
Phosphoproteomic Analysis of the Highly-Metastatic Hepatocellular Carcinoma Cell Line, MHCC97-H



Abstract
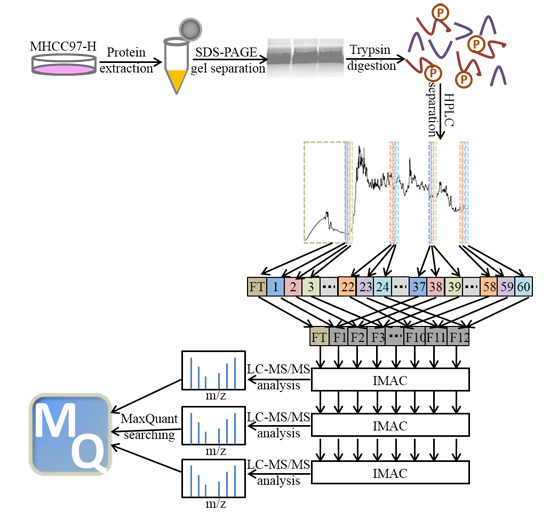
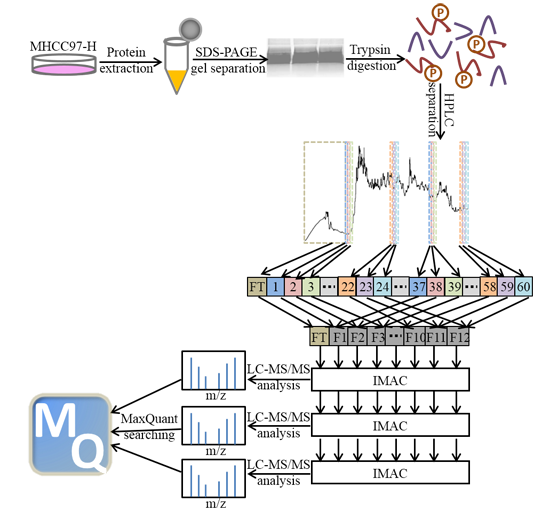
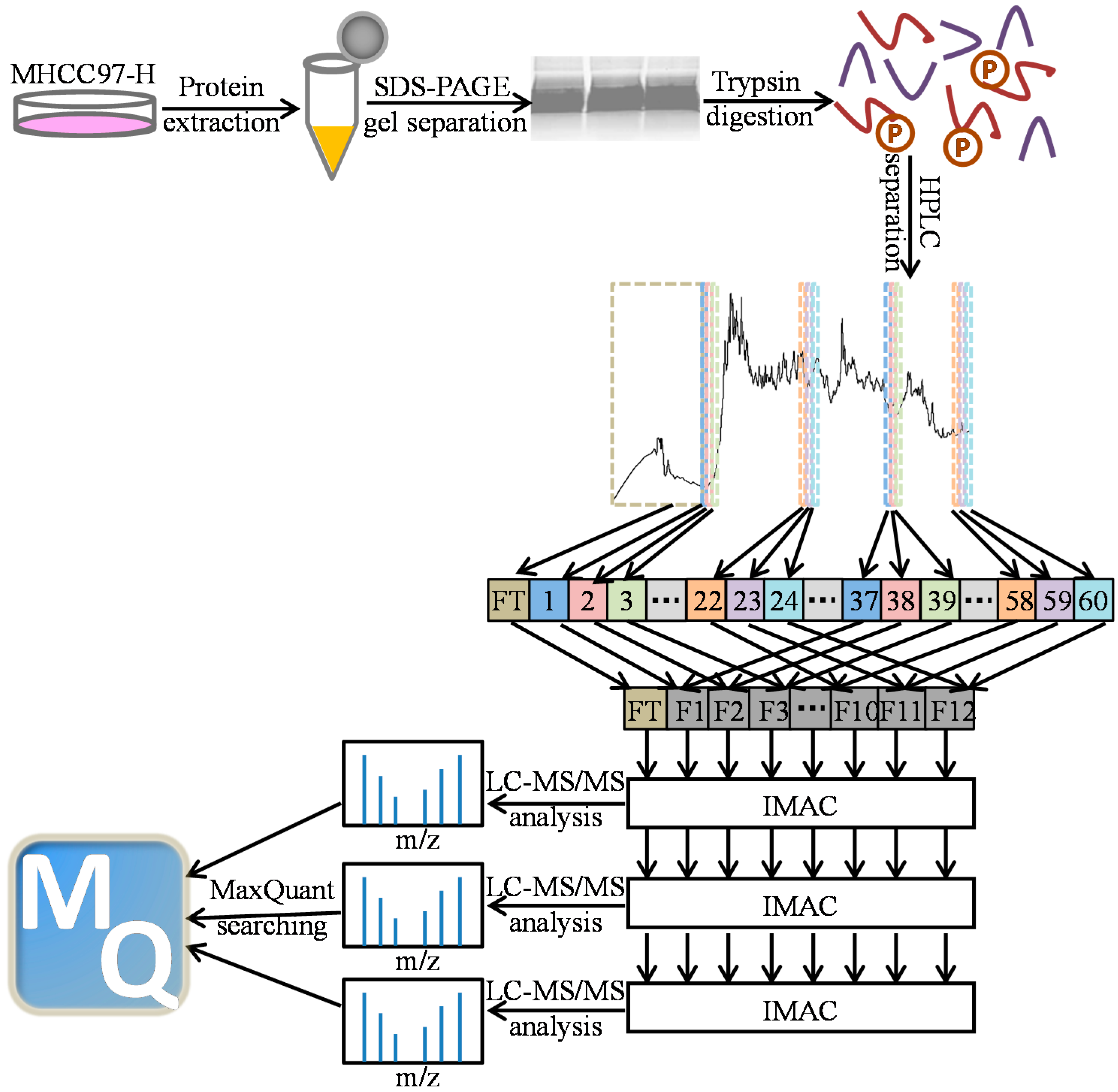
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
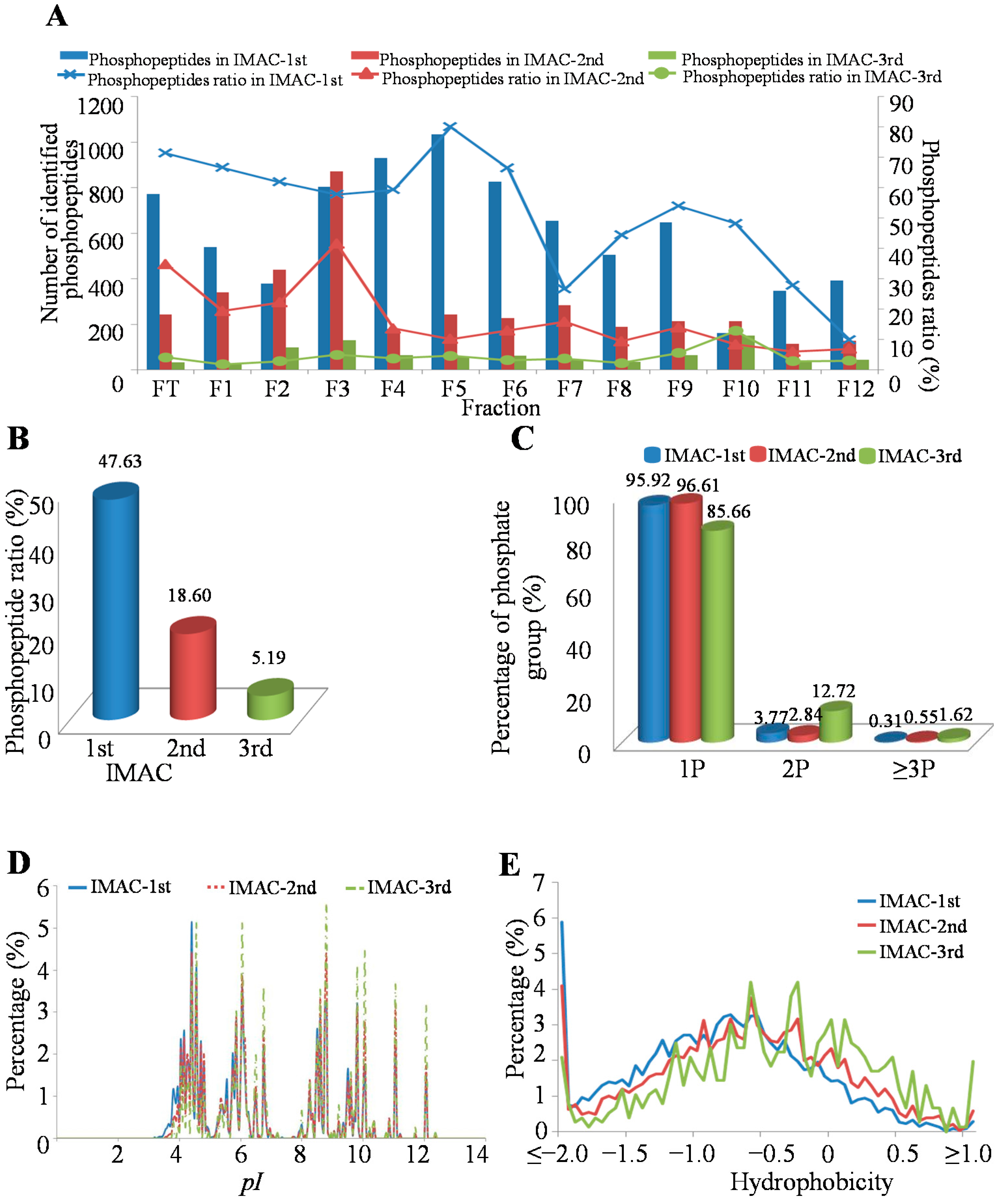
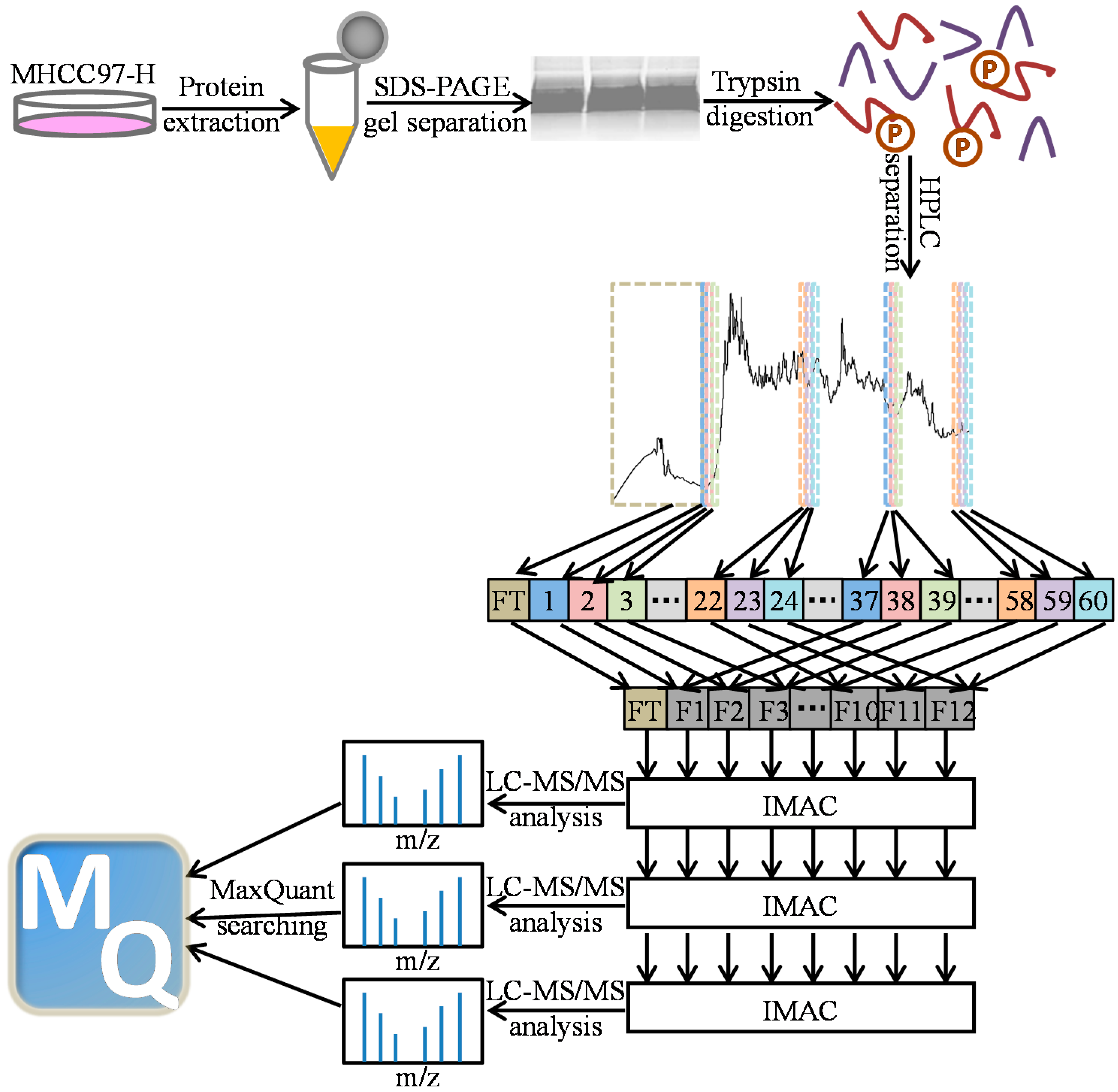
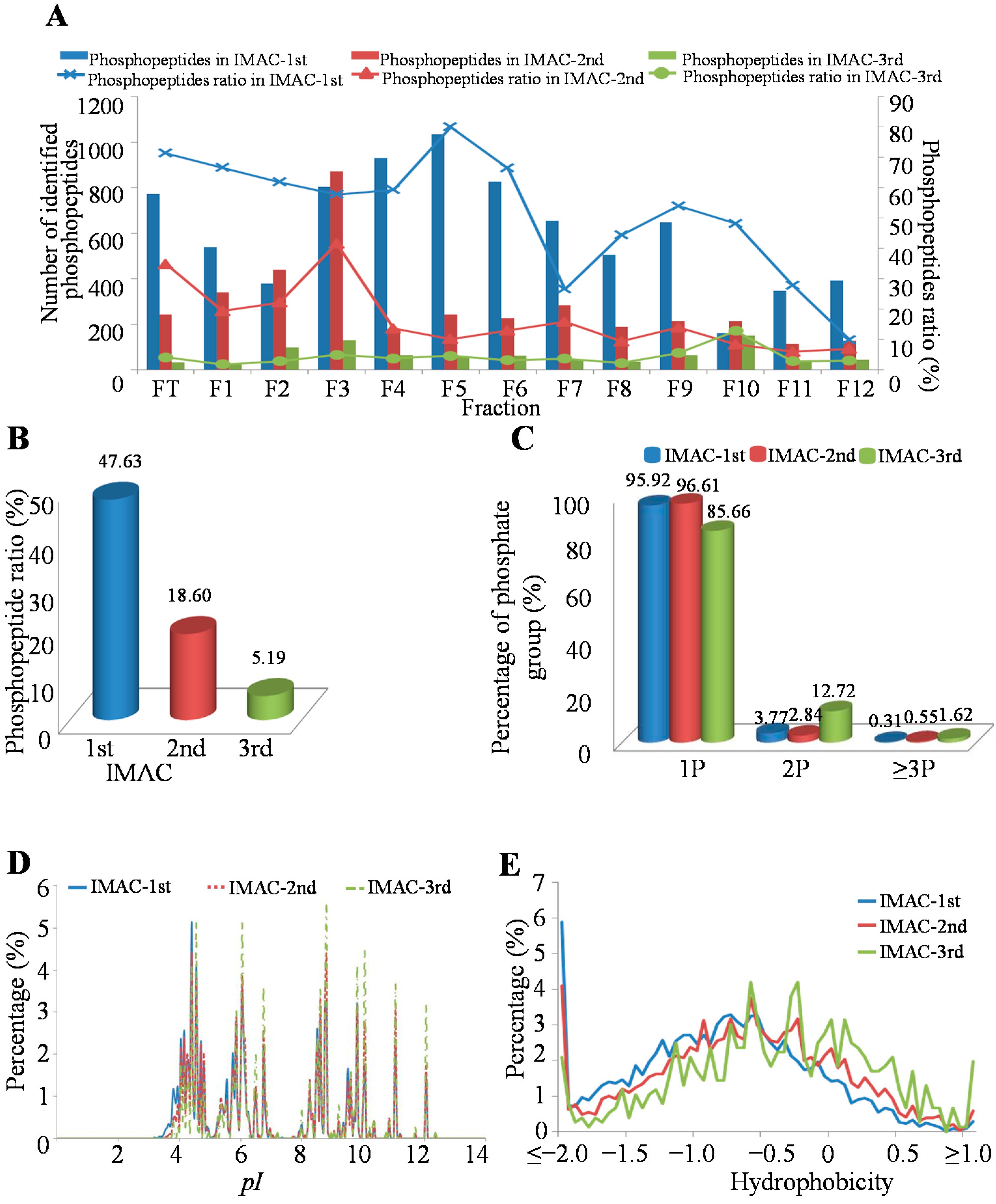
2.1. Phosphopeptides in MHCC97-H by Off-Line High-pH HPLC Separation with Multi-Step IMAC (Immobilized Metal ion Affinity Chromatography)
2.2. Characteristics of Identified Unique Phosphorylation Events in the MHCC97-H Cell Line
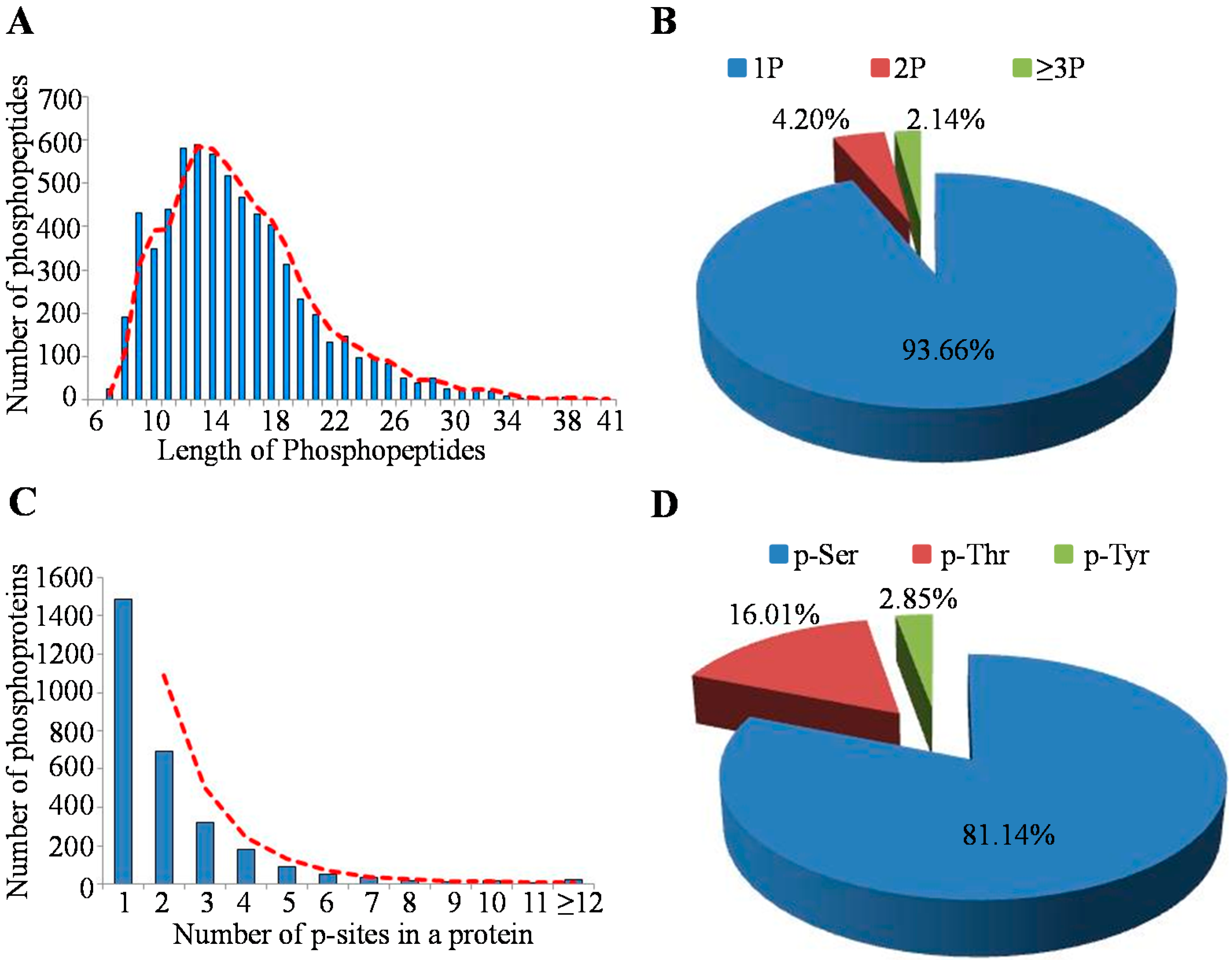
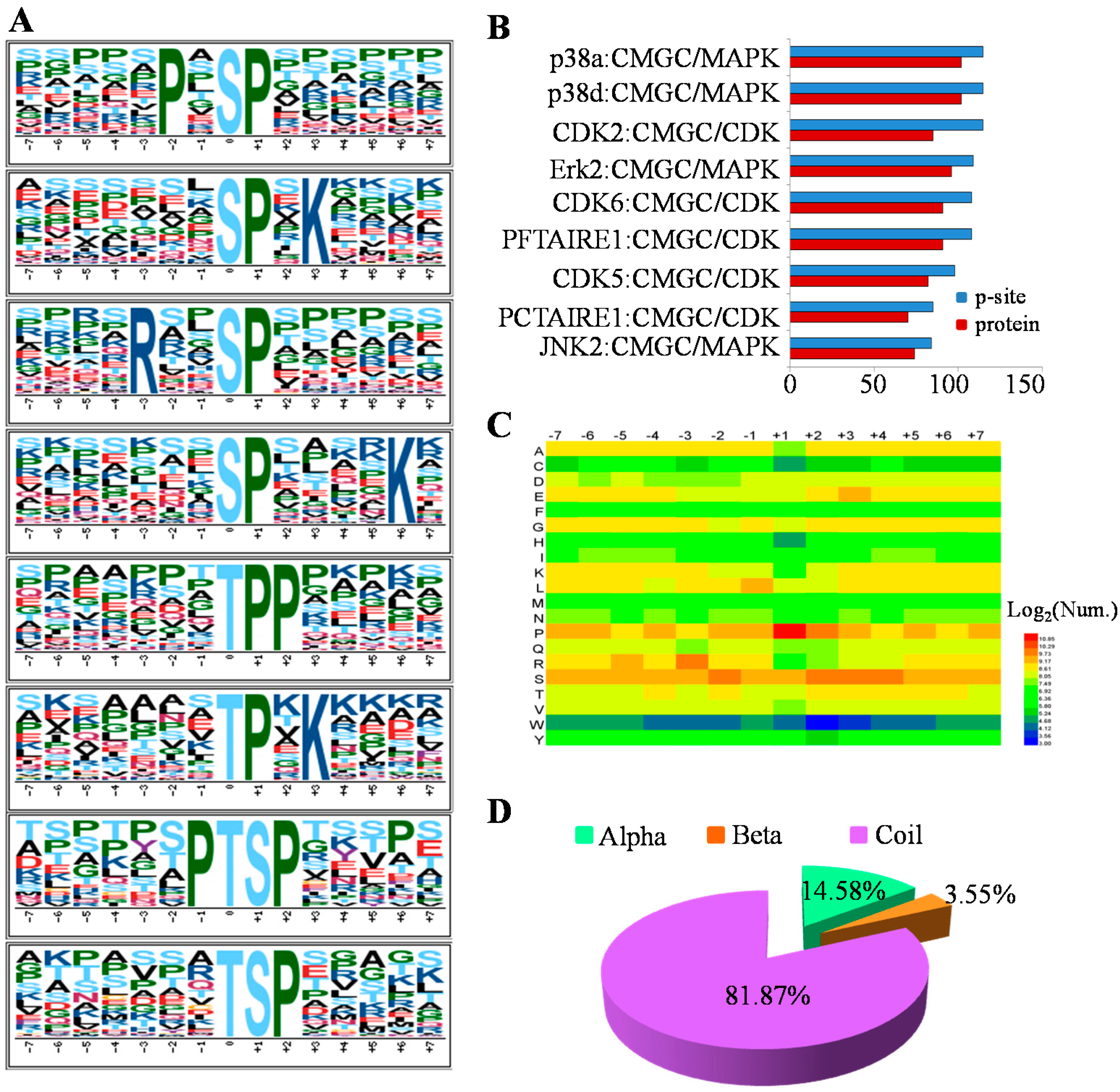
2.3. The Preferences for Sequence and Structure Features of Phosphorylation Sites (p-Sites) in the MHCC97-H Cell Line
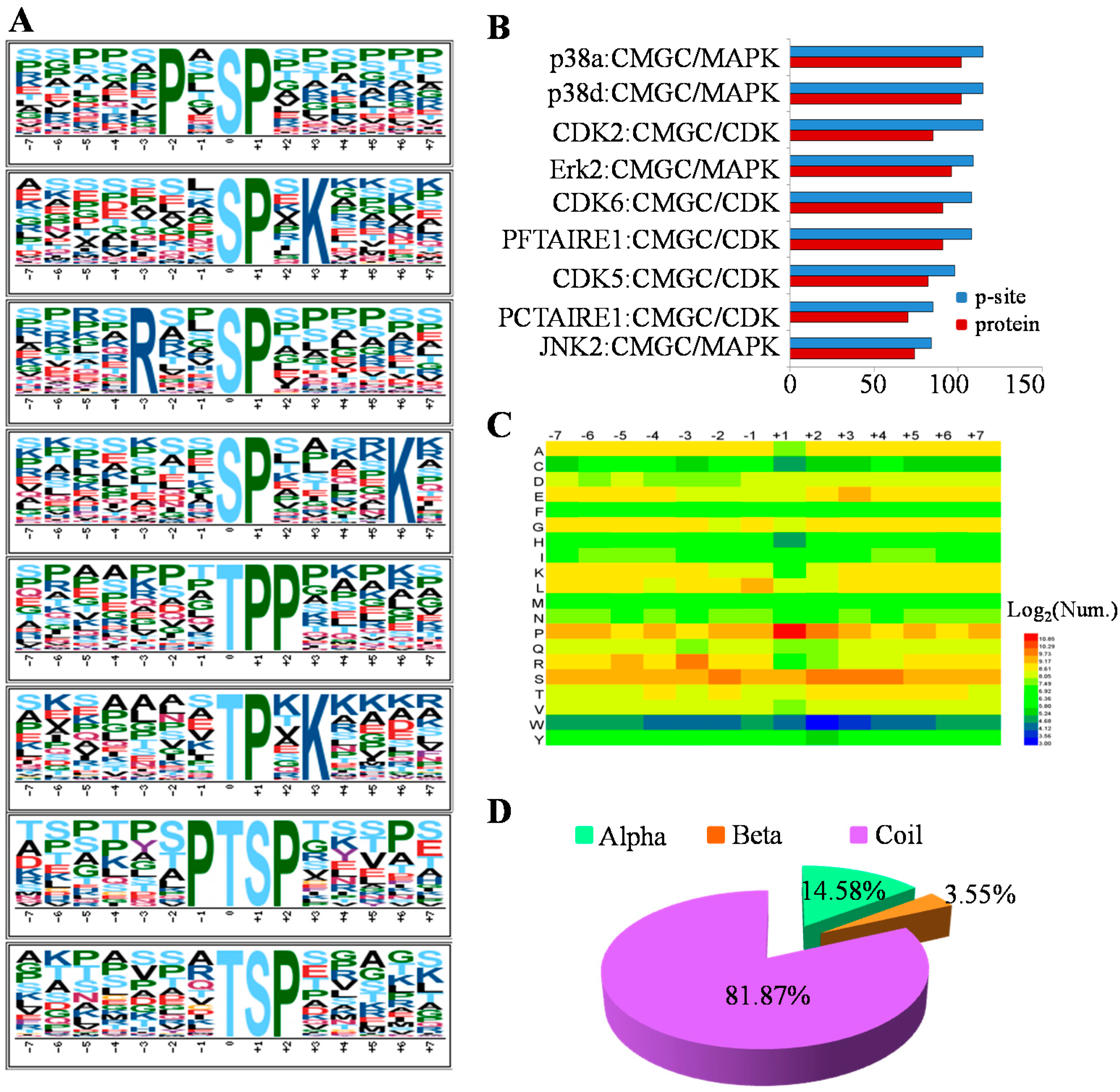
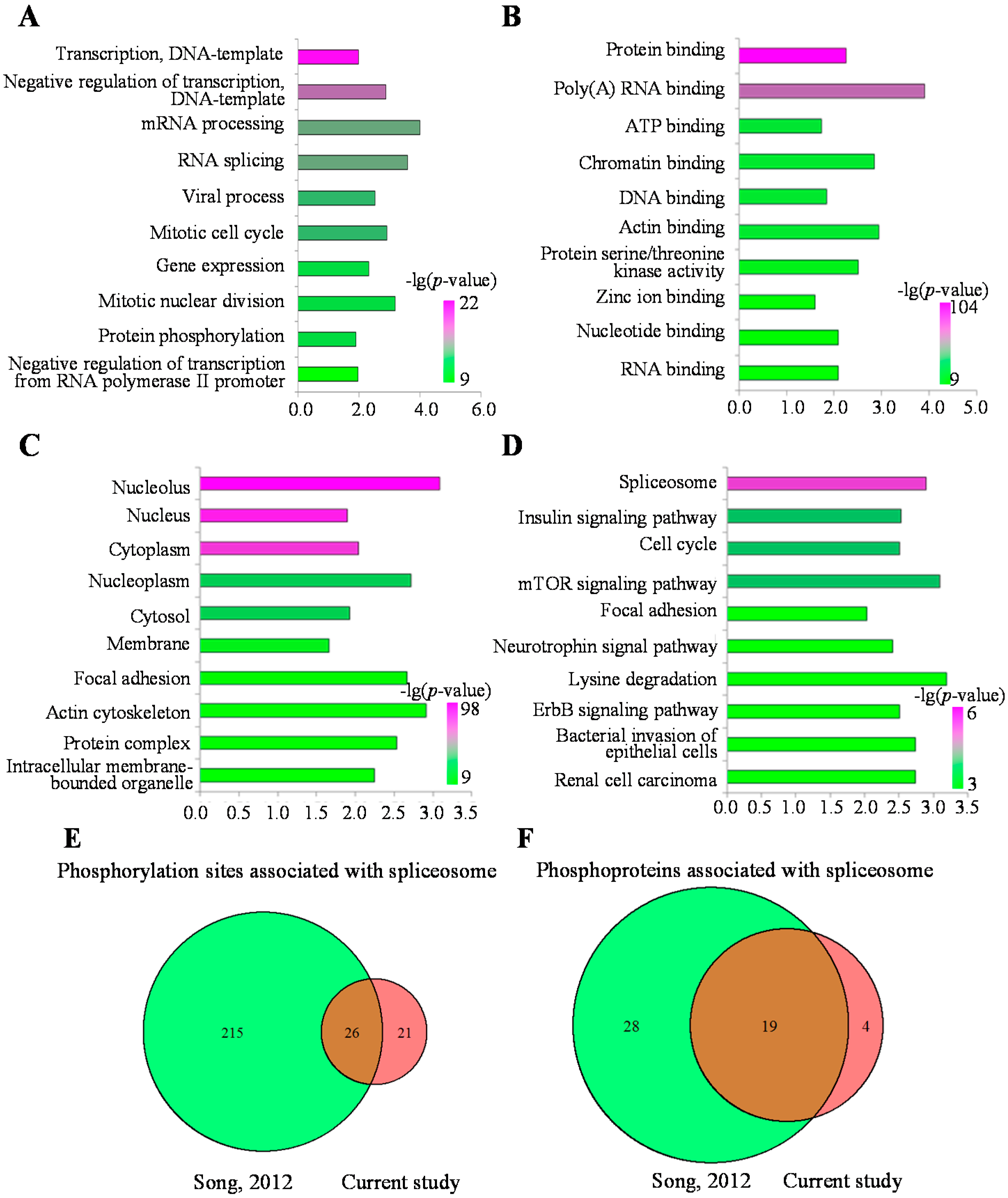
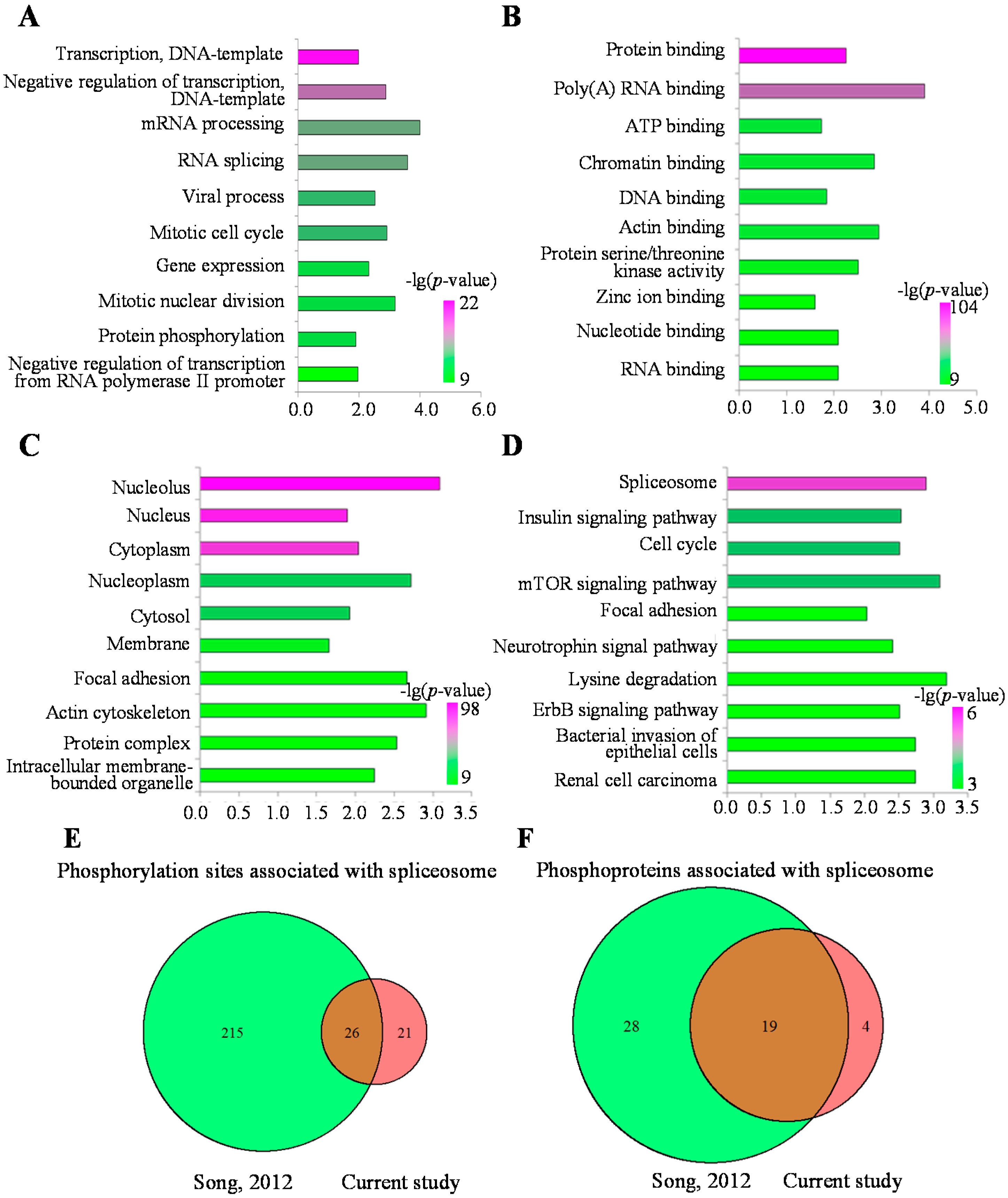
2.4. Functional Analysis of the MHCC97-H Phosphoproteomic Data
3. Experimental Section
3.1. Cell Culture
3.2. Protein Extraction and Digestion
3.3. Peptide Desalting
3.4. An Off-Line High-pH HPLC Separation
3.5. IMAC Enrichment
3.6. Mass Spectrometric Analysis and Data Analysis
3.7. Phosphorylation Motif Analysis
3.8. Group-Based Prediction System (GPS) Algorithm with the Interaction Filter, or in Vivo GPS (iGPS) Analysis
3.9. Analysis of Secondary Structures
3.10. The Functional Enrichment Analysis of the GO (Gene Ontology)/KEGG (the Kyoto Encyclopedia of Genes and Genomes) Pathway
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Brzezianska, E.; Pastuszak-Lewandoska, D. A minireview: The role of MAPK/ERK and PI3K/Akt pathways in thyroid follicular cell-derived neoplasm. Front. Biosci. 2011, 16, 422–439. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Mehta, A. CFTR: A hub for kinases and crosstalk of cAMP and Ca2+. FEBS J. 2013, 280, 4417–4429. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.W.; Margulies, J.E.; Coulter, P.M., 2nd.; Watson, J.B. Functional consequences of expression of the neuron-specific, protein kinase C substrate RC3 (neurogranin) in Xenopus oocytes. Brain Res. 1993, 627, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, R.; Krakstad, C.; Selheim, F.; Kopperud, R.; Doskeland, S.O. The cAMP-dependent protein kinase pathway as therapeutic target: Possibilities and pitfalls. Curr. Top. Med. Chem. 2011, 11, 1393–1405. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-White, J.L.; Andersen, N.J.; Duesbery, N.S. Mek genomics in development and disease. Brief. Funct. Genomics 2012, 11, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Otero, C.; Penaloza, J.P.; Rodas, P.I.; Fernandez-Ramires, R.; Velasquez, L.; Jung, J.E. Temporal and spatial regulation of cAMP signaling in disease: Role of cyclic nucleotide phosphodiesterases. Fundam. Clin. Pharmacol. 2014, 28, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Morizane, T.; Watanabe, T.; Kagawa, T.; Iwabuchi, M.N.; Kumagai, N.; Inagaki, Y.; Tsuchimoto, K.; Tsuchiya, M. Establishment of a human cell line (HCC-T) from a patient with hepatoma bearing no evidence of hepatitis B or A virus infection. Cancer 1989, 64, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Morizane, T.; Tsuchimoto, K.; Inagaki, Y.; Munakata, Y.; Nakamura, T.; Kumagai, N.; Tsuchiya, M. Establishment of a cell line (HCC-M) from a human hepatocellular carcinoma. Int. J. Cancer 1983, 32, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, Z.Y.; Ye, S.L.; Liu, Y.K.; Chen, J.; Xue, Q.; Gao, D.M.; Bao, W.H. Establishment of cell clones with different metastatic potential from the metastatic hepatocellular carcinoma cell line MHCC97. World J. Gastroenterol. 2001, 7, 630–636. [Google Scholar] [PubMed]
- Hu, Y.J.; Li, H.Y.; Qiu, K.J.; Li, D.C.; Zhou, J.H.; Hu, Y.H.; Zhang, F.M. Downregulation of Notch1 inhibits the invasion of human hepatocellular carcinoma Hepg2 and MHCC97H cells through the regulation of PTEN and FAK. Int. J. Mol. Med. 2014, 34, 1081–1086. [Google Scholar] [PubMed]
- Tu, K.; Dou, C.; Zheng, X.; Li, C.; Yang, W.; Yao, Y.; Liu, Q. Fibulin-5 inhibits hepatocellular carcinoma cell migration and invasion by down-regulating matrix metalloproteinase-7 expression. BMC Cancer 2014, 14, 938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Duan, W.D.; Leng, J.J.; Zhou, L.; Wang, X.; Xu, Y.Z.; Wang, X.D.; Zhang, A.Q.; Dong, J.H. STAT3 regulates the migration and invasion of a stemlike subpopulation through microRNA21 and multiple targets in hepatocellular carcinoma. Oncol. Rep. 2015, 33, 1493–1498. [Google Scholar] [PubMed]
- Schwartz, D.; Gygi, S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005, 23, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.Elm: A database of phosphorylation sites—Update 2011. Nucleic Acids Res. 2011, 39, D261–D267. [Google Scholar] [CrossRef] [PubMed]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bian, Y.; Cheng, K.; Gu, L.F.; Ye, M.; Zou, H.; Sun, S.S.; He, J.X. A large-scale protein phosphorylation analysis reveals novel phosphorylation motifs and phosphoregulatory networks in Arabidopsis. J. Prot. 2013, 78, 486–498. [Google Scholar] [CrossRef]
- White, R.R.; Kwon, Y.G.; Taing, M.; Lawrence, D.S.; Edelman, A.M. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J. Biol. Chem. 1998, 273, 3166–3172. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Ye, M.; Liu, Z.; Cheng, H.; Jiang, X.; Han, G.; Songyang, Z.; Tan, Y.; Wang, H.; Ren, J.; et al. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol. Cell. Prot. 2012, 11, 1070–1083. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 map kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. Hemi: A toolkit for illustrating heatmaps. PLoS One 2014, 9, e111988. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Sette, C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Bell, B.; Revil, T.; Venables, J.P.; Prinos, P.; Elela, S.A.; Chabot, B. Cancer-associated perturbations in alternative pre-messenger RNA splicing. Cancer Treat. Res. 2013, 158, 41–94. [Google Scholar] [PubMed]
- Marzese, D.M.; Liu, M.; Huynh, J.L.; Hirose, H.; Donovan, N.C.; Huynh, K.T.; Kiyohara, E.; Chong, K.; Cheng, D.; Tanaka, R.; et al. Brain metastasis is predetermined in early stages of cutaneous melanoma by CD44v6 expression through epigenetic regulation of the spliceosome. Pigment Cell Melanoma Res. 2015, 28, 82–93. [Google Scholar] [CrossRef]
- Tarallo, R.; Bamundo, A.; Nassa, G.; Nola, E.; Paris, O.; Ambrosino, C.; Facchiano, A.; Baumann, M.; Nyman, T.A.; Weisz, A. Identification of proteins associated with ligand-activated estrogen receptor alpha in human breast cancer cell nuclei by tandem affinity purification and nano LC–MS/MS. Proteomics 2011, 11, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, T.; Tange, T.O.; Sonenberg, N.; Moore, M.J. eIF4AIII binds spliced mRNA in the exon junction complex and is essential for nonsense-mediated decay. Nat. Struct. Mol. Biol. 2004, 11, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Kong, X.T.; Zhang, G.A.; Hou, X.J.; Qiang, H.; Zhong, R.Q. Proteomics-based identification of dead-box protein 48 as a novel autoantigen, a prospective serum marker for pancreatic cancer. Biochem. Biophys. Res. Commun. 2005, 330, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Graub, R.; Lancero, H.; Pedersen, A.; Chu, M.; Padmanabhan, K.; Xu, X.Q.; Spitz, P.; Chalkley, R.; Burlingame, A.L.; Stokoe, D.; et al. Cell cycle-dependent phosphorylation of human CDC5 regulates RNA processing. Cell Cycle 2008, 7, 1795–1803. [Google Scholar] [CrossRef]
- Sanidas, I.; Polytarchou, C.; Hatziapostolou, M.; Ezell, S.A.; Kottakis, F.; Hu, L.; Guo, A.; Xie, J.; Comb, M.J.; Iliopoulos, D.; et al. Phosphoproteomics screen reveals Akt isoform-specific signals linking RNA processing to lung cancer. Mol. Cell 2014, 53, 577–590. [Google Scholar] [CrossRef]
- Dapat, C.; Saito, R.; Suzuki, H.; Horigome, T. Quantitative phosphoproteomic analysis of host responses in human lung epithelial (A549) cells during influenza virus infection. Virus Res. 2014, 179, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Na, K.; Kwon, M.S.; Kim, H.; Kim, K.S.; Paik, Y.K. Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics 2009, 9, 3395–3408. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ren, Z.; Kang, X.; Zhang, L.; Li, X.; Wang, Y.; Xue, T.; Shen, Y.; Liu, Y. Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer 2009, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Lu, Y.; Zhao, Y.J.; Jaeweon, K.; Kang, J.; Xiao-Nan, L.; Ge, G.; Meyer, R.; Perlaky, L.; Hicks, J.; et al. Cell cycle regulator gene CDC5L, a potential target for 6p12-p21 amplicon in osteosarcoma. Mol. Cancer Res. 2008, 6, 937–946. [Google Scholar] [CrossRef]
- Talbot, K.; Miguel-Aliaga, I.; Mohaghegh, P.; Ponting, C.P.; Davies, K.E. Characterization of a gene encoding survival motor neuron (SMN)-related protein, a constituent of the spliceosome complex. Hum. Mol. Genet. 1998, 7, 2149–2156. [Google Scholar] [CrossRef] [PubMed]
- Giri, K.; Shameer, K.; Zimmermann, M.T.; Saha, S.; Chakraborty, P.K.; Sharma, A.; Arvizo, R.R.; Madden, B.J.; McCormick, D.J.; Kocher, J.P.; et al. Understanding protein-nanoparticle interaction: A new gateway to disease therapeutics. Bioconj. Chem. 2014, 25, 1078–1090. [Google Scholar] [CrossRef]
- Villen, J.; Gygi, S.P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008, 3, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Ficarro, S.B.; Adelmant, G.; Tomar, M.N.; Zhang, Y.; Cheng, V.J.; Marto, J.A. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal. Chem. 2009, 81, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cao, J.; Ma, Q.; Gao, X.; Ren, J.; Xue, Y. GPS-YNO2: Computational prediction of tyrosine nitration sites in proteins. Mol. Biosyst. 2011, 7, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, D.; Caruso, M.; Lewis, M.; Qi, Y.; Yi, Z. Quantitative phosphoproteomics reveals novel phosphorylation events in insulin signaling regulated by protein phosphatase 1 regulatory subunit 12A. J. Proteomics 2014, 109, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Loroch, S.; Zahedi, R.P.; Sickmann, A. Highly sensitive phosphoproteomics by tailoring solid-phase extraction to electrostatic repulsion-hydrophilic interaction chromatography. Anal. Chem. 2015, 87, 1596–1604. [Google Scholar] [CrossRef]
- Batth, T.S.; Francavilla, C.; Olsen, J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. [Google Scholar] [CrossRef] [PubMed]
- Engholm-Keller, K.; Larsen, M.R. Technologies and challenges in large-scale phosphoproteomics. Proteomics 2013, 13, 910–931. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, M.; Cheng, H.; Wang, Z.; Su, N.; Liu, Z.; Sun, C.; Zhen, B.; Hong, X.; Xue, Y.; Xu, P. Phosphoproteomic Analysis of the Highly-Metastatic Hepatocellular Carcinoma Cell Line, MHCC97-H. Int. J. Mol. Sci. 2015, 16, 4209-4225. https://doi.org/10.3390/ijms16024209
Tian M, Cheng H, Wang Z, Su N, Liu Z, Sun C, Zhen B, Hong X, Xue Y, Xu P. Phosphoproteomic Analysis of the Highly-Metastatic Hepatocellular Carcinoma Cell Line, MHCC97-H. International Journal of Molecular Sciences. 2015; 16(2):4209-4225. https://doi.org/10.3390/ijms16024209
Chicago/Turabian StyleTian, Miaomiao, Han Cheng, Zhiqiang Wang, Na Su, Zexian Liu, Changqing Sun, Bei Zhen, Xuechuan Hong, Yu Xue, and Ping Xu. 2015. "Phosphoproteomic Analysis of the Highly-Metastatic Hepatocellular Carcinoma Cell Line, MHCC97-H" International Journal of Molecular Sciences 16, no. 2: 4209-4225. https://doi.org/10.3390/ijms16024209
APA StyleTian, M., Cheng, H., Wang, Z., Su, N., Liu, Z., Sun, C., Zhen, B., Hong, X., Xue, Y., & Xu, P. (2015). Phosphoproteomic Analysis of the Highly-Metastatic Hepatocellular Carcinoma Cell Line, MHCC97-H. International Journal of Molecular Sciences, 16(2), 4209-4225. https://doi.org/10.3390/ijms16024209