
Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
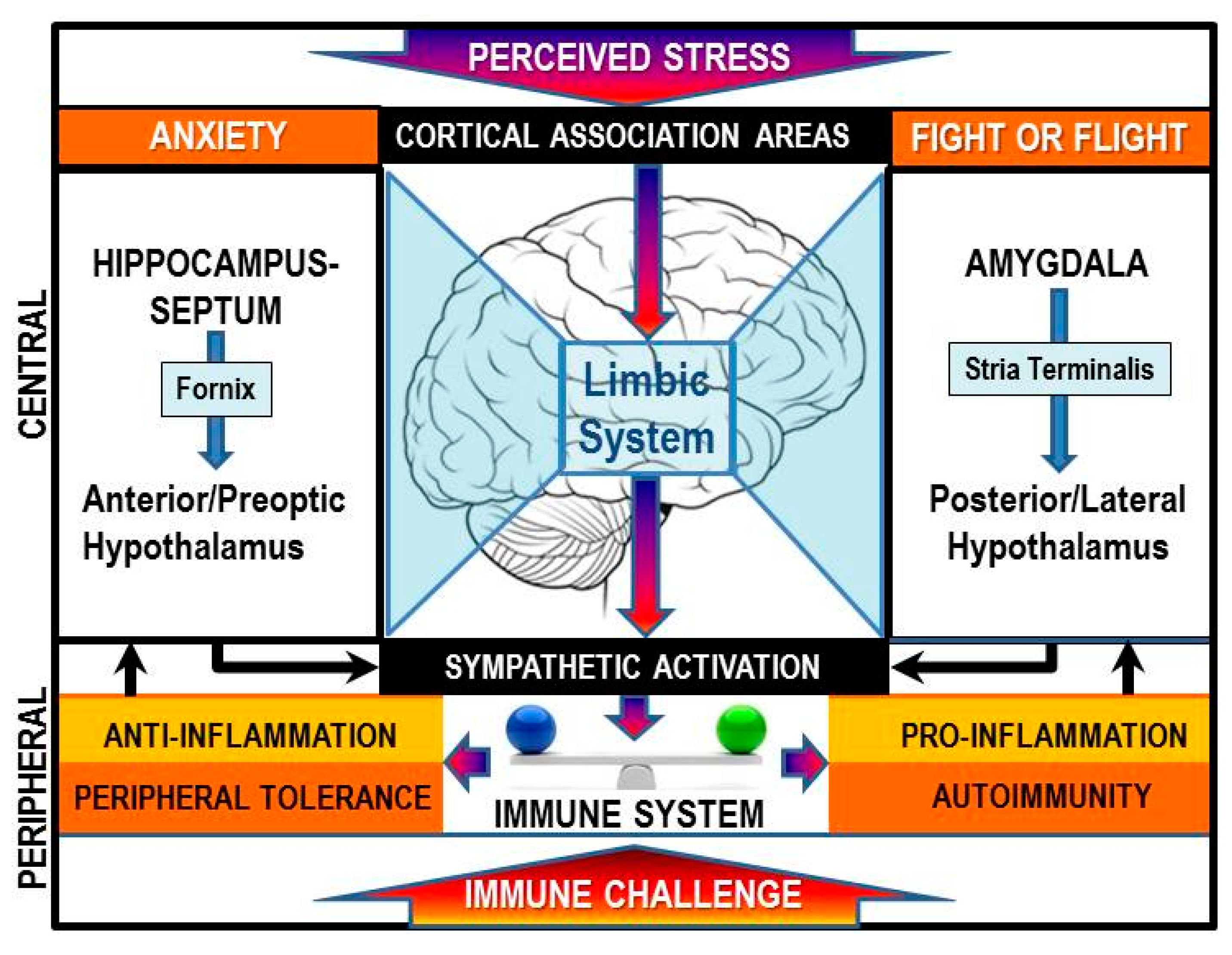
:1. Introduction
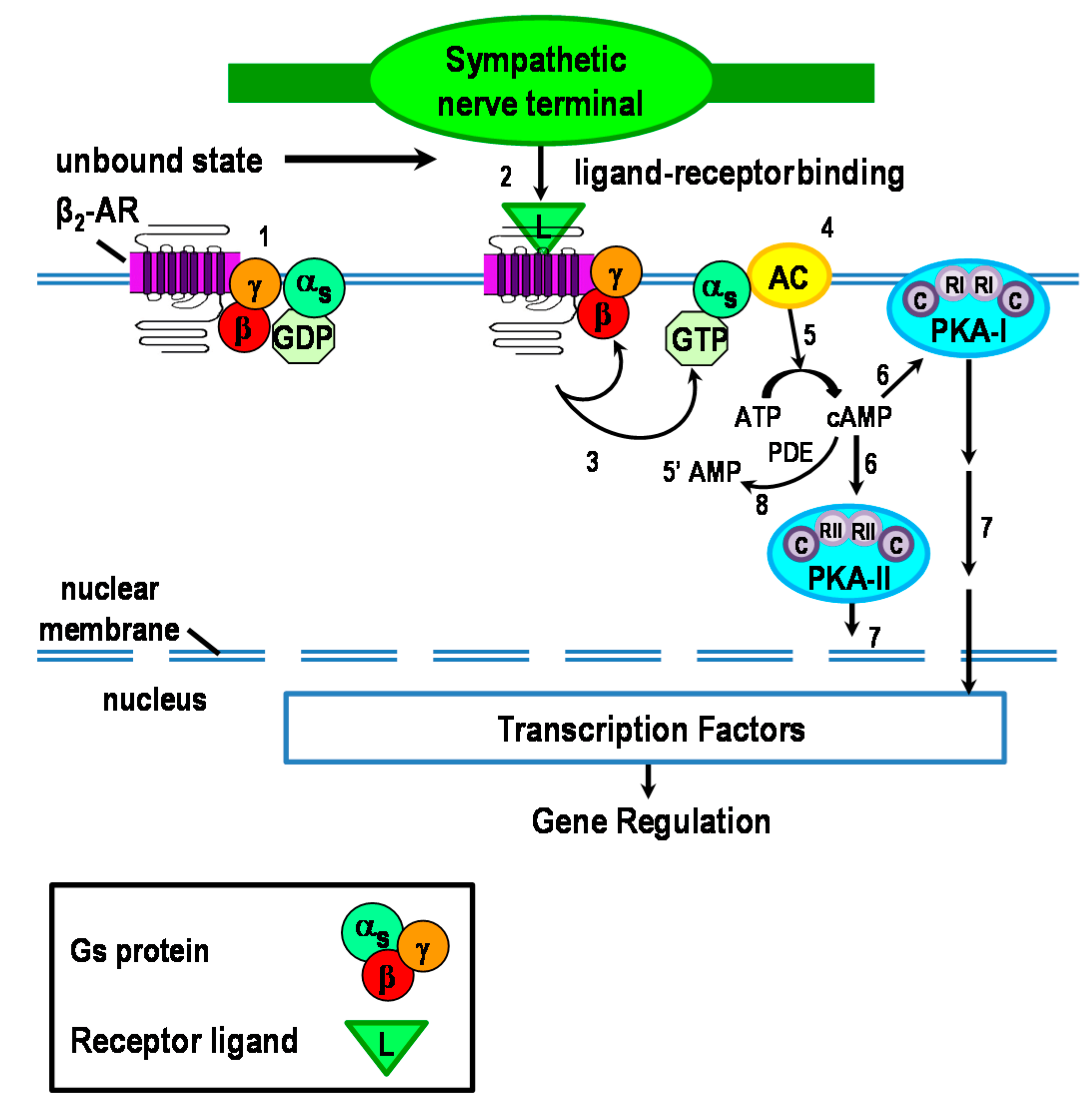
2. Canonical Intracellular Signaling by β2-Adrenergic Receptors (ARs)
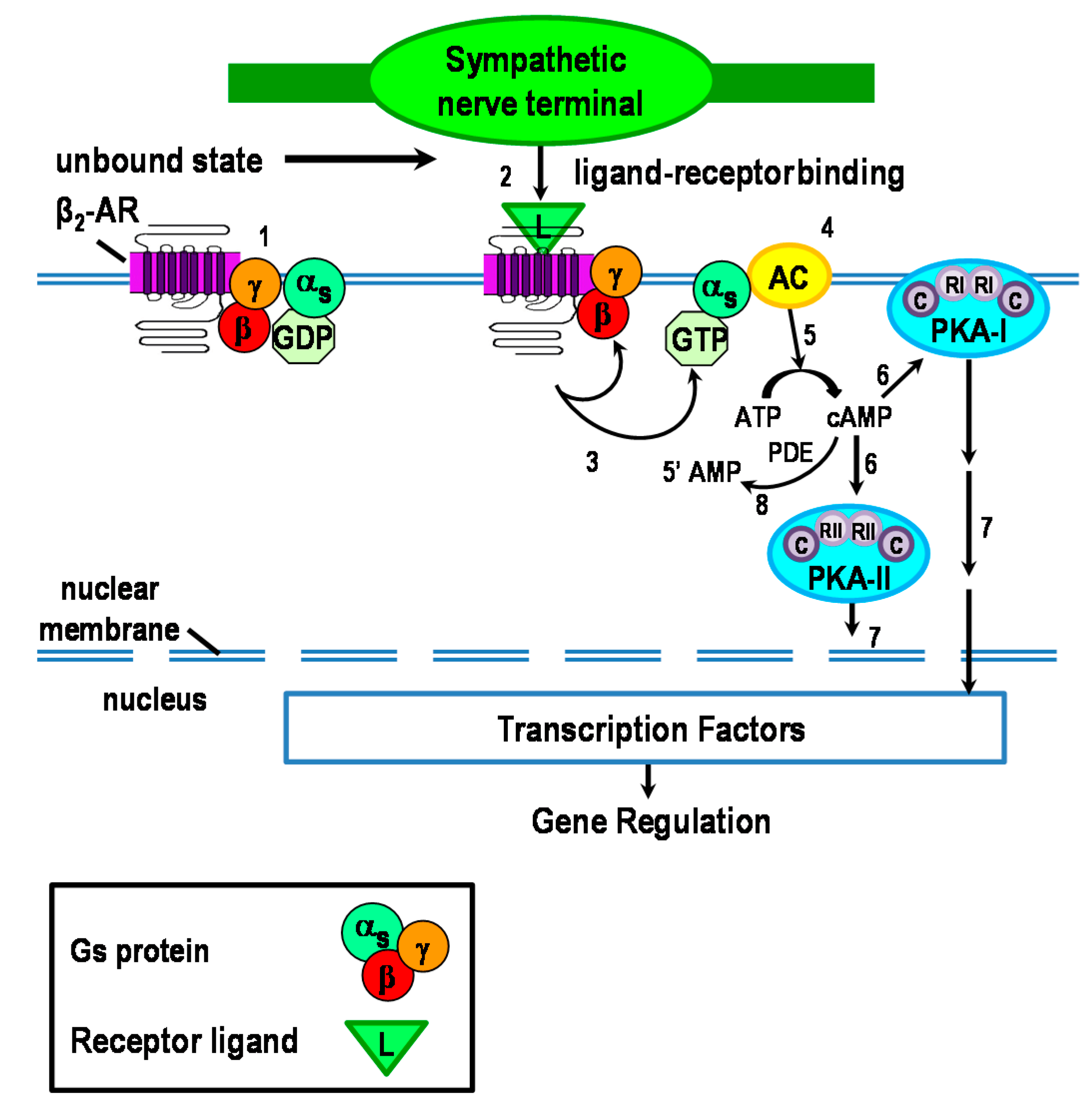
2.1. cAMP: The Second Messenger in the β2-AR Signaling Pathway
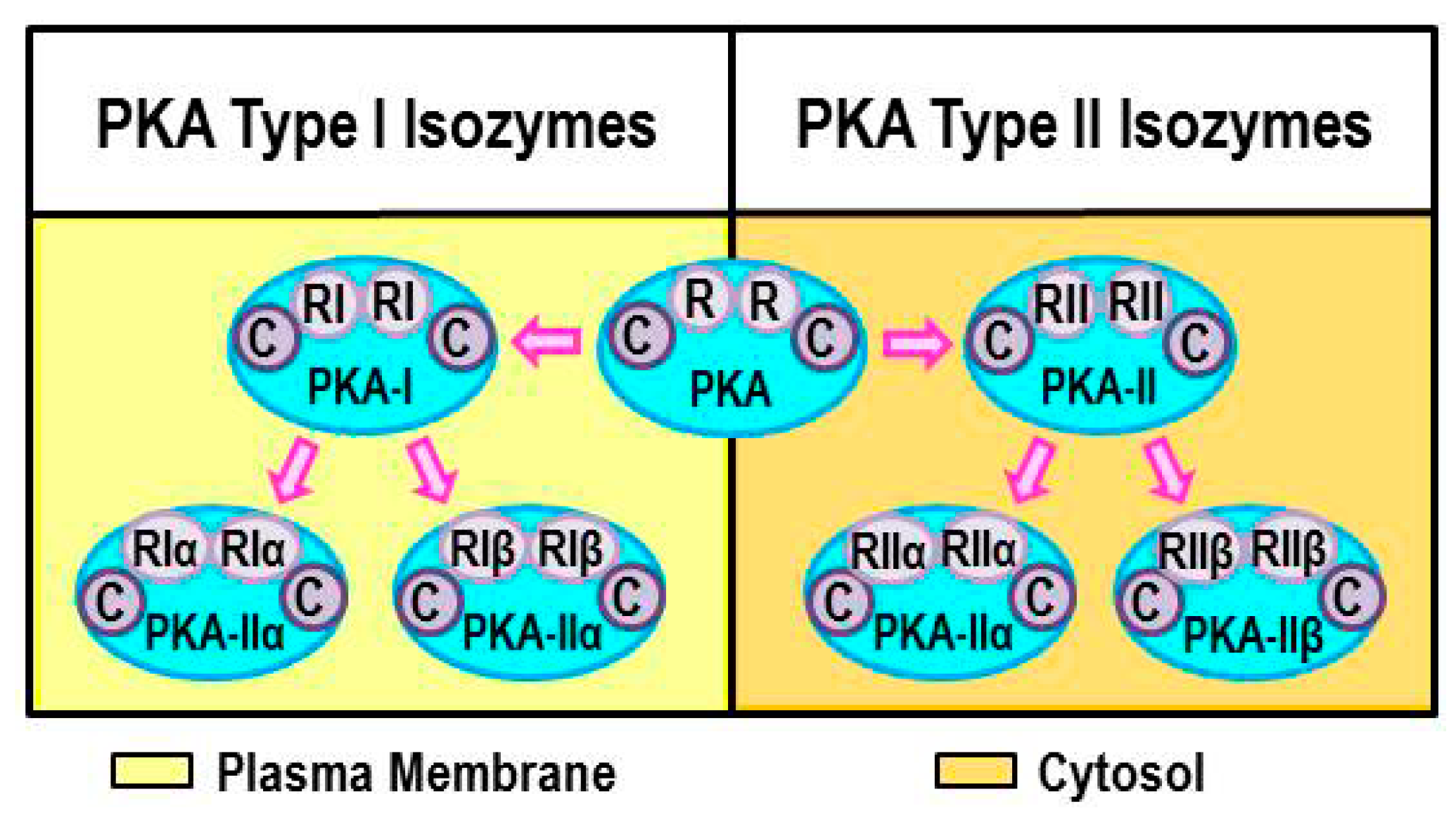
2.2. Intracellular Protein Kinase A (PKA) Localization Determines Specificity of Response
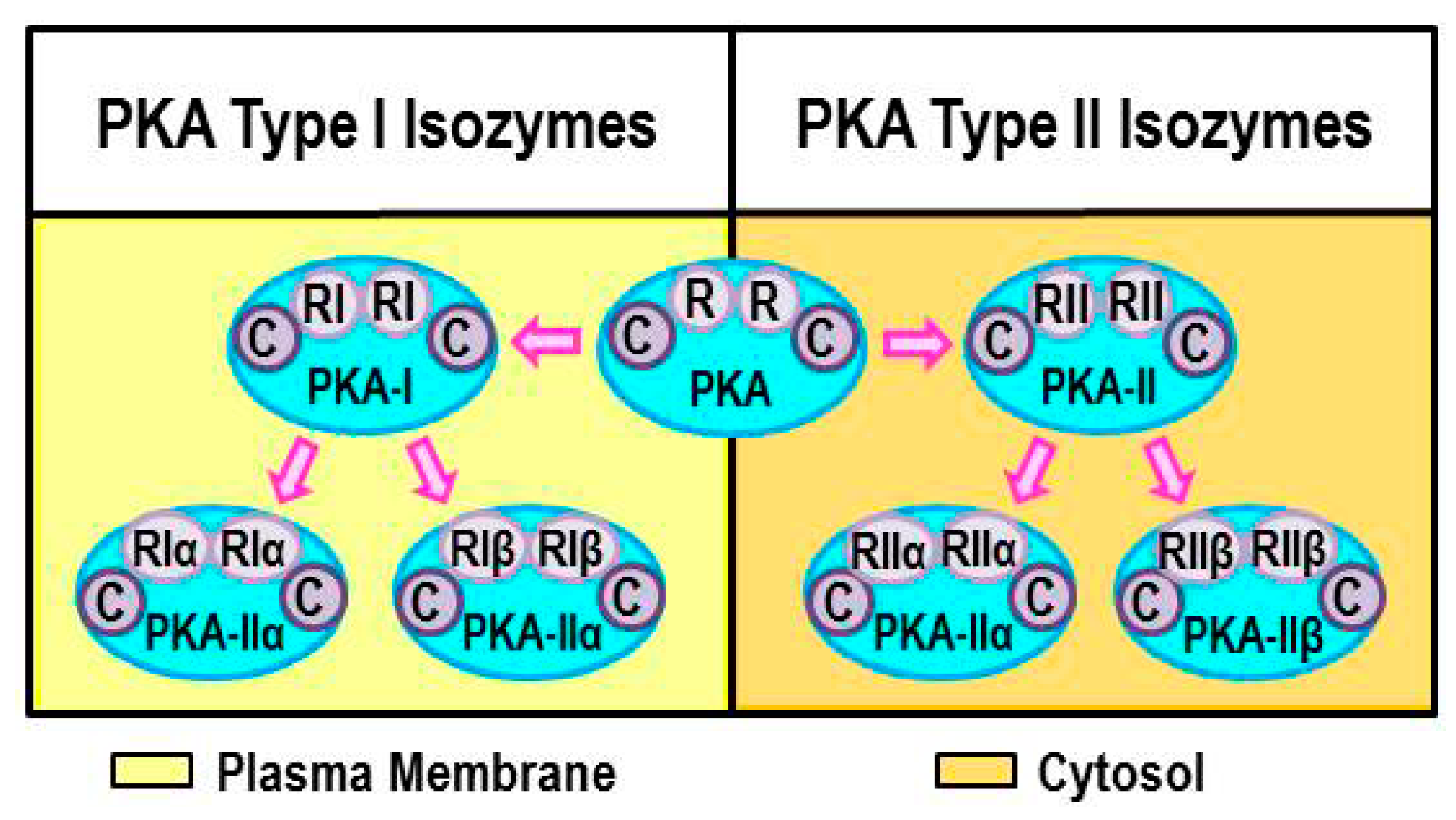
2.3. Regulation of Canonical β2-AR Signaling
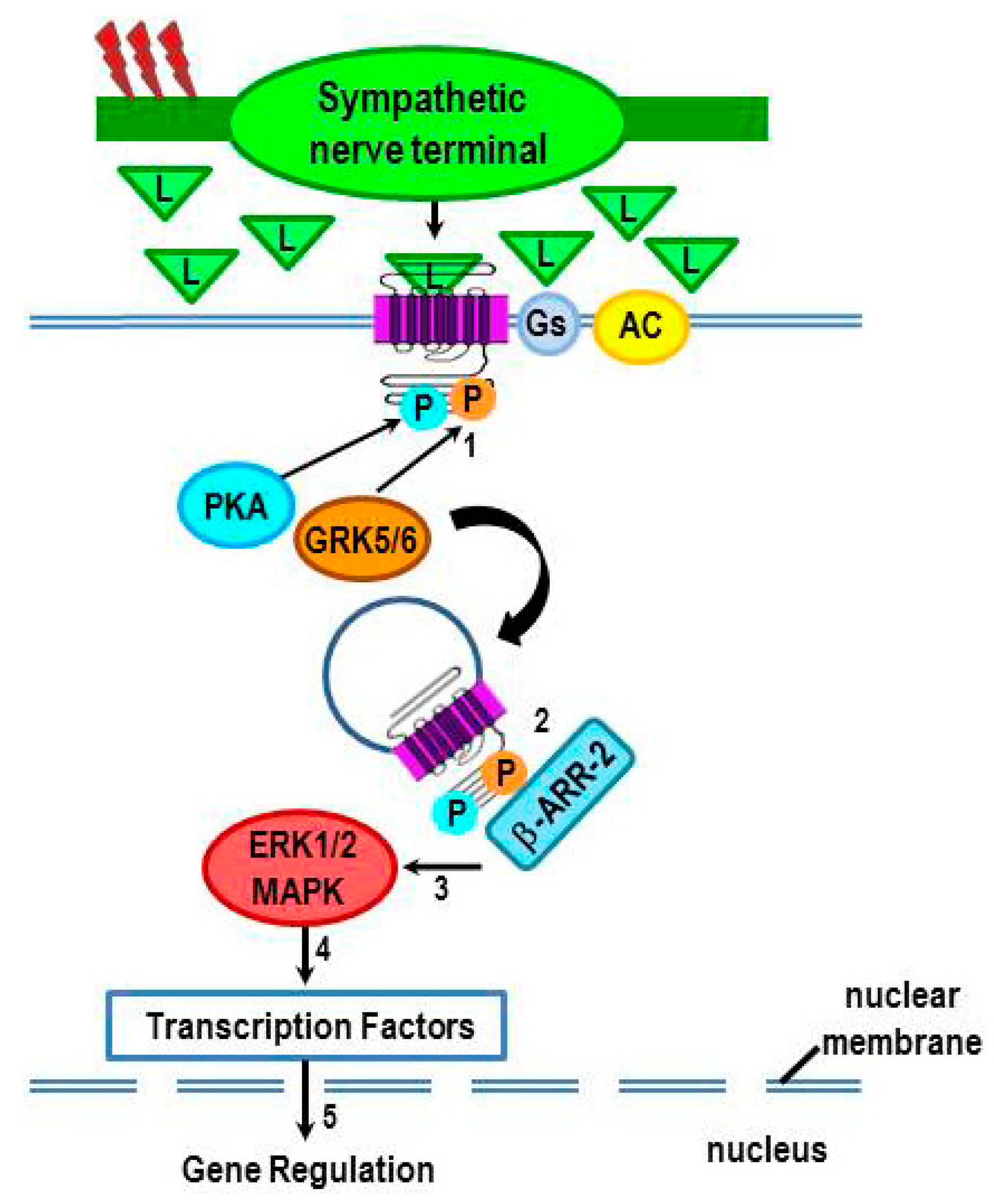
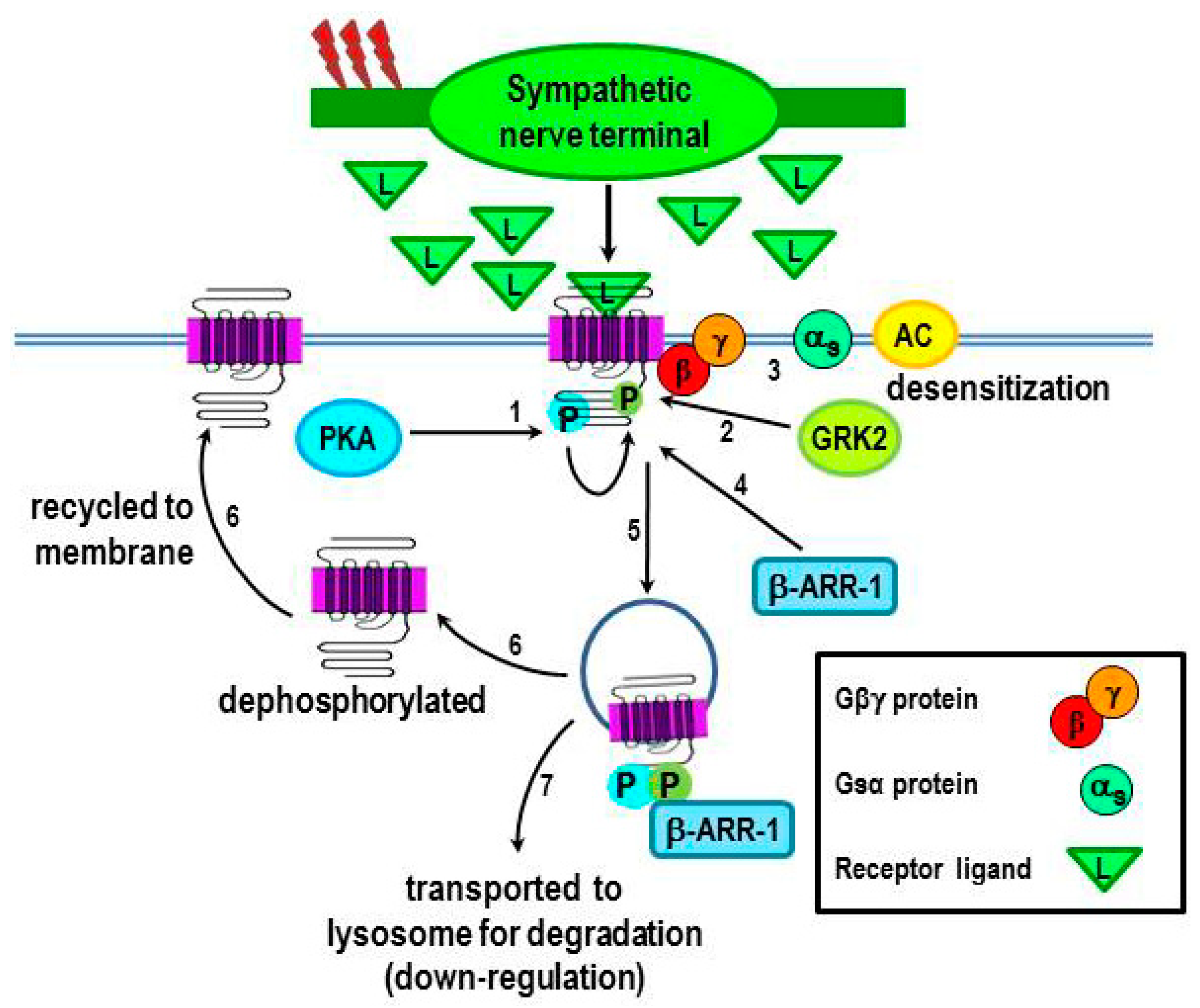
2.3.1. β2-AR Desensitization
2.3.2. β2-AR internalization and Functional Consequences
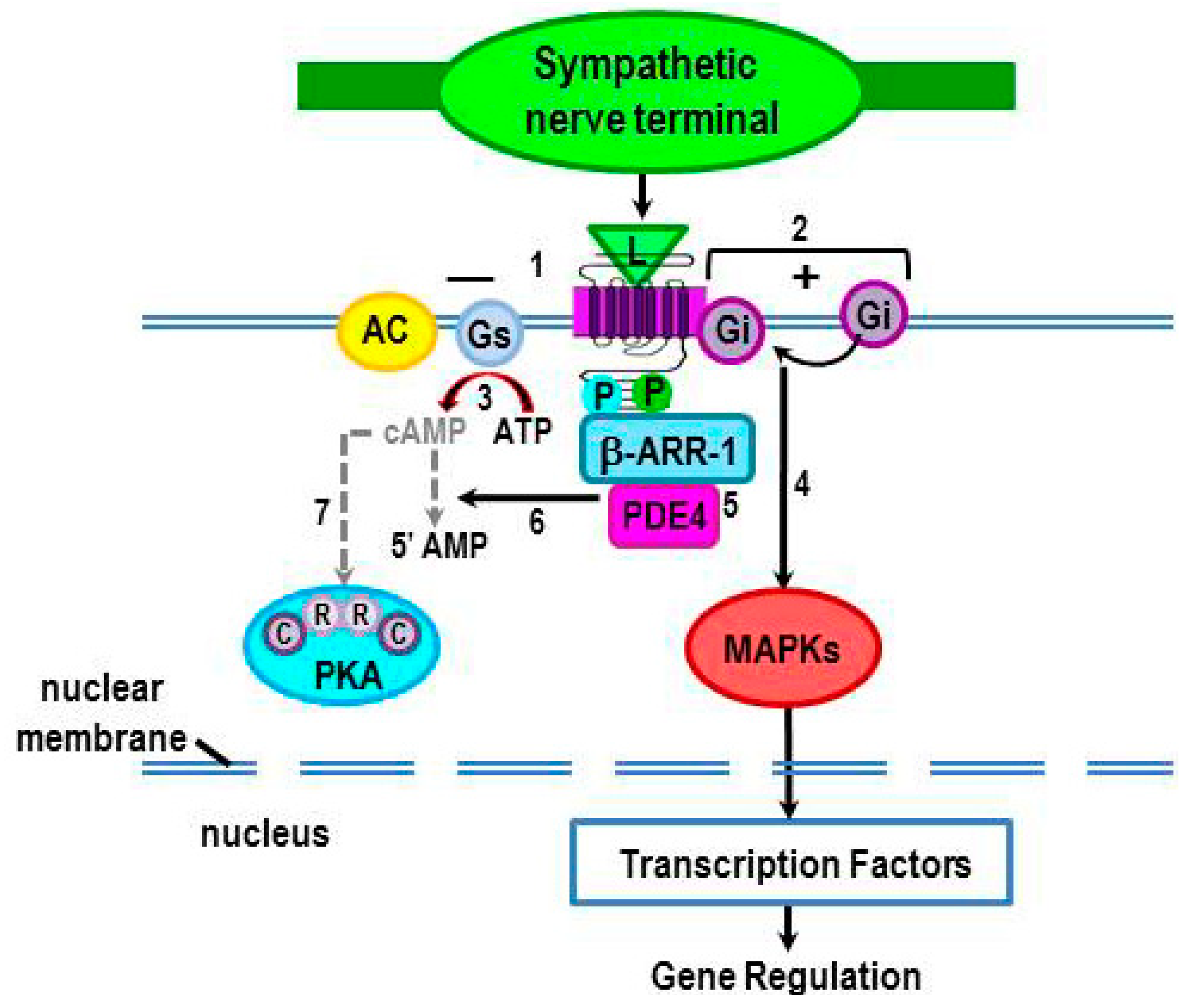
2.3.3. PKA Phosphorylation Induces a β2-AR Switch in Coupling from Gs to Gi Protein
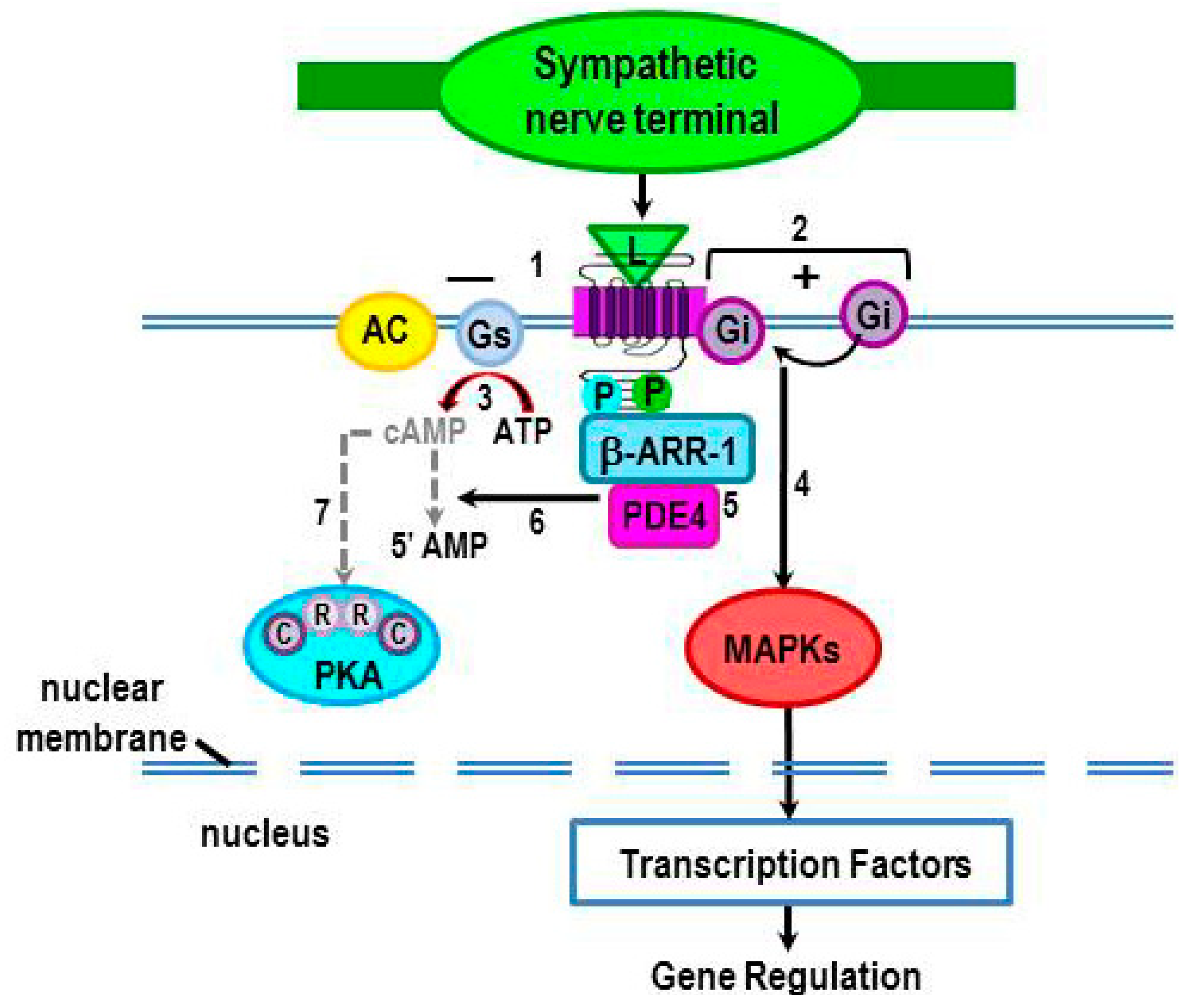
2.3.4. Terminating cAMP Signal Transduction
3. Non-Canonical Intracellular Signaling by β2-ARs: G Protein-Independent Signaling
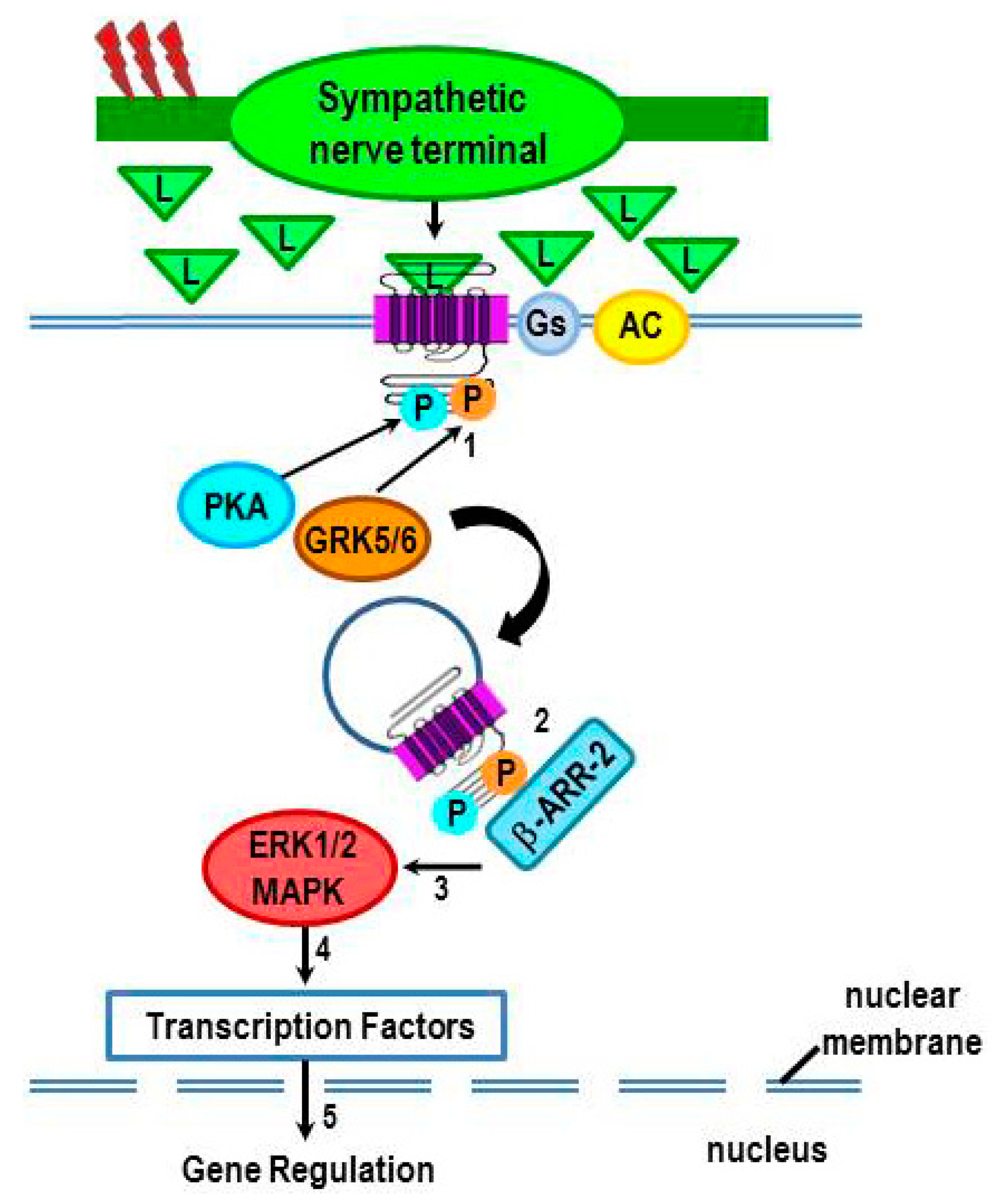
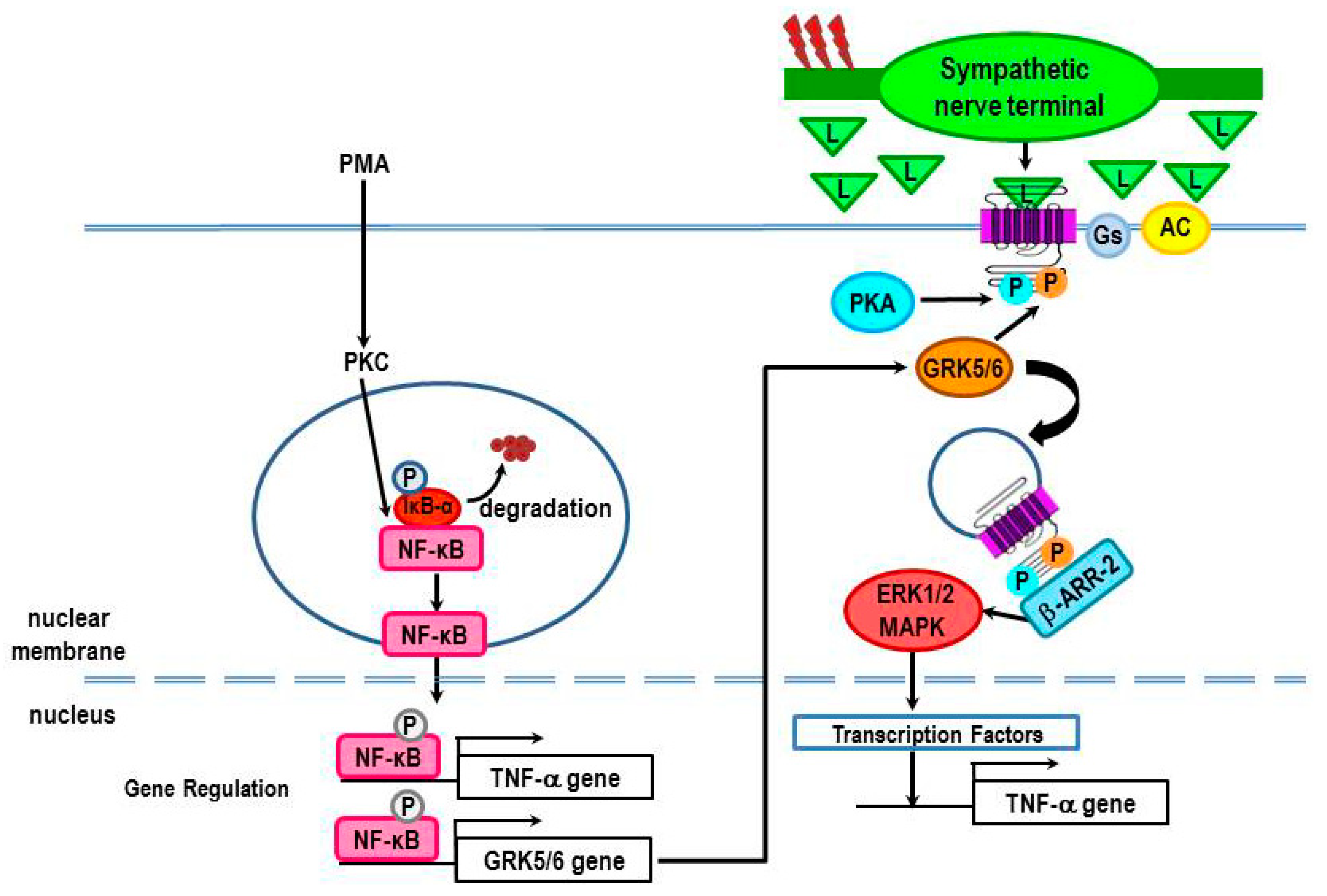
4. Immune System–SNS “Cross-Talk”
4.1. Cross-Talk and the Canonical Pathway: Traditional Viewpoint
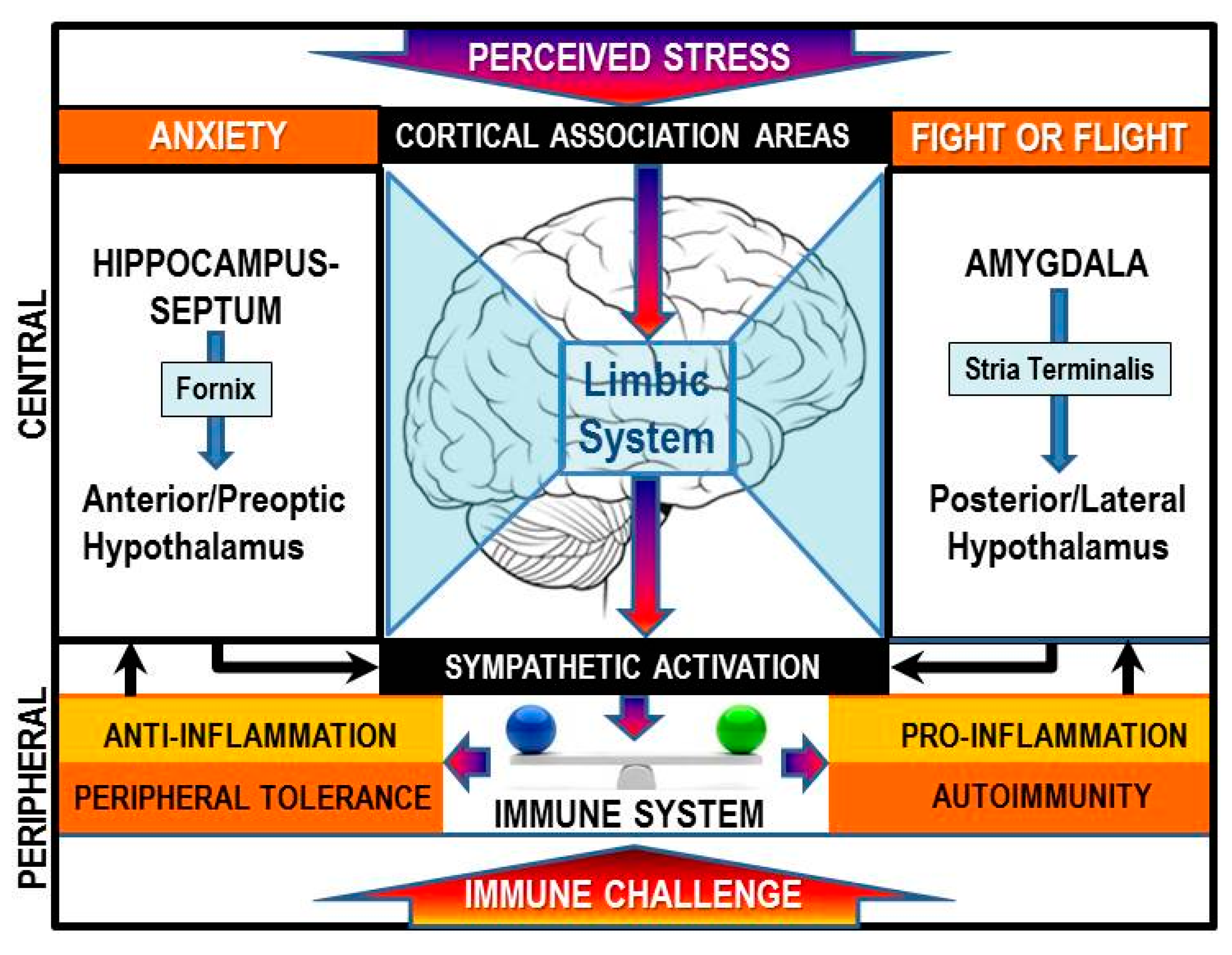
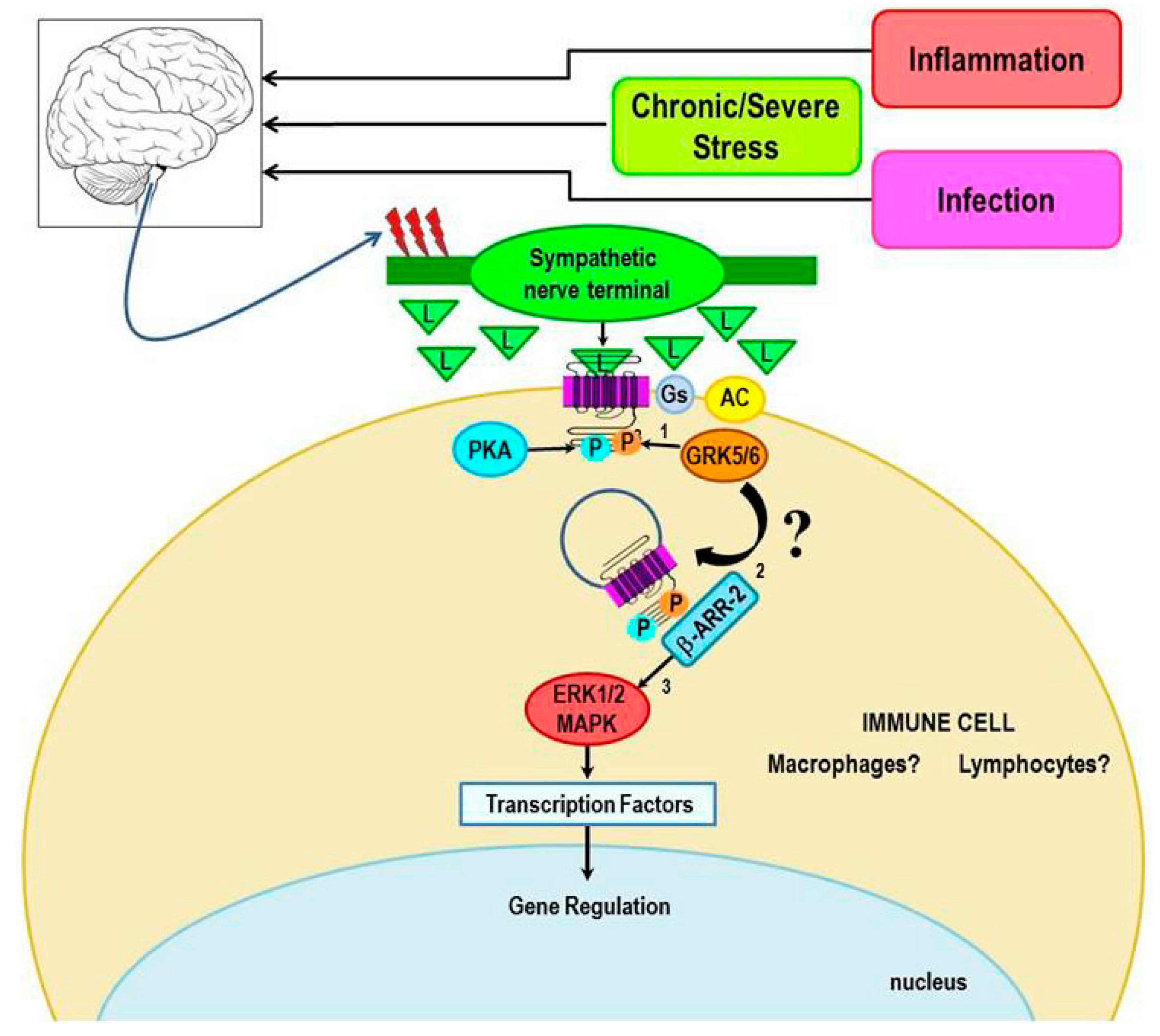
4.2. Evidence for Non-Canonical Signaling with Context-Dependent Inflammatory Responses
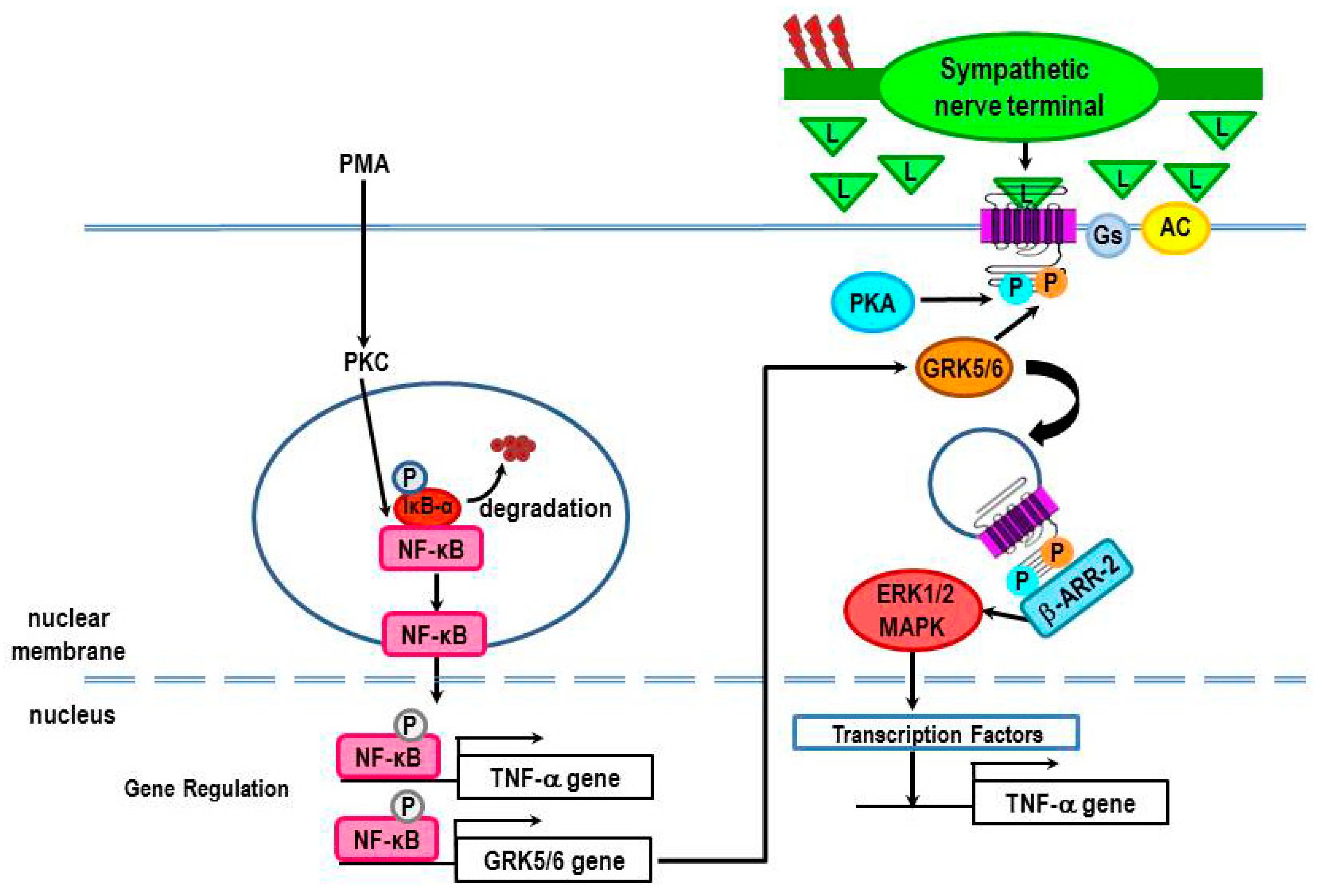
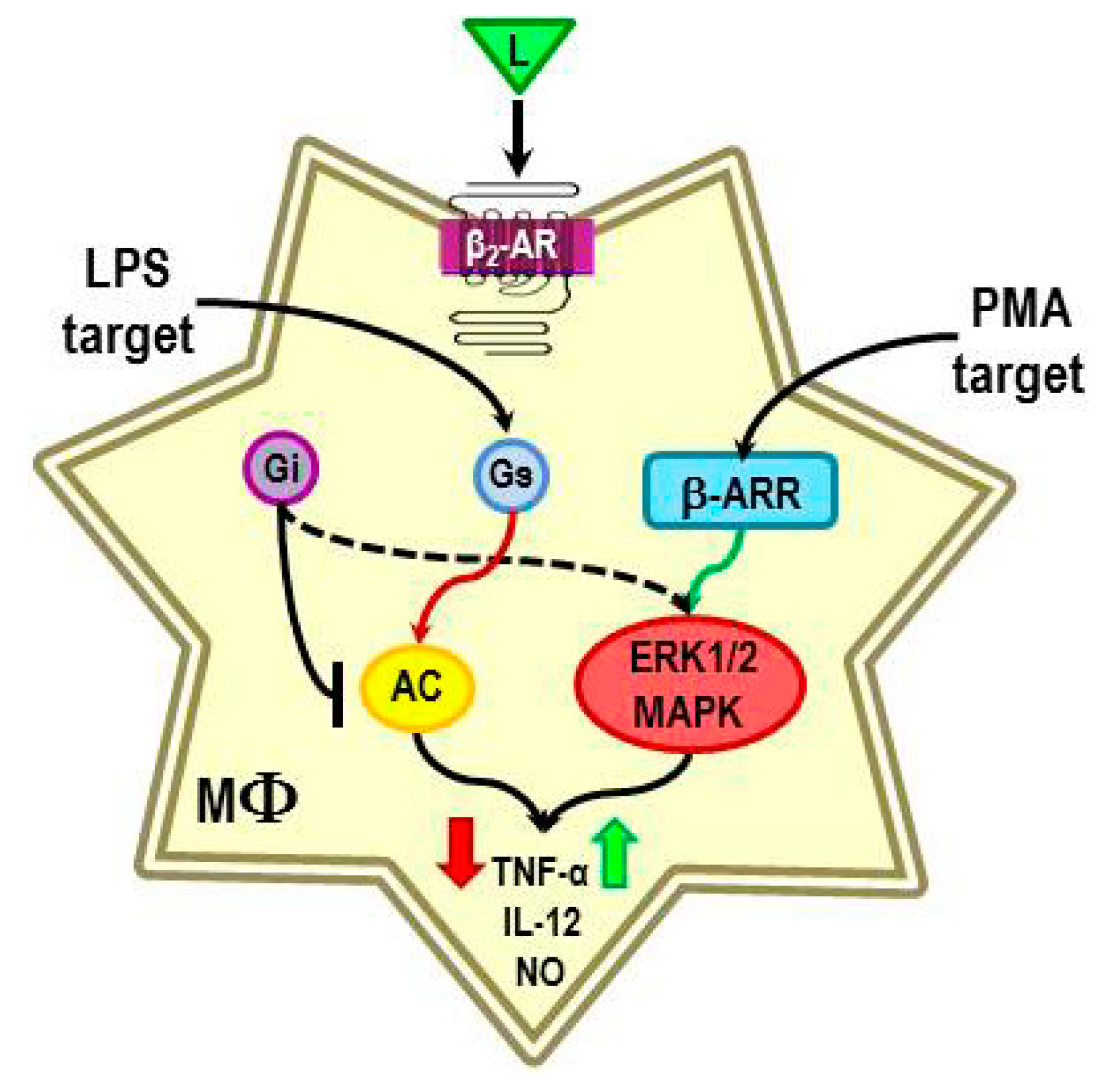
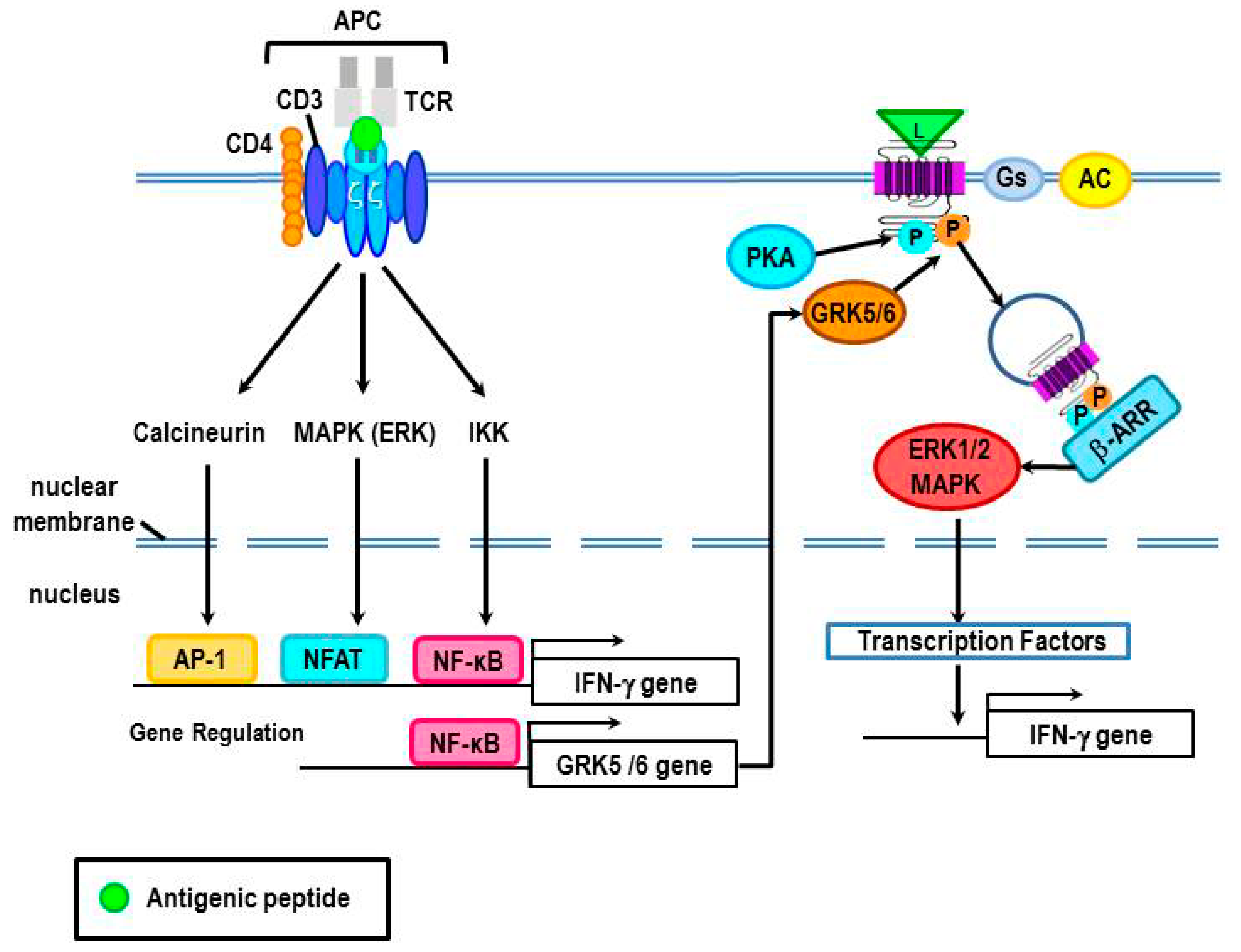
4.3. Evidence for β2-AR Non-Canonical Signaling in Adaptive Immune Responses
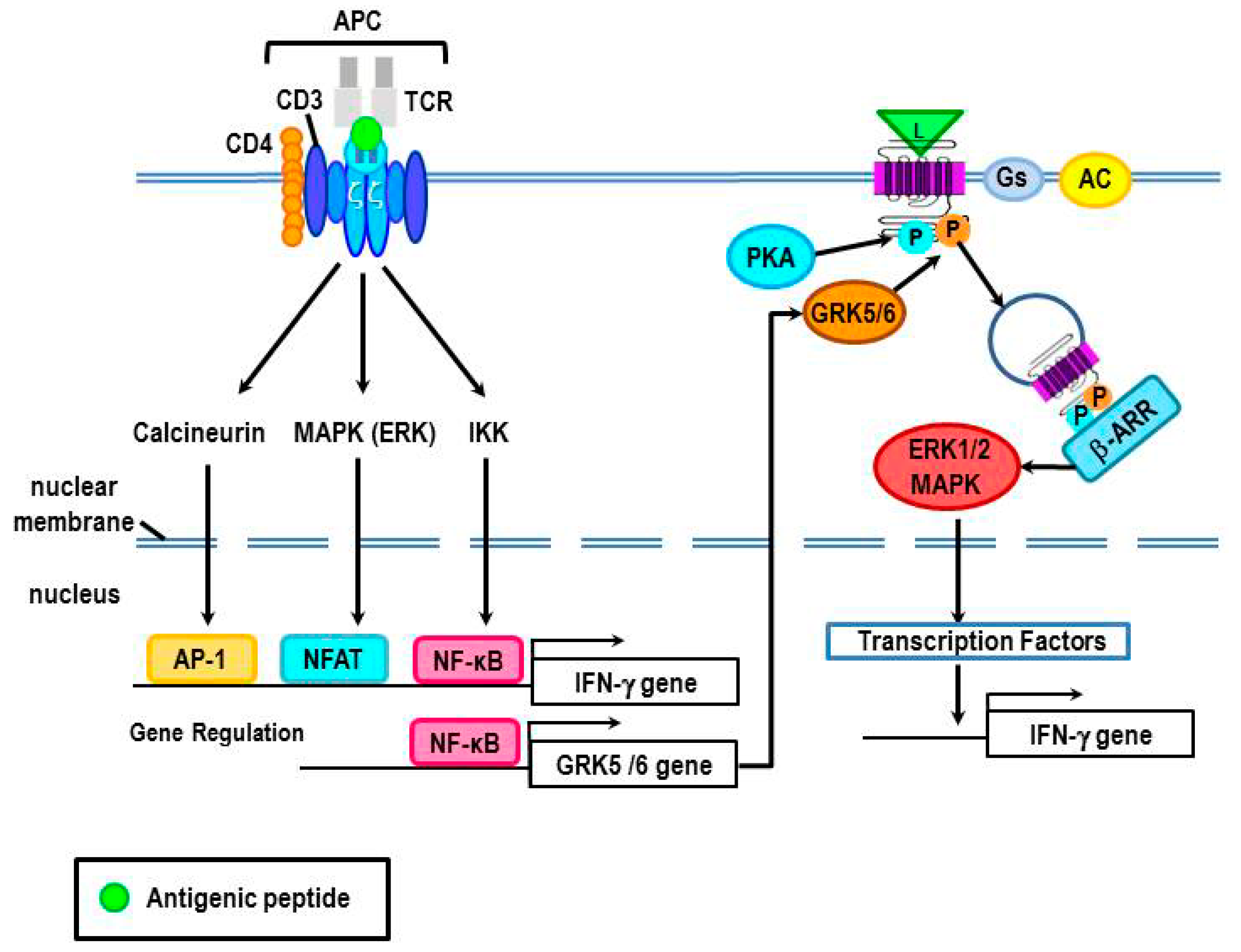
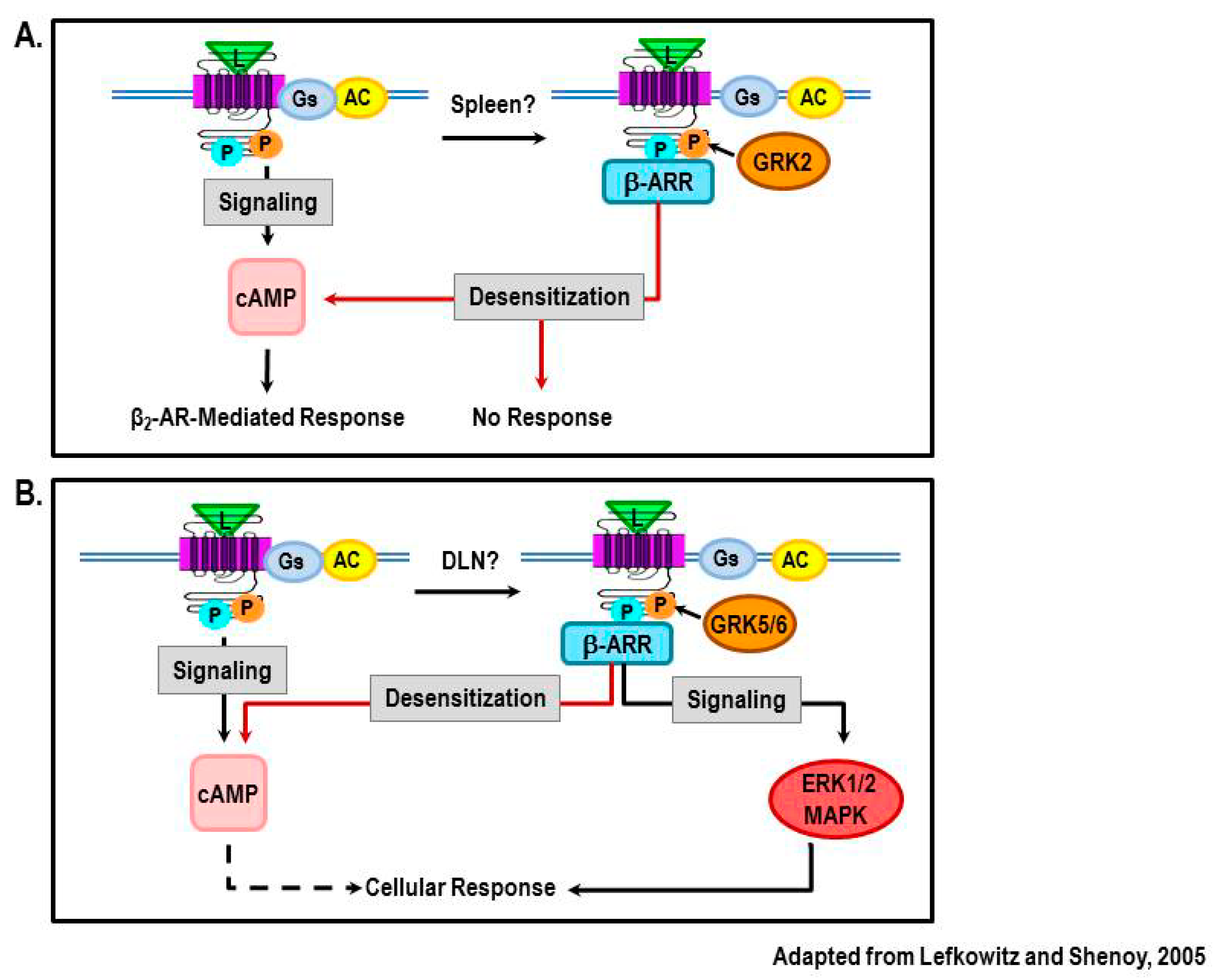
Acknowledgments
Author Contributions
Abbreviations
| 5'-AMP | 5'-monophosphate |
| β-ARR-2 | β-arrestin 2 |
| pβ2-ARPKA | β2-AR phosphorylation by PKA |
| ATF-2 | activating transcription factor 2 |
| AC | activator protein 1 (AP-1, adenylate cyclase |
| ARs | adrenergic receptors |
| AKAPs | A-kinase anchor proteins |
| APC | antigen-presenting cell |
| C | catalytic subunit |
| CREB | cAMP response element-binding |
| CNS | central nervous system |
| JNK | c-Jun N-terminal kinase |
| DLNs | draining lymph nodes (of arthritic joints) |
| Epac | exchange protein directly activated by cAMP |
| EAE | experimental autoimmune encephalomyelitis |
| ERK | extracellular signal-regulated protein kinase |
| Gαs | G protein alpha s subunit |
| Gβγ | G protein beta/gamma subunit |
| GRKs | G protein-coupled kinases |
| GDP | guanosine diphosphate |
| GTP | guanosine triphosphate |
| GPCRs | G protein-coupled receptors |
| HEK | human embryonic kidney |
| IκBα | inhibitor of kappa B |
| IFN | interferon |
| IL | interleukin |
| cAMPi | intracellular cAMP |
| ISO | isoproterenol |
| L | ligand |
| LPS | lipopolysaccharide |
| MLNs | mesenteric lymph nodes |
| MAPK | mitogen-activated protein kinase |
| MS | multiple sclerosis |
| NE | norepinephrine |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | nuclear factor of activated T cells |
| PBMCs | peripheral blood mononuclear cells |
| PMA | phorbol 12-myristate 13-acetate |
| PDE | phosphodiesterases |
| PKA | protein kinase A |
| PKC | protein kinase C |
| TNF-α | tumor necrosis factor-α |
| R | regulatory subunit dimer |
| RA | rheumatoid arthritis |
| SNS | sympathetic nervous system |
| TCRs | T cell receptors |
| Th | T helper |
Conflicts of Interest
References
- Bellinger, D.L; Lorton, D. Autonomic regulation of cellular immune function. Auton. Neurosci. 2014, 182, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav. Immun. 2007, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Sivertsen, B.; Holliday, N.; Madsen, A.N.; Holst, B. Functionally biased signaling properties of 7TM receptors—Opportunities for drug development for the ghrelin receptor. Br. J. Pharmacol. 2013, 170, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G.; Levy, F.O. Novel aspects of G-protein-coupled receptor signaling—Different ways to achieve specificity. Acta Physiol. (Oxf.) 2007, 190, 33–38. [Google Scholar] [CrossRef]
- Dessauer, C.W. Adenylyl cyclase–A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol. Pharmacol. 2009, 76, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.S. Lipid rafts: Now you see them, now you don’t. Nat. Immunol. 2006, 7, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta. Physiol. (Oxf.) 2007, 190, 9–19. [Google Scholar] [CrossRef]
- Vandamme, J.; Castermans, D.; Thevelein, J.M. Molecular mechanisms of feedback inhibition of protein kinase A on intracellular cAMP accumulation. Cell Signal. 2012, 24, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Sanders, V.M. Norepinephrine and β2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol. Rev. 2001, 53, 487–525. [Google Scholar] [PubMed]
- Baillie, G.S.; Houslay, M.D. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr. Opin. Cell Biol. 2005, 17, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Shirshev, S.V. Role of Epac proteins in mechanisms of cAMP-dependent immunoregulation. Biochemistry (Mosc.) 2011, 76, 981–998. [Google Scholar] [CrossRef]
- Duan, B.; Davis, R.; Sadat, E.L.; Collins, J.; Sternweis, P.C.; Yuan, D.; Jiang, L.I. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J. Immunol. 2010, 185, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.E.; Gu, C.; Fagan, K.A.; Hu, B.; Cooper, D.M. Residence of adenylyl cyclase type 8 in caveolae is necessary but not sufficient for regulation by capacitative Ca2+ entry. J. Biol. Chem. 2002, 277, 6025–6031. [Google Scholar] [CrossRef] [PubMed]
- Pontier, S.M.; Percherancier, Y.; Galandrin, S.; Breit, A.; Galés, C.; Bouvier, M. Cholesterol-dependent separation of the β2-adrenergic receptor from its partners determines signaling efficacy: Insight into nanoscale organization of signal transduction. J. Biol. Chem. 2008, 283, 24659–24672. [Google Scholar] [CrossRef]
- Hertz, A.L.; Beavo, J.A. Cyclic nucleotides and phosphodiesterases in monocytic differentiation. Handb. Exp. Pharmacol. 2011, 204, 365–390. [Google Scholar] [PubMed]
- Griffiths, G.; Hollinshead, R.; Hemmings, B.A.; Nigg, E.A. Ultrastructural localization of the regulatory (RII) subunit of cyclic AMP-dependent protein kinase to subcellular compartments active in endocytosis and recycling of membrane receptors. J. Cell Sci. 1990, 96, 691–703. [Google Scholar] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic AMP: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Yang, W.L.; Ravatn, R.; Kita, T.; Reitman, E.; Vettori, D.; Cvijic, M.E.; Shin, M.; Iacono, L. Reinventing the wheel of cyclic AMP: Novel mechanisms of cAMP signaling. Ann. N. Y. Acad. Sci. 2002, 968, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Schillace, R.V.; Andrews, S.F.; Galligan, S.G.; Burton, K.A.; Starks, H.J.; Bouwer, H.G.; McKnight, G.S.; Davey, M.P.; Carr, D.W. The role of protein kinase a anchoring via the RIIα regulatory subunit in the murine immune system. J. Immunol. 2005, 174, 6847–6853. [Google Scholar] [CrossRef] [PubMed]
- Skålhegg, B.S.; Taskén, K.; Hansson, V.; Huitfeldt, H.S.; Jahnsen, T.; Lea, T. Location of cAMP-dependent protein kinase type I with the TCR-CD3 complex. Science 1994, 263, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Skålhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.B.; Knoll, B.J.; Barber, R. Partial agonists and G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 1999, 20, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.M.; Friedman, J.; Qunaibi, E.; Baameur, F.; Moore, R.H.; Clark, R.B. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the β2-adrenergic receptor using phosphoserine-specific antibodies. Mol. Pharmacol. 2004, 65, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Hausdorff, W.P.; Bouvier, M.; O’Dowd, B.F.; Irons, G.P.; Caron, M.G.; Lefkowitz, R.J. Phosphorylation sites on two domains of the β2-adrenergic receptor are involved in distinct pathways of receptor desensitization. J. Biol. Chem. 1989, 264, 12657–12665. [Google Scholar] [PubMed]
- Lohse, M.J.; Benovic, J.L.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin: A protein that regulates β-adrenergic receptor function. Science 1990, 248, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. G protein-coupled receptors III. New roles for receptor kinases and β-arrestins in receptor signaling and desensitization. J. Biol. Chem. 1998, 273, 18677–18680. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [PubMed]
- Benovic, J.L.; Kuhn, H.; Weyand, I.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Functional desensitization of the isolated beta-adrenergic receptor by the β-adrenergic receptor kinase: Potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc. Natl. Acad. Sci. USA 1987, 84, 8879–8882. [Google Scholar] [CrossRef] [PubMed]
- Hausdorff, W.P.; Lohse, M.J.; Bouvier, M.; Liggett, S.B.; Caron, M.G.; Lefkowitz, R.J. Two kinases mediate agonist-dependent phosphorylation and desensitization of the β2-adrenergic receptor. Symp. Soc. Exp. Biol. 1990, 44, 225–240. [Google Scholar] [PubMed]
- Inglese, J.; Freedman, N.J.; Koch, W.J.; Lefkowitz, R.J. Structure and mechanism of the G protein-coupled receptor kinases. J. Biol. Chem. 1993, 268, 23735–23738. [Google Scholar] [PubMed]
- Freedman, N.J.; Lefkowitz, R.J. Desensitization of G protein-coupled receptors. Recent Prog. Horm. Res. 1996, 51, 319–351. [Google Scholar] [PubMed]
- Krupnick, J.G.; Benovic, J.L. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.S.; Ménard, L.; Barak, L.S.; Koch, W.J.; Colapietro, A.M.; Caron, M.G. Role of phosphorylation in agonist-promoted β2-adrenergic receptor sequestration. Rescue of a sequestration-defective mutant receptor by βARK1. J. Biol. Chem. 1995, 270, 24782–24789. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Lefkowitz, R.J. GRKs and beta-arrestins: Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 2006, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B., Jr.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. β-arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Chien, K.R.; Choi, D.J.; Iaccarino, G.; Hunter, J.J.; Ross, J., Jr.; Lefkowitz, R.J.; Koch, W.J. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc. Natl. Acad. Sci. USA. 1998, 95, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, H.; Naga Prasad, S.V.; Lefkowitz, R.J.; Koch, W.J.; Rockman, H.A. Level of β-adrenergic receptor kinase 1 inhibition determines degree of cardiac dysfunction after chronic pressure overload-induced heart failure. Circulation 2005, 111, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Mihlbachler, K.A.; Brunnett, A.C.; Liggett, S.B. Targeted transgenesis reveals discrete attenuator functions of GRK and PKA in airway β2-adrenergic receptor physiologic signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 15007–15012. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Pablos, J.L.; Laurent, L.; Planchenault, T.; Verola, O.; Lebbe, C.; Kerob, D.; Dupuy, A.; Hermine, O.; et al. WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood 2005, 105, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.L.; Schaller, J.A.; Shewmaker, E.; Osredkar, T.; Lubahn, C. Altered sympathetic-to-immune cell signaling via β2-adrenergic receptors in adjuvant arthritis. Clin. Dev. Immunol. 2013, 2013, 764395. [Google Scholar] [CrossRef] [PubMed]
- Baillie, G.S.; Sood, A.; McPhee, I.; Gall, I.; Perry, S.J.; Lefkowitz, R.J.; Houslay, M.D. β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi. Proc. Natl. Acad. Sci. USA. 2003, 100, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, SK.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the β2-adrenergic receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; New, L.; Jiang, Y.; Han, J.; Su, B. Molecular cloning and characterization of a human protein kinase that specifically activates c-Jun N-terminal kinase. Gene 1998, 212, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. Inflammatory signaling in macrophages: Transitions from acute to tolerant and alternative activation states. Eur. J. Immunol. 2011, 41, 2477–2481. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 2006, 6, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Furler, R.L; Uittenbogaart, C.H. Signaling through the P38 and ERK pathways: A common link between HIV replication and the immune response. Immunol. Res. 2010, 48, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Negrin, R.S. Interleukin-2 receptor downstream events in regulatory T cells: Implications for the choice of immunosuppressive drug therapy. Cell Cycle 2008, 7, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Benczik, M.; Gaffen, S.L. The interleukin (IL)-2 family cytokines: Survival and proliferation signaling pathways in T lymphocytes. Immunol. Investig. 2004, 33, 109–142. [Google Scholar] [CrossRef]
- Alberola-Ila, J.; Hernández-Hoyos, G. The Ras/MAPK cascade and the control of positive selection. Immunol. Rev. 2003, 191, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Essayan, D.M. Cyclic nucleotide phosphodiesterases. J. Allergy Clin. Immunol. 2001, 108, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, H.; Baillie, G.; Ngai, J.; Vang, T.; Nika, K.; Ruppelt, A.; Mustelin, T.; Zaccolo, M.; Houslay, M.; Taskén, K. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J. Immunol. 2004, 173, 4847–4858. [Google Scholar] [CrossRef] [PubMed]
- Bjørgo, E.; Solheim, S.A.; Abrahamsen, H.; Baillie, G.S.; Brown, K.M.; Berge, T.; Okkenhaug, K.; Houslay, M.D.; Taskén, K. Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase a signaling pathways at the level of a protein kinase B/β-arrestin/cAMP phosphodiesterase 4 complex. Mol. Cell. Biol. 2010, 30, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Houslay, M.D. Challenge of human Jurkat T-cells with the adenylate cyclase activator forskolin elicits major changes in cAMP phosphodiesterase (PDE) expression by up-regulating PDE3 and inducing PDE4D1 and PDE4D2 splice variants as well as down-regulating a novel PDE4A splice variant. Biochem. J. 1997, 321, 165–175. [Google Scholar] [PubMed]
- Giembycz, M.A. Phosphodiesterase 4 and tolerance to β2-adrenoceptor agonists in asthma. Trends Pharmacol. Sci. 1996, 17, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Page, C.P.; Spina, D. Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb. Exp. Pharmacol. 2011, 204, 391–414. [Google Scholar] [PubMed]
- Chuang, T.T.; Sallese, M.; Ambrosini, G.; Parruti, G.; de Blasi, A. High expression of β-adrenergic receptor kinase in peripheral human blood leukocytes. J. Biol. Chem. 1992, 267, 6886–6892. [Google Scholar] [PubMed]
- Loudon, R.P.; Perussia, B.; Benovic, J.L. Differentially regulated expression of the G protein-coupled receptor kinases, βARK and GRK6, during myelomonocytic cell development in vitro. Blood 1996, 88, 4547–4557. [Google Scholar] [PubMed]
- Oppermann, M.; Mack, M.; Proudfoot, A.E.; Olbrich, H. Differential effects of CC chemokines on CC chemokine receptor 5 (CCR5) phosphorylation and identification of phosphorylation sites on the CCR5 carboxyl terminus. J. Biol. Chem. 1999, 274, 8875–8885. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.X.; Shan, J.; Qi, X.Y.; Zhang, S.J.; Ma, S.P.; Wang, N.; Li, J.P.; Xue, H.; Wu, M. The catecholamine-β-adrenoreceptor-cAMP system and prediction of cardiovascular events in hypertension. Clin. Exp. Pharmacol. Physiol. 2006, 33, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Bernardin, G.; Strosberg, A.D.; Bernard, A.; Mattei, M.; Marullo, S. β-Adrenergic receptor-dependent and -independent stimulation of adenylate cyclase is impaired during severe sepsis in humans. Intensive Care Med. 1998, 24, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Notterman, D.A.; Metakis, L. Tumor necrosis factor produces homologous desensitization of lymphocyte β2-adrenergic responses. Circ. Shock 1993, 39, 275–278. [Google Scholar] [PubMed]
- Silverman, H.J.; Lee, N.H.; el-Fakahany, E.E. Effects of canine endotoxin shock on lymphocytic β-adrenergic receptors. Circ. Shock 1990, 32, 293–306. [Google Scholar] [PubMed]
- Baerwald, C.; Graefe, C.; von Wichert, P.; Krause, A. Decreased density of β-adrenergic receptors on peripheral blood mononuclear cells in patients with rheumatoid arthritis. J. Rheumatol. 1992, 19, 204–210. [Google Scholar] [PubMed]
- Baerwald, C.G.; Wahle, M.; Ulrichs, T.; Jonas, D.; von Bierbrauer, A.; von Wichert, P.; Burmester, G.R.; Krause, A. Reduced catecholamine response of lymphocytes from patients with rheumatoid arthritis. Immunobiology 1999, 200, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Wahle, M.; Hanefeld, G.; Brunn, S.; Straub, R.H.; Wagner, U.; Krause, A.; Häntzschel, H.; Baerwald, C.G. Failure of catecholamines to shift T-cell cytokine responses toward a Th2 profile in patients with rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R138. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Xue, Q.; Lin, Y.; Wang, L; Zhu, G; Zhao, Q; Chen, Y. Role of β-adrenoceptor at different stages of bronchial asthma. Chin. Med. J. (Engl.) 2001, 114, 1317–1319. [Google Scholar]
- Hataoka, I.; Okayama, M.; Sugi, M.; Inoue, H.; Takishima, T.; Shirato, K. Decrease in β-adrenergic receptors of lymphocytes in spontaneously occurring acute asthma. Chest 1993, 104, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Yoshie, Y.; Nakazawa, T. Hormone-sensitive adenylate cyclase system in lymphocytes from asthmatic patients: Possible defects at the postreceptor sites. Ann. Allergy 1991, 66, 167–172. [Google Scholar] [PubMed]
- Oyama, N.; Urasawa, K.; Kaneta, S.; Sakai, H.; Saito, T.; Takagi, C.; Yoshida, I.; Kitabatake, A.; Tsutsui, H. Chronic β-adrenergic receptor stimulation enhances the expression of G-Protein coupled receptor kinases, GRK2 and GRK5, in both the heart and peripheral lymphocytes. Circ. J. 2005, 69, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Gros, R.; Benovic, J.L.; Tan, C.M.; Feldman, R.D. G-protein-coupled receptor kinase activity is increased in hypertension. J. Clin. Investig. 1997, 99, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.S.; Kavelaars, A.; Schedlowski, M.; Bijlsma, J.W.; Okihara, K.L.; van de Pol, M.; Ochsmann, S.; Pawlak, C.; Schmidt, R.E.; Heijnen, C.J. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999, 13, 715–725. [Google Scholar] [PubMed]
- Vroon, A.; Kavelaars, A.; Limmroth, V.; Lombardi, M.S.; Goebel, M.U.; van dam, A.M.; Caron, M.G.; Schedlowski, M.; Heijnen, C.J. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Immunol. 2005, 174, 4400–4406. [Google Scholar] [CrossRef] [PubMed]
- Giorelli, M.; Livrea, P.; Trojano, M. Post-receptorial mechanisms underlie functional disregulation of β2-adrenergic receptors in lymphocytes from Multiple Sclerosis patients. J. Neuroimmunol. 2004, 155, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Zoukos, Y.; Thomaides, T.N.; Kidd, D.; Cuzner, M.L.; Thompson, A. Expression of β2-adrenoreceptors on peripheral blood mononuclear cells in patients with primary and secondary progressive multiple sclerosis: A longitudinal six month study. J. Neurol. Neurosurg. Psychiatry 2003, 74, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Zoukos, Y.; Kidd, D.; Woodroofe, M.N.; Kendall, B.E.; Thompson, A.J.; Cuzner, M.L. Increased expression of high affinity IL-2 receptors and beta-adrenoceptors on peripheral blood mononuclear cells is associated with clinical and MRI activity in multiple sclerosis. Brain 1994, 117, 307–315. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple functions of G protein-coupled receptor kinases. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [PubMed]
- Trester-Zedlitz, M.; Burlingame, A.; Kobilka, B.; von Zastrow, M. Mass spectrometric analysis of agonist effects on posttranslational modifications of the β2-adrenoceptor in mammalian cells. Biochemistry 2005, 44, 6133–6143. [Google Scholar] [CrossRef] [PubMed]
- Millman, E.E.; Rosenfeld, J.L.; Vaughan, D.J.; Nguyen, J.; Dai, W.; Alpizar-Foster, E.; Clark, R.B.; Knoll, B.J.; Moore, R.H. Endosome sorting of β2-adrenoceptors is GRK5 independent. Brit. J. Pharmacol. 2004, 141, 277–284. [Google Scholar] [CrossRef]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Claing, A.; LaPorte, S.A.; Caron, M.G.; Lefkowitz, R.J. Endocytosis of G protein-coupled receptors: Roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog. Neurobiol. 2002, 66, 61–79. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; LaPorte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. β-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Ferguson, S.S.; Daaka, Y.; Miller, W.E.; Maudsley, S.; Della Rocca, G.J.; Lin, F.; Kawakatsu, H.; Owada, K.; Luttrell, D.K.; et al. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 1999, 283, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Park, D.H.; Wessel, T.C.; Song, B.; Wagner, J.A.; Joh, T.H. A dual role for the cAMP-dependent protein kinase in tyrosine hydroxylase gene expression. Proc. Natl. Acad. Sci. USA 1993, 90, 3471–3475. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.; Lohse, M.J.; Codina, J.; Caron, M.G.; Lefkowitz, R.J. Desensitization of the isolated β2-adrenergic receptor by beta-adrenergic receptor kinase, cAMP-dependent protein kinase, and protein kinase C occurs via distinct molecular mechanisms. Biochemistry 1992, 31, 3193–3197. [Google Scholar] [CrossRef]
- Meltzer, J.C.; MacNeil, B.J.; Sanders, V.; Pylypas, S.; Jansen, A.H.; Greenberg, A.H.; Nance, D.M. Contribution of the adrenal glands and splenic nerve to LPS-induced splenic cytokine production in the rat. Brain Behav. Immun. 2003, 17, 482–497. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, B.J.; Jansen, A.H.; Greenberg, A.H.; Nance, D.M. Activation and selectivity of splenic sympathetic nerve electrical activity response to bacterial endotoxin. Am. J. Physiol. 1996, 270, R264–R270. [Google Scholar] [PubMed]
- Szelenyi, J.; Selmeczy, Z.; Brozik, A.; Medgyesi, D.; Magocsi, M. Dual β-adrenergic modulation in the immune system. Stimulus-dependent effect of isoproterenol on MAPK activation and inflammatory mediator production in macrophages. Neurochem. Int. 2006, 49, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Gerlo, S.; Kooijman, R.; Beck, I.M.; Kolmus, K.; Spooren, A.; Haegeman, G. Cyclic AMP: A selective modulator of NF-κB action. Cell. Mol. Life Sci. 2011, 68, 3823–3841. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Hasko, G.; Kovacs, K.J.; Vizi, E.S. Modulation of lipopolysaccharide-induced tumor necrosis factor-α production by selective α- and β-adrenergic drugs in mice. J. Neuroimmunol. 1995, 61, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Ramer-Quinn, D.S.; Swanson, M.A.; Lee, W.T.; Sanders, V.M. Cytokine production by naive and primary effector CD4+ T cells exposed to norepinephrine. Brain Behav. Immun. 2000, 14, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, A.J.; Stam, W.B.; Vanderschueren, R.G.; Nijkamp, F.P. Effects of cytokines on β-adrenoceptor function of human peripheral blood mononuclear cells and guinea pig trachea. J. Allergy Clin. Immunol. 1992, 90, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Seyedi, S.; Andalib, A.; Rezaei, A.; Hosseini, S.M.; Mohebbi, S.R.; Zali, M.R.; Vafai, M.; Behboo, R.; Tabatabaei, S.A.; Shahabi, S. The effects of isoproterenol and propranolol on cytokine profile secretion by cultured tumor-infiltrating lymphocytes derived from colorectal cancer patients. Cell J. 2012, 13, 281–289. [Google Scholar] [PubMed]
- Mohede, I.C.; van Ark, I.; Brons, F.M.; van Oosterhout, A.J.; Nijkamp, F.P. Salmeterol inhibits interferon-gamma and interleukin-4 production by human peripheral blood mononuclear cells. Int. J. Immunopharmacol. 1996, 18, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Osawa, Y; Lee, H.T.; Hirshman, C.A.; Xu, D.; Emala, C.W. Lipopolysaccharide-induced sensitization of adenylyl cyclase activity in murine macrophages. Am. J. Physiol. Cell Physiol. 2006, 290, C143–C151. [Google Scholar] [PubMed]
- Liu, Z.; Jiang, Y.; Li, Y.; Wang, J.; Fan, L.; Scott, M.J; Xiao, G.; Li, S.; Billiar, T,R.; Wilson, M.A.; Fan, J. TLR4 Signaling augments monocyte chemotaxis by regulating G protein-coupled receptor kinase 2 translocation. J. Immunol. 2013, 191, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Loniewski, K.; Shi, Y.; Pestka, J.; Parameswaran, N. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol. Immunol. 2008, 45, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Clark, R.B.; Friedman, J.; Dixon, R.A.; Strader, C.D. Identification of a specific domain in the β-adrenergic receptor required for phorbol ester-induced inhibition of catecholamine-stimulated adenylyl cyclase. Mol. Pharmacol. 1990, 38, 289–293. [Google Scholar] [PubMed]
- Yuan, N.; Friedman, J.; Whaley, B.S.; Clark, R.B. cAMP-dependent protein kinase and protein kinase C consensus site mutations of the β-adrenergic receptor. Effect on desensitization and stimulation of adenylyl cyclase. J. Biol. Chem. 1994, 269, 23032–23038. [Google Scholar] [PubMed]
- Carmena, M.J.; García-Paramio, P.; Solano, R.M.; Prieto, J.C. Protein kinase C regulation of the adenylyl cyclase system in rat prostatic epithelium. Prostate 1995, 27, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Deiss, K.; Kisker, C.; Lohse, M.J.; Kristina Lorenz, K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein-coupled receptor kinase (GRK) 2. J. Biol. Chem. 2012, 287, 23407–23417. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef] [PubMed]
- De Blasi, A.; Parruti, G.; Sallese, M. Regulation of G protein-coupled receptor kinase subtypes in activated T lymphocytes. Selective increase of beta-adrenergic receptor kinase 1 and 2. J. Clin. Investig. 1995, 95, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Benovic, J.L. Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J. Biol. Chem. 1997, 272, 3806–3812. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, IM. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, M.; Chaudhari, S.; Ding, Y.; Yuan, J.; Stankowska, D.; He, S.; Krishnamoorthy, R.; Cunningham, J.T.; Ma, R. Nuclear factor κB mediates suppression of canonical transient receptor potential 6 expression by reactive oxygen species and protein kinase C in kidney cells. J. Biol. Chem. 2013, 288, 12852–12865. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, M.S.; Rhew, J.H.; Park, R.W.; de Crombrugghe, B.; Kim, I.S. Transcriptional regulation of fibronectin gene by phorbol myristate acetate in hepatoma cells: A negative role for NF-κB. J. Cell. Biochem. 2000, 76, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.N.; Koch, W.J. Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J. Biol. Chem. 2012, 287, 12771–12778. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Nackley, A.G.; Satterfield, K.; Maixner, W.; Diatchenko, L.; Flood, P.M. β2-adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-κB-independent mechanisms. Cell Signal. 2007, 19, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Ahn, S.; Rominger, D.H.; Gowen-MacDonald, W.; Lam, C.M.; Dewire, S.M.; Violin, J.D.; Lefkowitz, R.J. Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol. 2011, 80, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Violin, J.D.; Whalen, E.J.; Wisler, J.W.; Shenoy, S.K.; Lefkowitz, R.J. β-Arrestin-biased agonism at the β2-adrenergic receptor. J. Biol. Chem. 2008, 283, 5669–5676. [Google Scholar] [CrossRef] [PubMed]
- Kin, N.W.; Sanders, V.M. It takes nerve to tell T and B cells what to do. J. Leukoc. Biol. 2006, 79, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.A.; Lee, W.T.; Sanders, V.M. IFN-γ production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J. Immunol. 2001, 166, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Badou, A.; Savignac, M.; Moreau, M.; Leclerc, C.; Foucras, G.; Cassar, G.; Paulet, P.; Lagrange, D.; Druet, P.; Guéry, J.C.; et al. Weak TCR stimulation induces a calcium signal that triggers IL-4 synthesis, stronger TCR stimulation induces MAP kinases that control IFN-gamma production. Eur. J. Immunol. 2001, 31, 2487–2496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kaplan, M.H. The p38 mitogen-activated protein kinase is required for IL-12-induced IFN-γ expression. J. Immunol. 2000, 165, 1374–1380. [Google Scholar] [CrossRef] [PubMed]
- Lubahn, C.L.; Lorton, D.; Schaller, J.A.; Sweeney, S.J.; Bellinger, D.L. Targeting α- and β-adrenergic receptors differentially shifts Th1, Th2, and inflammatory cytokine profiles in immune organs to attenuate adjuvant arthritis. Front. Immunol. 2014, 5, 346. [Google Scholar] [CrossRef] [PubMed]
- Vroon, A; Heijnen, C.J.; Kavelaars, A. GRKs and arrestins: Regulators of migration and inflammation. J. Leukoc. Biol. 2006, 80, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.S.; Kavelaars, A.; Cobelens, P.M.; Schmidt, R.E.; Schedlowski, M.; Heijnen, C.J. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J. Immunol. 2001, 166, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Tajima, M.; Kosako, H.; Nakaya, T.; Hashimoto, A.; Watari, K.; Nishihara, H.; Ohba, M.; Komiya, S.; Tani, N.; et al. GRK6 deficiency in mice causes autoimmune disease due to impaired apoptotic cell clearance. Nat. Commun. 2013, 4, 1532. [Google Scholar] [CrossRef] [PubMed]
- Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Thomas, A.J.; Sutton, N.; Hu, Y.; Mukhopadhyay, S; Chen, X.L.; Leung, T.; Richardson, R.M. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J. Immunol. 2013, 190, 5329–5336. [Google Scholar] [CrossRef] [PubMed]
- Murga, C.; Mayor, F., Jr. GRK6, a gatekeeper of visceral hyperalgesia. Brain Behav. Immun. 2009, 23, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Eijkelkamp, N.; Heijnen, C.J.; Carbajal, A.G.; Willemen, H.L.; Wang, H.; Minett, M.S.; Wood, J.N.; Schedlowski, M.; Dantzer, R.; Kelley, K.W.; et al. G protein-coupled receptor kinase 6 acts as a critical regulator of cytokine-induced hyperalgesia by promoting phosphatidylinositol 3-kinase and inhibiting p38 signaling. Mol. Med. 2012, 18, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Managò, F.; Espinoza, S.; Salahpour, A.; Sotnikova, T.D.; Caron, M.G.; Premont, R.T.; Gainetdinov, R.R. The role of GRK6 in animal models of Parkinson’s disease and L-DOPA treatment. Sci. Rep. 2012, 2, 301. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorton, D.; Bellinger, D.L. Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. Int. J. Mol. Sci. 2015, 16, 5635-5665. https://doi.org/10.3390/ijms16035635
Lorton D, Bellinger DL. Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. International Journal of Molecular Sciences. 2015; 16(3):5635-5665. https://doi.org/10.3390/ijms16035635
Chicago/Turabian StyleLorton, Dianne, and Denise L. Bellinger. 2015. "Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells" International Journal of Molecular Sciences 16, no. 3: 5635-5665. https://doi.org/10.3390/ijms16035635
APA StyleLorton, D., & Bellinger, D. L. (2015). Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. International Journal of Molecular Sciences, 16(3), 5635-5665. https://doi.org/10.3390/ijms16035635