrinsically disordered proteins. J. Biomol. NMR 2013, 57, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; de Simone, A.; Schenk, D.; Toth, G.; Dobson, C.M.; Vendruscolo, M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013, 139, 035101. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed]
- Akoury, E.; Gajda, M.; Pickhardt, M.; Biernat, J.; Soraya, P.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Inhibition of tau filament formation by conformational modulation. J. Am. Chem. Soc. 2013, 135, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.A.; Platt, G.W.; Hellewell, A.L.; Hewitt, E.W.; Homans, S.W.; Ashcroft, A.E.; Radford, S.E. Ligand binding to distinct states diverts aggregation of an amyloid-forming protein. Nat. Chem. Biol. 2011, 7, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Convertino, M.; Magno, A.; Caflisch, A.; Corazza, A.; Haridas, H.; Esposito, G.; Cataldo, S.; Pignataro, B.; Milardi, D.; et al. Carnosine inhibits Aβ42 aggregation by perturbing the H-bond network in and around the central hydrophobic cluster. Chembiochem 2013, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Bienkiewicz, E.A.; Adkins, J.N.; Lumb, K.J. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle Inhibitor p27Kip1. Biochemistry 2002, 41, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Bharatham, N.; Bharatham, K.; Shelat, A.A.; Bashford, D. Ligand binding mode prediction by docking: MDM2/MDMX inhibitors as a case study. J. Chem. Inf. Model. 2014, 54, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX loop for cancer therapy. Sub-Cell. Biochem. 2014, 85, 281–319. [Google Scholar]
- Unal, A.; Pray, T.R.; Lagunoff, M.; Pennington, M.W.; Ganem, D.; Craik, C.S. The protease and the assembly protein of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8). J. Virol. 1997, 71, 7030–7038. [Google Scholar] [PubMed]
- Shahian, T.; Lee, G.M.; Lazic, A.; Arnold, L.A.; Velusamy, P.; Roels, C.M.; Guy, R.K.; Craik, C.S. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat. Chem. Biol. 2009, 5, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Bower, K.E.; Beuscher, A.E., 4th; Zhou, B.; Bobkov, A.A.; Olson, A.J.; Vogt, P.K. Stabilizers of the Max homodimer identified in virtual ligand screening inhibit Myc function. Mol. Pharmacol. 2009, 76, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Cohen, S.B.; Desharnais, J.; Sonderegger, C.; Maslyar, D.J.; Goldberg, J.; Boger, D.L.; Vogt, P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 3830–3835. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, J.; Yamamoto, N.; Moss, J.A.; Vogt, P.K.; Janda, K.D. A credit-card library approach for disrupting protein–protein interactions. Bioorg. Med. Chem. 2006, 14, 2660–2673. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.; Prochownik, E.V.; Metallo, S.J. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, Y.; Morimoto, K.; Yamamoto, T.; Chaki, H.; Hashiramoto, A.; Narita, H.; Hirono, S.; Shiozawa, S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cesena, T.I.; Ohnishi, Y.; Burger-Caplan, R.; Lam, V.; Kirchhoff, P.D.; Larsen, S.D.; Larsen, M.J.; Nestler, E.J.; Rudenko, G. Small molecule screening identifies regulators of the transcription factor ΔFosB. ACS Chem. Neurosci. 2012, 3, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Sadar, M.D.; Williams, D.E.; Mawji, N.R.; Patrick, B.O.; Wikanta, T.; Chasanah, E.; Irianto, H.E.; Soest, R.V.; Andersen, R.J. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org. Lett. 2008, 10, 4947–4950. [Google Scholar] [CrossRef] [PubMed]
- Choy, W.Y.; Forman-Kay, J.D. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J. Mol. Biol. 2001, 308, 1011–1032. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Tompa, P.; Simon, I.; Dosztanyi, Z. Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sainis, I.; Fokas, D.; Vareli, K.; Tzakos, A.G.; Kounnis, V.; Briasoulis, E. Cyanobacterial cyclopeptides as lead compounds to novel targeted cancer drugs. Mar. Drugs 2010, 8, 629–657. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marasco, D.; Scognamiglio, P.L. Identification of Inhibitors of Biological Interactions Involving Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2015, 16, 7394-7412. https://doi.org/10.3390/ijms16047394
Marasco D, Scognamiglio PL. Identification of Inhibitors of Biological Interactions Involving Intrinsically Disordered Proteins. International Journal of Molecular Sciences. 2015; 16(4):7394-7412. https://doi.org/10.3390/ijms16047394
Chicago/Turabian StyleMarasco, Daniela, and Pasqualina Liana Scognamiglio. 2015. "Identification of Inhibitors of Biological Interactions Involving Intrinsically Disordered Proteins" International Journal of Molecular Sciences 16, no. 4: 7394-7412. https://doi.org/10.3390/ijms16047394
APA StyleMarasco, D., & Scognamiglio, P. L. (2015). Identification of Inhibitors of Biological Interactions Involving Intrinsically Disordered Proteins. International Journal of Molecular Sciences, 16(4), 7394-7412. https://doi.org/10.3390/ijms16047394