3.1. The SCO Archetype Compound [Fe(phen)2(NCS)2]
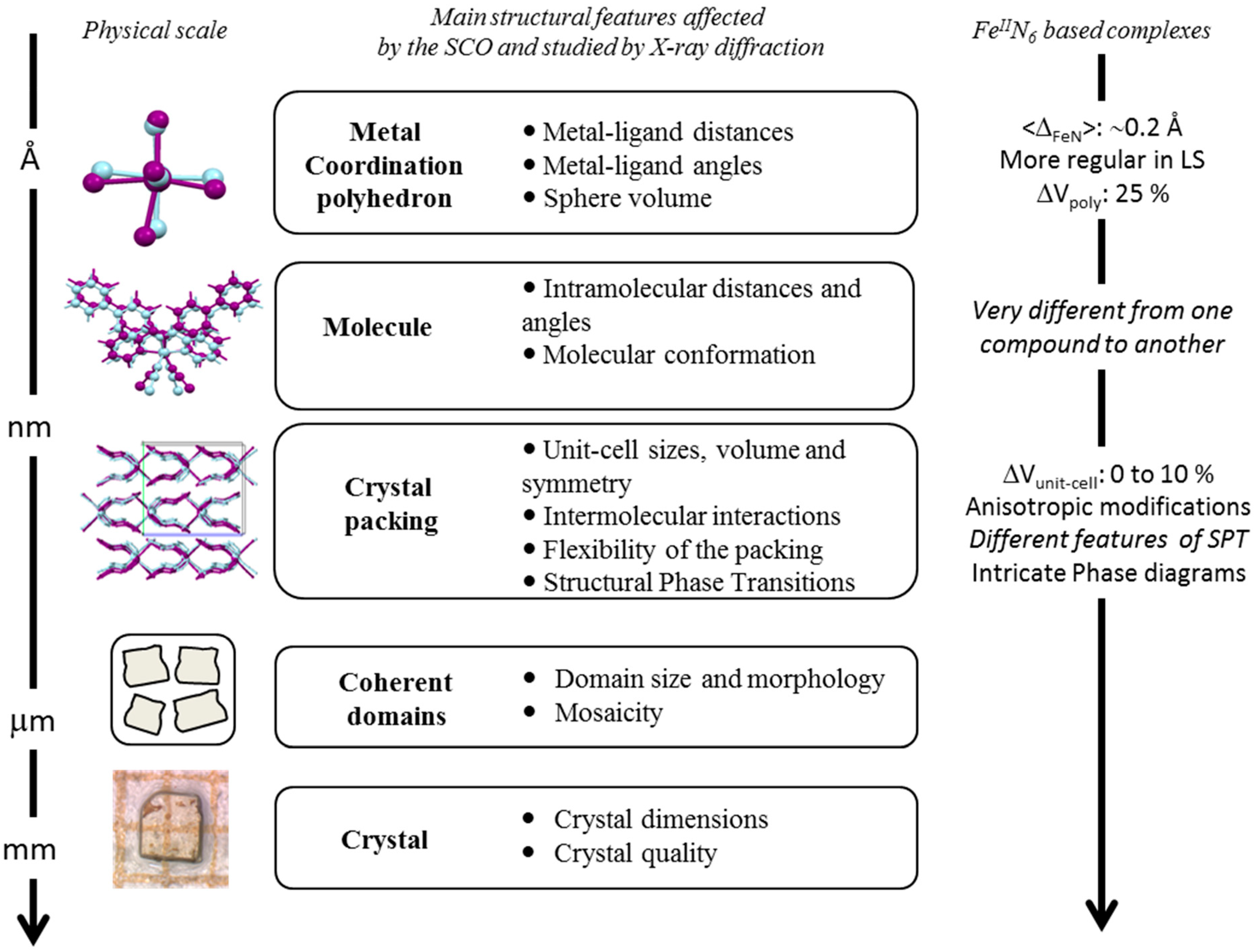
Most SCO compounds form extended organized solids, meaning that they crystallize in different space groups with long range atomic order. For instance the SCO compound [Fe(phen)
2(NCS)
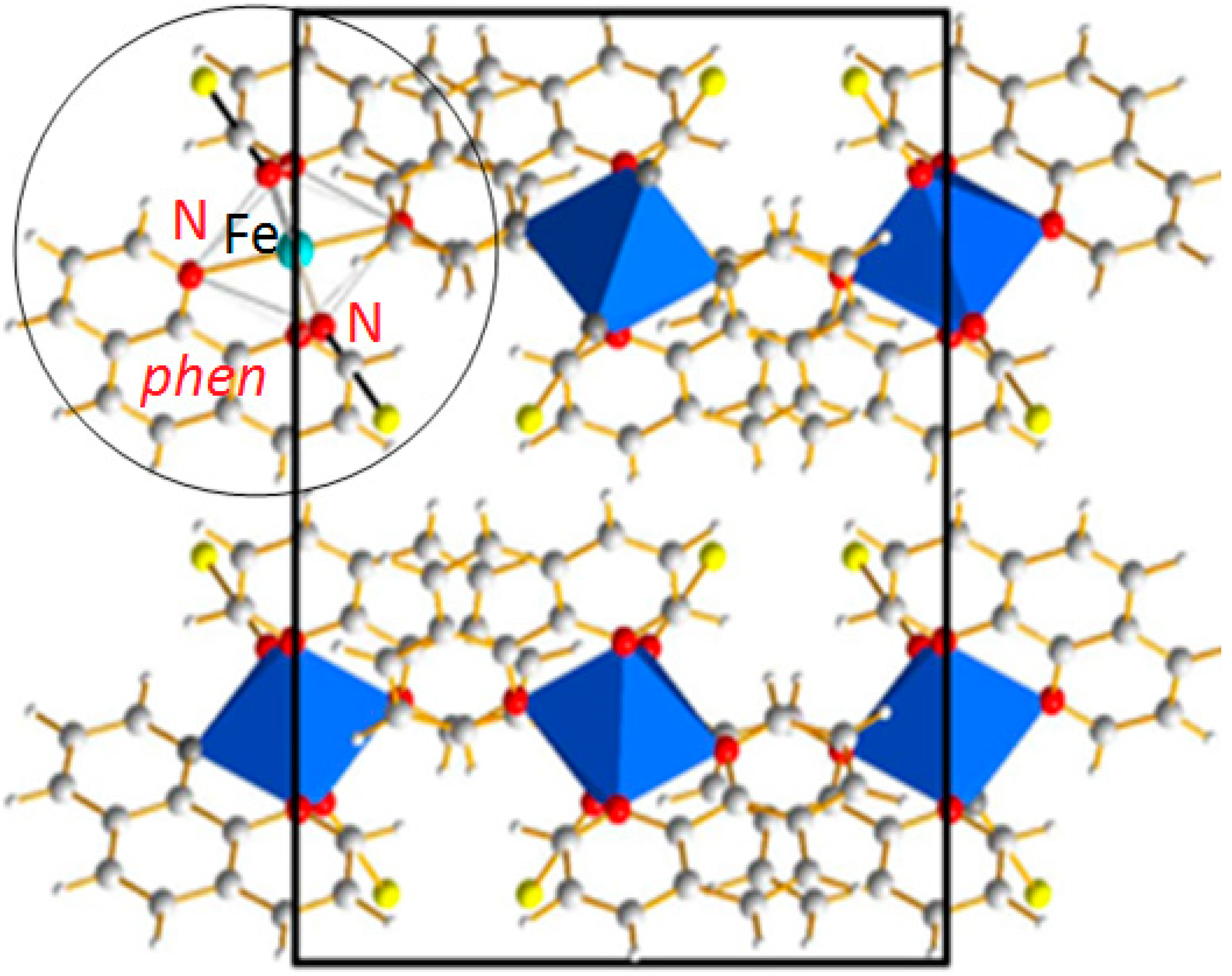
2] adopts an orthorhombic crystal system with four molecular complexes per unit cell. A projection of the crystal packing is given in
Figure 2. This compound is one of the most studied among the Fe(II) SCO complexes and is very often cited as the archetype for SCO materials. [Fe(phen)
2(NCS)
2] exhibits a first-order phase transition LS → HS reversible in the solid state. The magnetic and photo-magnetic properties reflect an abrupt thermal SCO with
T1/2 = 176 K at which equal HS and LS fractions are found and a narrow thermal hysteresis of Δ
T ~ 1 K [
19] and a complete light-induced SCO characterized by T (LIESST) = 62 K [
20]. The abrupt first-order phase transition associated to SCO phenomenon does not affect the symmetry of the crystal packing, being orthorhombic
Pbcn in both HS and LS, whatever the stimulus used to induce the SCO. At room temperature, the pressure-induced SCO occurs at about 0.6 GPa. The (P, T, light) phase diagram has been experimentally determined by means of variable temperature, pressure X-ray diffraction [
21] and photo crystallography [
22,
23]. Elsewhere this compound has even been used as the model compound to develop photo-crystallographic high-accuracy experimental protocols and, at this occasion, the need for DFT investigation was underlined [
24]. [Fe(phen)
2(NCS)
2] was also used to investigate the multiscale sequence of the structural modifications associated to the SCO as described above in
Section 3.1.
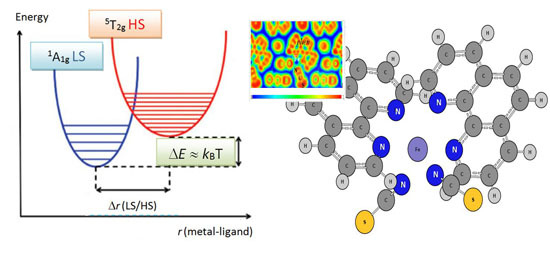
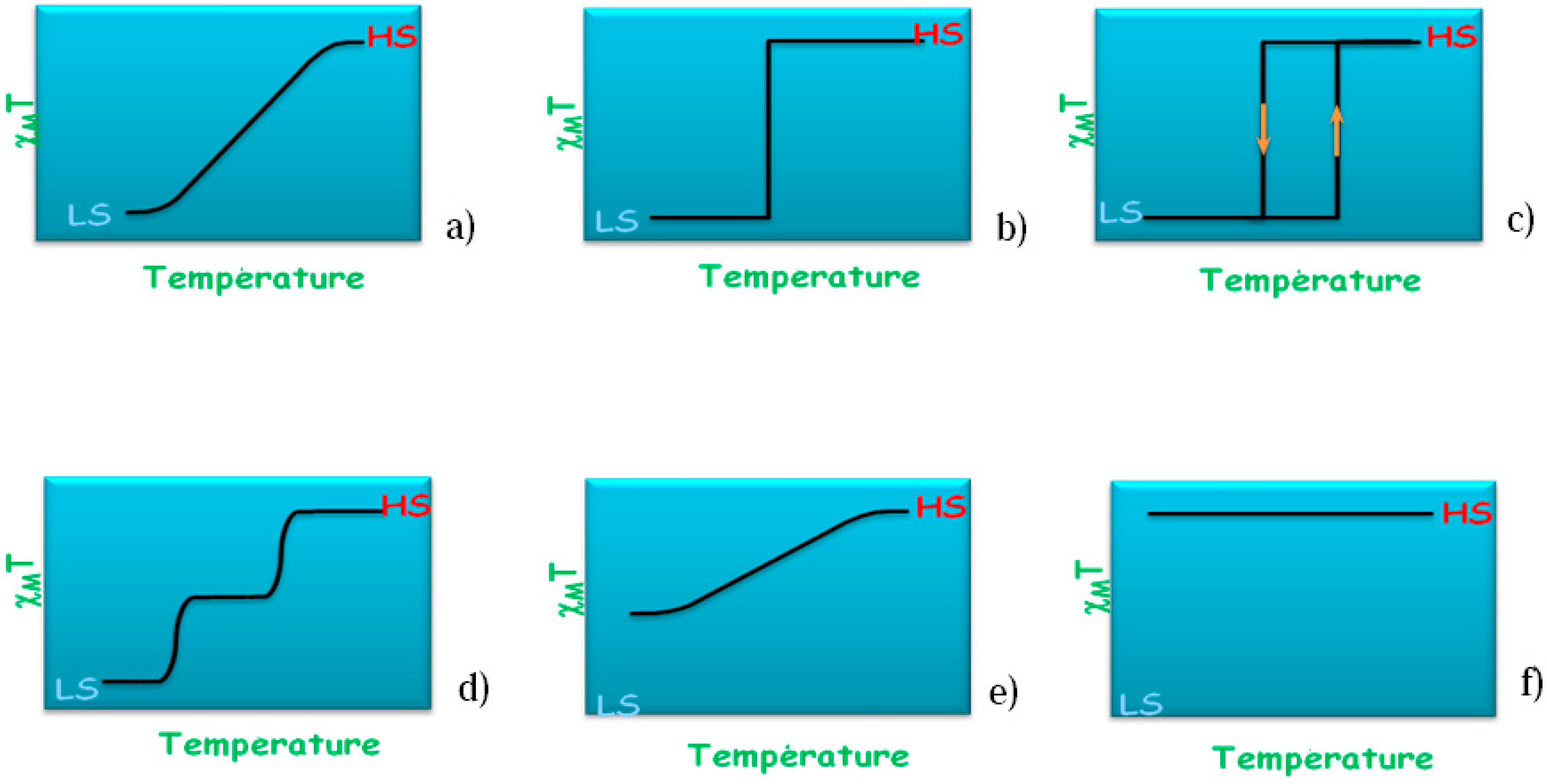
As stated above the thermal transition in SCO compounds can exhibit different behaviors. These scenarios are sketched in
Figure 3 where the magnetic susceptibility is plotted against temperature schematically. In the case of the [Fe(phen)
2(NCS)
2] SCO complex the behavior is abrupt with a small T hysteresis width as depicted in
Figure 3c. During the transition the average Fe–N distance changes from ~1.98 Å (LS) to ~2.20 Å (HS) and the jump between the two spin states occurs at the temperature of
T1/2 = 176 K.
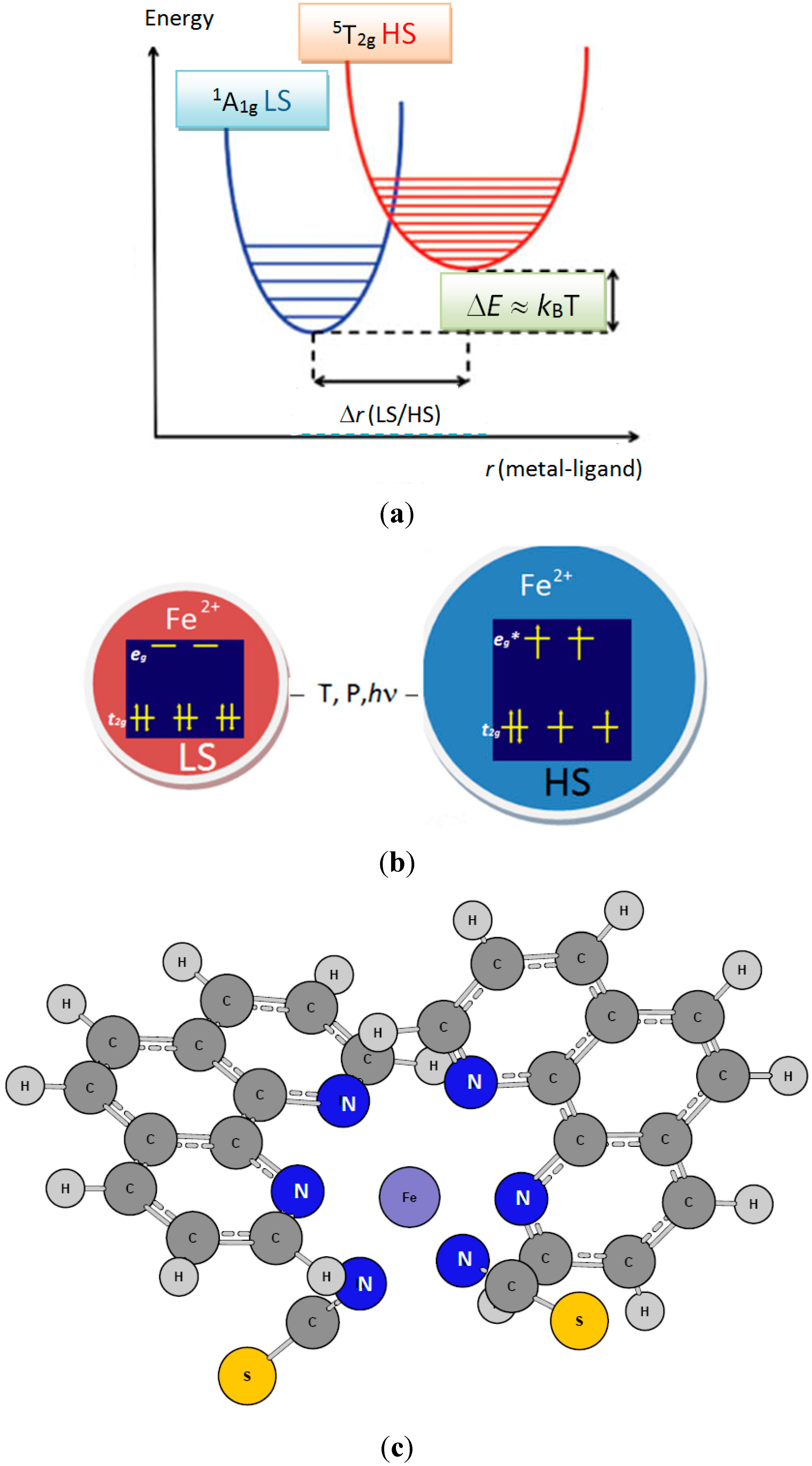
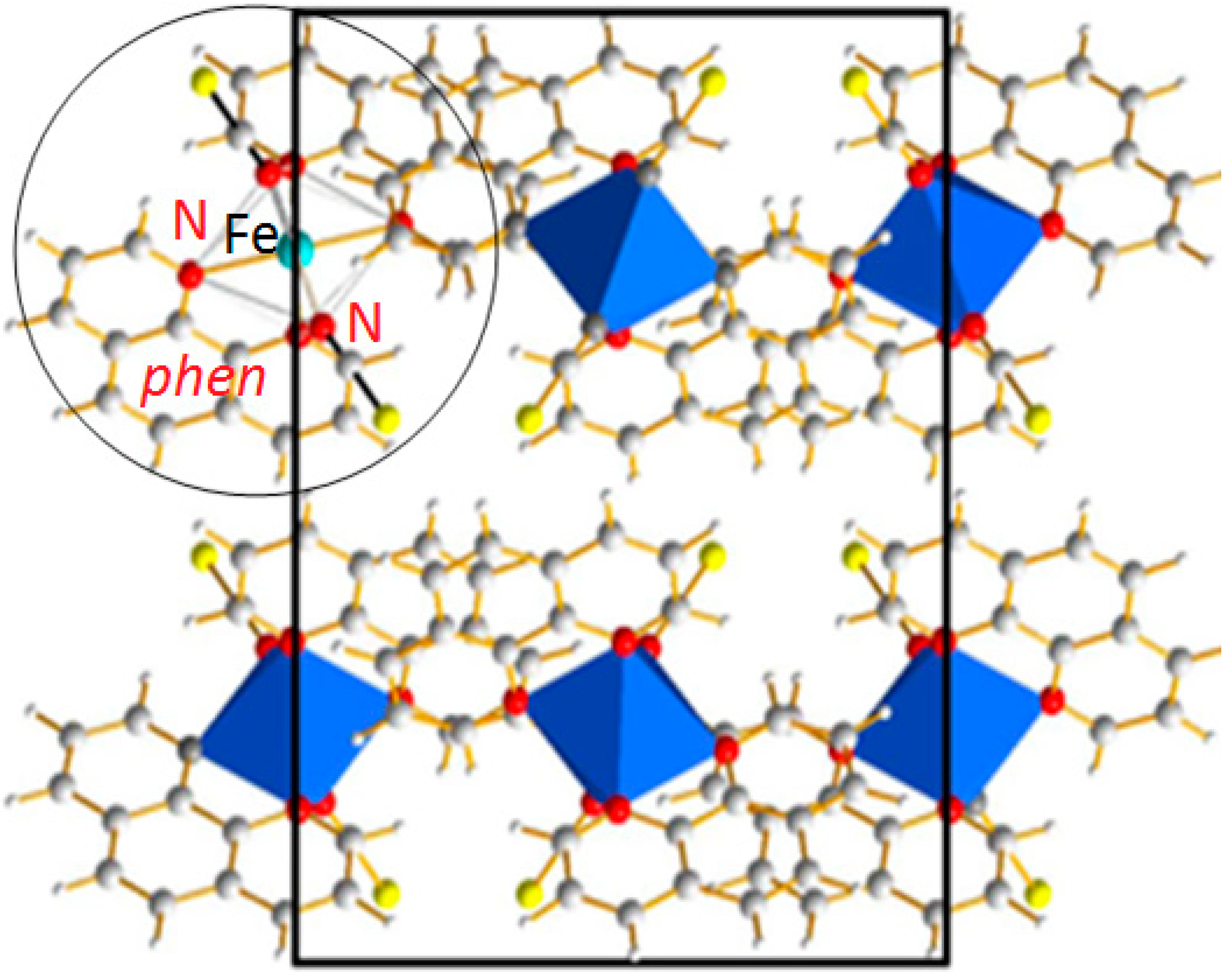
Figure 2.
[Fe(phen)
2(NCS)
2] SCO complex: Projection of the orthorhombic structure containing 4 formula units. The molecular entity as sketched in
Figure 1c is highlighted in the circle.
Figure 2.
[Fe(phen)
2(NCS)
2] SCO complex: Projection of the orthorhombic structure containing 4 formula units. The molecular entity as sketched in
Figure 1c is highlighted in the circle.
Figure 3.
Schematic temperature variation of molar magnetic susceptibility χM of a SCO compound showing the different types of thermal spin transitions encountered: (a) Gradual; (b) Abrupt; (c) Hysteretic; (d) Multistep; (e) Incomplete; and (f) No transition.
Figure 3.
Schematic temperature variation of molar magnetic susceptibility χM of a SCO compound showing the different types of thermal spin transitions encountered: (a) Gradual; (b) Abrupt; (c) Hysteretic; (d) Multistep; (e) Incomplete; and (f) No transition.
The photo-induced state is characterized by a short lifetime and is subjected to ultrafast photo-switching dynamics. In this context, it is necessary to address the basic mechanisms allowing light to photoswitch a spin-crossover molecular crystal from LS to HS. For this purpose a recent study of [Fe(phen)
2(NCS)
2] at T = 140 K (
i.e., in the LS state) was done with a femto-second (10
−15 s) laser pulse at λ = 650 nm leading to identify a LS → HS photo-switching through a metal-to-ligand charge transfer process (MLCT) [
25]. A two-step structural trapping occurs: Firstly molecular breathing vibrations are activated but rapidly damped when energy is partly and sequentially transferred to the molecular bending vibrations. This was identified based on time-dependent fast Fourier transform (FFT) of optical transmission showing the activation of a breathing mode at 113 cm
−1, calculated at 128 cm
−1 and a delayed activation of a torsion mode at 85 cm
−1, calculated at 91 cm
−1. For an illustration,
Figure 4 shows two snapshots of the breathing mode with the arrows indicating the remarkable motions of the phen ligands around central Fe. This is also accompanied by a twisting of the two NCS
− ligands.
Figure 4.
HS [Fe(phen)2(NCS)2]: Snapshots of the breathing mode of the molecule at 128 cm−1 reproducing the experimentally observed mode.
Figure 4.
HS [Fe(phen)2(NCS)2]: Snapshots of the breathing mode of the molecule at 128 cm−1 reproducing the experimentally observed mode.
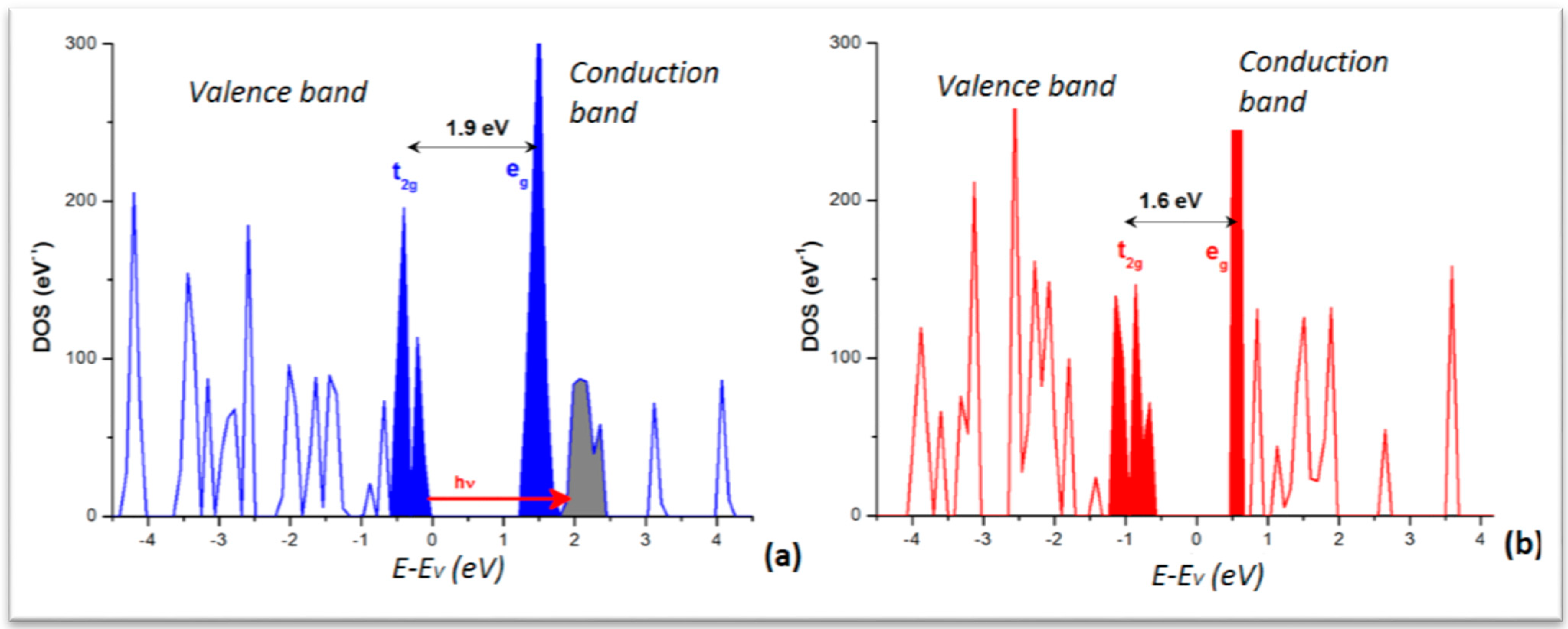
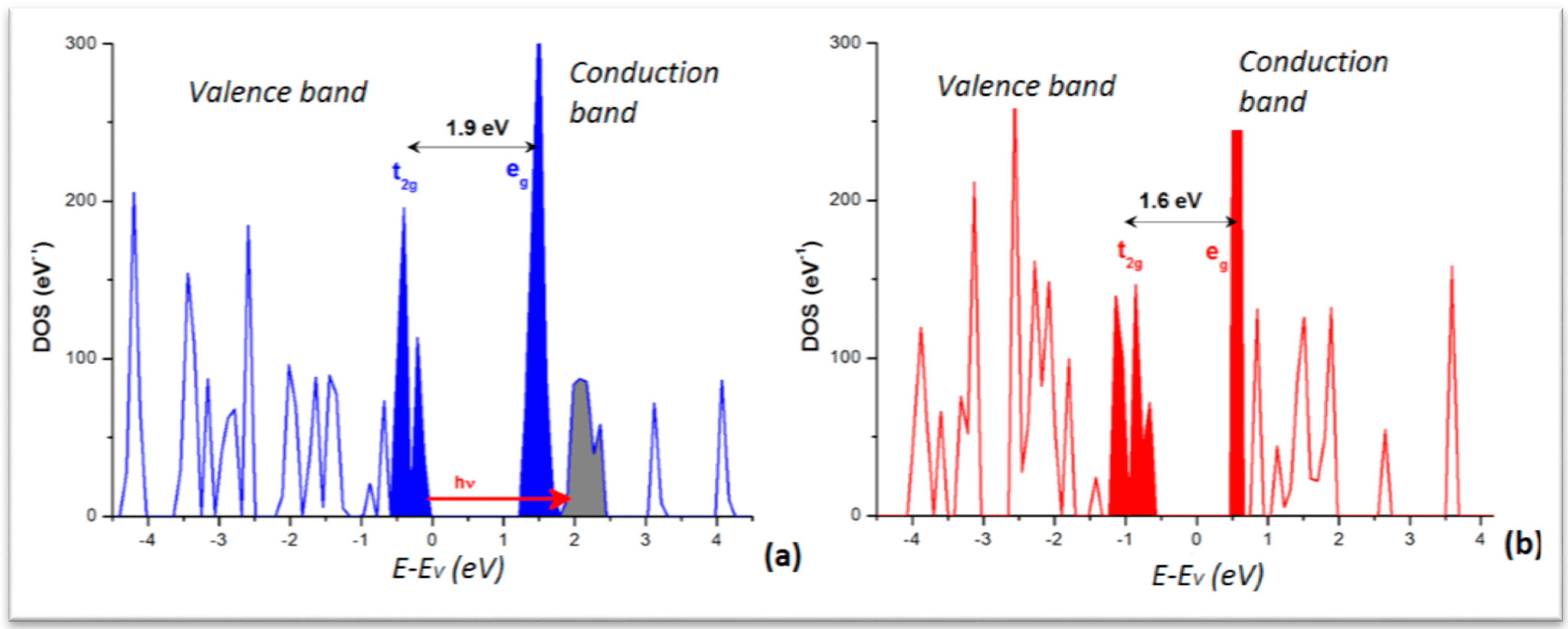
From the calculations in the extended crystal, the density of states (DOS) was obtained for the LS and HS phases. The plots are shown in
Figure 5 with the major feature of low dispersive and separated DOS peaks in both panels. This is significant of a molecular crystal, as opposed to metals or intermetallics characterized by broader and overlapping DOS. The lower part of the valence band (VB) is dominated by the ligand DOS whereas the VB top and conduction band (CB) bottom are dominated by the transition element Fe
d states split in the FeN
6 “nitrogenated” O
h field into t
2g- and e
g-like manifolds. For the sake of clarity they are shown in solid filled color peaks and separated by the band gap, larger for the LS (~1.9 eV) than for HS (~1.6 eV) as it can be inferred from the crystal field splitting, stronger for LS with empty e
g (t
2g6e
g0)
versus HS (t
2g4e
g2) with occupied e
g. Note that such gap magnitudes are not to be taken quantitatively in so far that the calculations within DFT regular functionals (local LDA and gradient GGA) do not provide accurate band gaps generally. Nevertheless the relevant information regarding the time-resolved optical studies is the correspondence of the DOS with the MLCT process pointed to above and shown here with the red arrow labeled “hν” going from the blue Fe DOS to the grey ligand’s. Also the DOS explain the stronger absorption of the HS state observed around 760 nm (1.6 eV) as being due to a decrease of the energy gap between t
2g and e
g bands as the molecular ligand field decreases. This narrowing of the gap, resulting from the longer Fe–N separation, occurs within ~140 fs and correlates well with the 170 fs elongation time obtained by X-ray absorption spectroscopy (XAS). Also in the course of the LS–HS transition, photo-excited singlet
1MLCT state (t
2g5e
g0L
1) and other potential intermediate states such as triplet ones as
3T
1, difficult to identify because of their short-lived lifetimes could not be excluded.
Figure 5.
Electronic Density of states (DOS) of [Fe(phen)2(NCS)2]: (a) LS; and (b) HS. Energy reference at the top of the valence band (EV), both varieties being insulators.
Figure 5.
Electronic Density of states (DOS) of [Fe(phen)2(NCS)2]: (a) LS; and (b) HS. Energy reference at the top of the valence band (EV), both varieties being insulators.
Lastly we point out another feature arising from the solid state calculations regarding total energies. From electronic structure calculations carried out for full geometry optimization of both LS and HS crystal varieties, we find the following total energies:
ELS = −1081.06 eV;
EHS = −1078.42 eV,
i.e., with a −2.637 eV/cell or ~ −0.7 eV/fu (formula unit) stabilization energy favoring the LS configuration system. This is interesting in view of the ∆
E ~ −0.95 eV/fu found from molecular calculations [
26]. The difference could have technical as well as fundamental origins. For the latter, lattice effects not accounted for in molecular calculations can be invoked. In this context it becomes interesting to see how the electrons are localized within the unit cell containing four formula units.
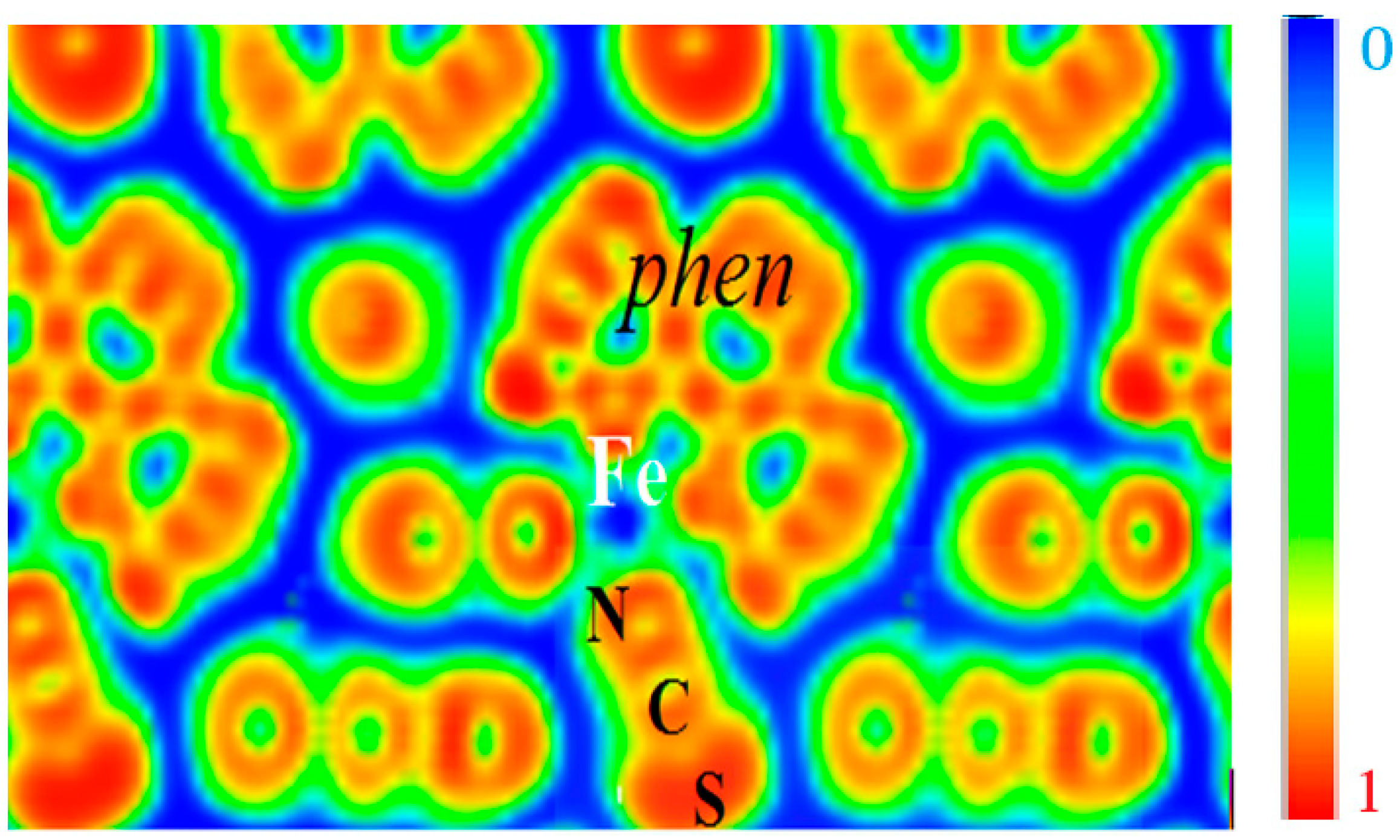
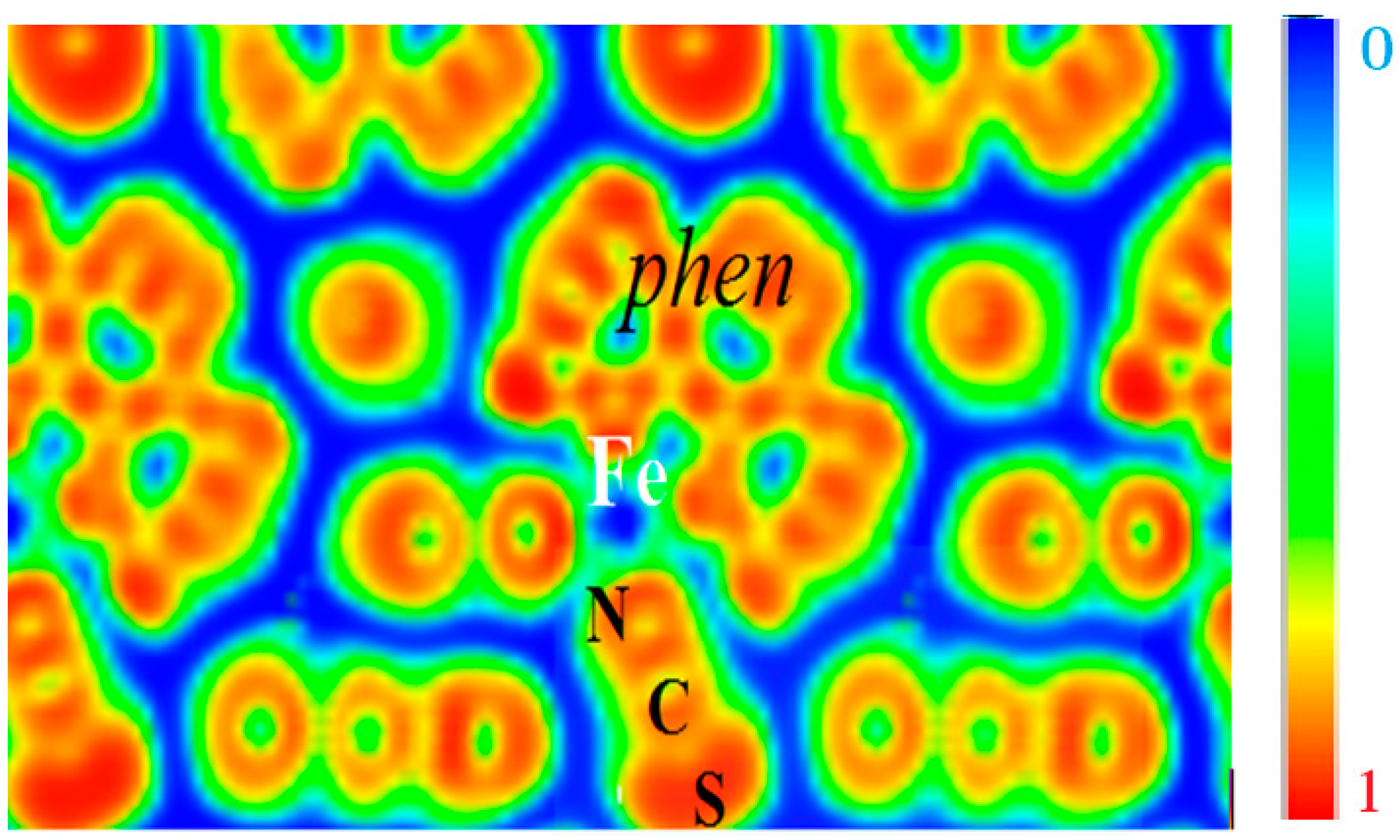
This can be visualized from the ELF contours given in
Figure 6 which can be seen to closely retrace the molecular entity of the complex with strong localization within the phen and NCS ligands, signaling the bonding within them. A marked feature is the presence of zero localization blue zones around the molecular entities, which allows the supposition that they are isolated from each other. Fe is centered on blue zero localization areas and surrounded by green free electron-like zones connecting with the
N-terminated ligands. These characteristics are indicative of its positive charge in agreement with expectations of Fe
2+ ion and of the different Fe–N bonding interactions.
Figure 6.
[Fe(phen)2(NCS)2] SCO complex: The molecular character in the solid state is illustrated by the electron localization function ELF slice dominated by intermolecular blue (zero localization) zones opposite to red intra-molecular zones with ELF = 1. Green areas correspond to free electron gas like medium localization (see text).
Figure 6.
[Fe(phen)2(NCS)2] SCO complex: The molecular character in the solid state is illustrated by the electron localization function ELF slice dominated by intermolecular blue (zero localization) zones opposite to red intra-molecular zones with ELF = 1. Green areas correspond to free electron gas like medium localization (see text).
3.2. From the Molecule to the Solid: Study of SCO [Fe(PM-BiA)2(NCS)2]
The [Fe(PM-BiA)
2(NCS)
2] compound (PM-BiA stands for
N-(2-pyridylmethylene)aminobiphenyl) was synthesized in 1997 by Létard
et al. [
27]. It is peculiar among the Fe(II) SCO complexes since it shows polymorphism. Initially, this compound was thought to replace [Fe(phen)
2(NCS)
2] as the SCO archetype material but the interplay it subsequently revealed between SCO and polymorphism prevented it to play this role [
28]. At room temperature, depending on the synthesis conditions, [Fe(PM-BIA)
2(NCS)
2] may crystallize in an orthorhombic
Pccn or a monoclinic
P2
1/
c space group. Both polymorphs, denoted respectively I and II (
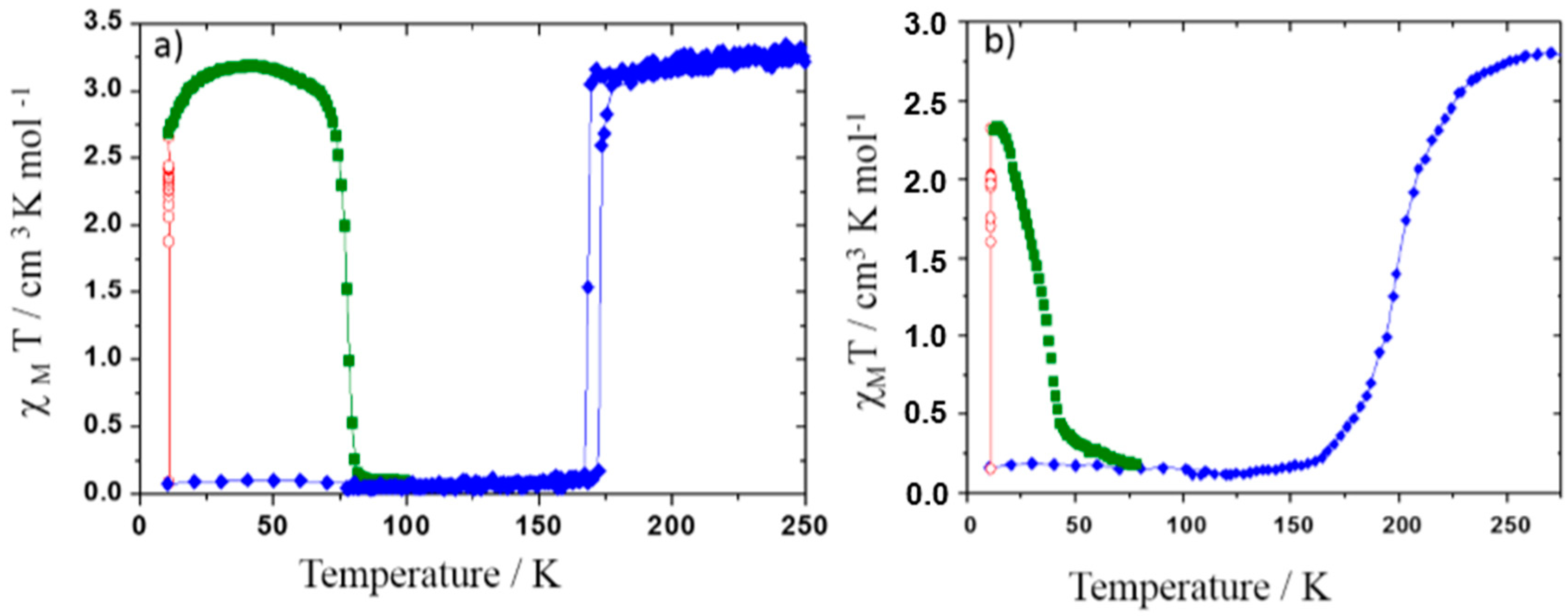
Figure 7), exhibit thermal SCO but the former undergoes an abrupt first-order spin transition while the latter undergoes a gradual SCO as shown in
Figure 7b. Magnetic and photo-magnetic behaviors as well as the structural studies of these polymorphs have been largely performed and related properties were compared [
29] allowing the tracking of the origin of their SCO feature differences. The latter is mainly explained by considerations on the crystal packing. More recently, preliminary experimental investigation of the (P, T) phase diagram by means of neutron diffraction have revealed that applying pressure to the orthorhombic polymorph induces a structural transition to the monoclinic polymorph [
30] leading to an intricate spin and structural phases diagram. As it is further shown below, molecular simulation has been used to determine the full (P, T) phase diagram, overcoming the high experimental difficulty to obtain reliable high pressure crystal structure at variable (low) temperature as well as its high cost [
31]. In addition, the theoretical investigation allows explaining the very unusual synergy between SCO and polymorphism.
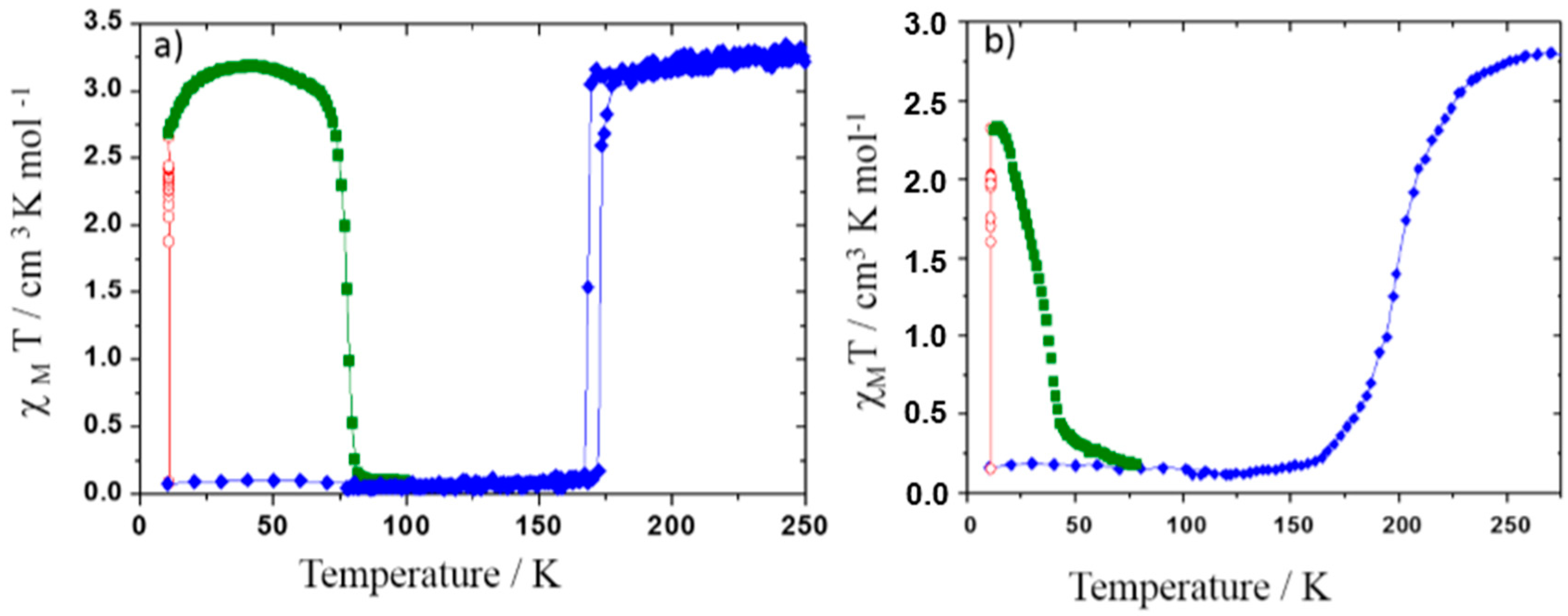
Figure 7.
Magnetic properties (molar susceptibility χM) function of temperature in the dark (blue diamond), under 830 nm irradiation (red open diamond) and in temperature in the dark after irradiation (dark green square) of phase I (a) and phase II (b) of [Fe(PM-BiA)2(NCS)2].
Figure 7.
Magnetic properties (molar susceptibility χM) function of temperature in the dark (blue diamond), under 830 nm irradiation (red open diamond) and in temperature in the dark after irradiation (dark green square) of phase I (a) and phase II (b) of [Fe(PM-BiA)2(NCS)2].
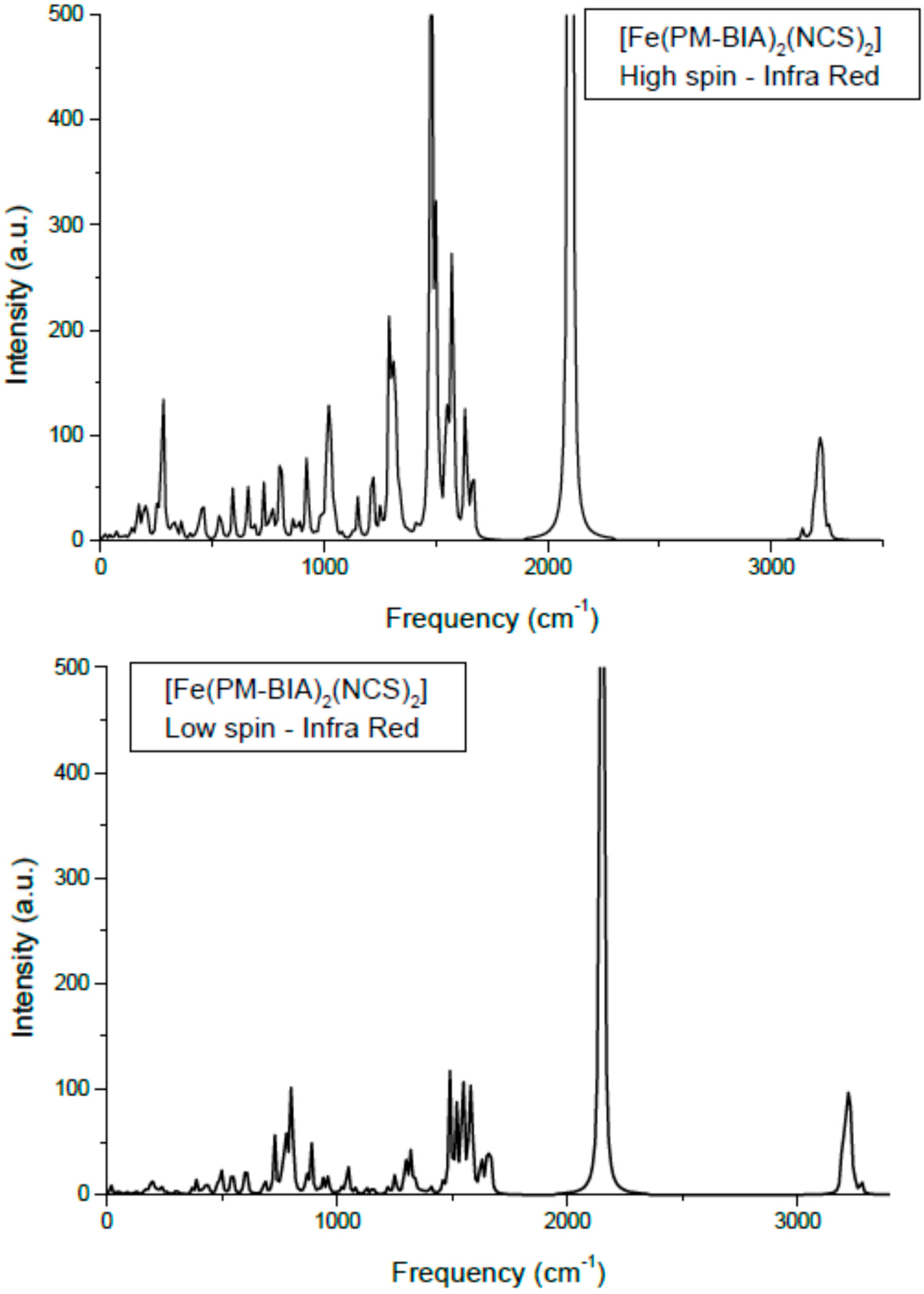
3.3. Vibrational Spectroscopy
One major signature of SCO complexes is their vibrational spectroscopy mainly through the infra red (IR) and Raman spectra [
32,
33,
34]. Using the hybrid functional B3LYP and an ECP (effective core potential) basis set, LanL2DZ, the molecular structures in LS and HS states are firstly geometrically optimized then the vibration modes are calculated based on the obtained minimum structure. Limiting our results to IR, we show in
Figure 8 the spectra for the low spin and the high spin states of phases. Also
Table 1 presents the main assignments together with a comparison between the calculated and the experimental frequencies [
35]. Although the two spectra show similar shapes and peak positions, the HS IR intensities are higher than LS ones, signaling higher activity in the former state. Also the absence of negative (imaginary) frequencies is a token of reliability for the calculations. The intensity values are relative to the highest value in the set (
i.e., follow the intensity of the NC in the NCS band at ~2100 cm
−1); therefore no reliable relationship to experimental intensities is allowed. Also no scaling was done since the main purpose was to assess the relative energy shifts of the band between HS and LS spin states.
Table 1.
[Fe(PM-BiA)2(NCS)2] experimental and calculated IR (infra red) frequencies.
Table 1.
[Fe(PM-BiA)2(NCS)2] experimental and calculated IR (infra red) frequencies.
| Infra Red ν (cm−1) | HS (High Spin) | LS (Low Spin) |
|---|
| Experiment | DFT | Experiment | DFT |
|---|
| Fe–N | 246 | 241 | 342 | 340 |
| 259 | 255 | 364 | 365 |
| 271 | 287 | 368 | 369 |
| δ(NCS) | 438 | 450 | 447 | 426 |
| 469 | 458 | 453 | 442 |
| 478 | 461 | 458 | 446 |
| C–S | Not determined | 775 | 809 | 768 |
| 779 | 771 |
| C–N | 2074 | 2030 | 2124 | 2123 |
| 2072 | 2133 |
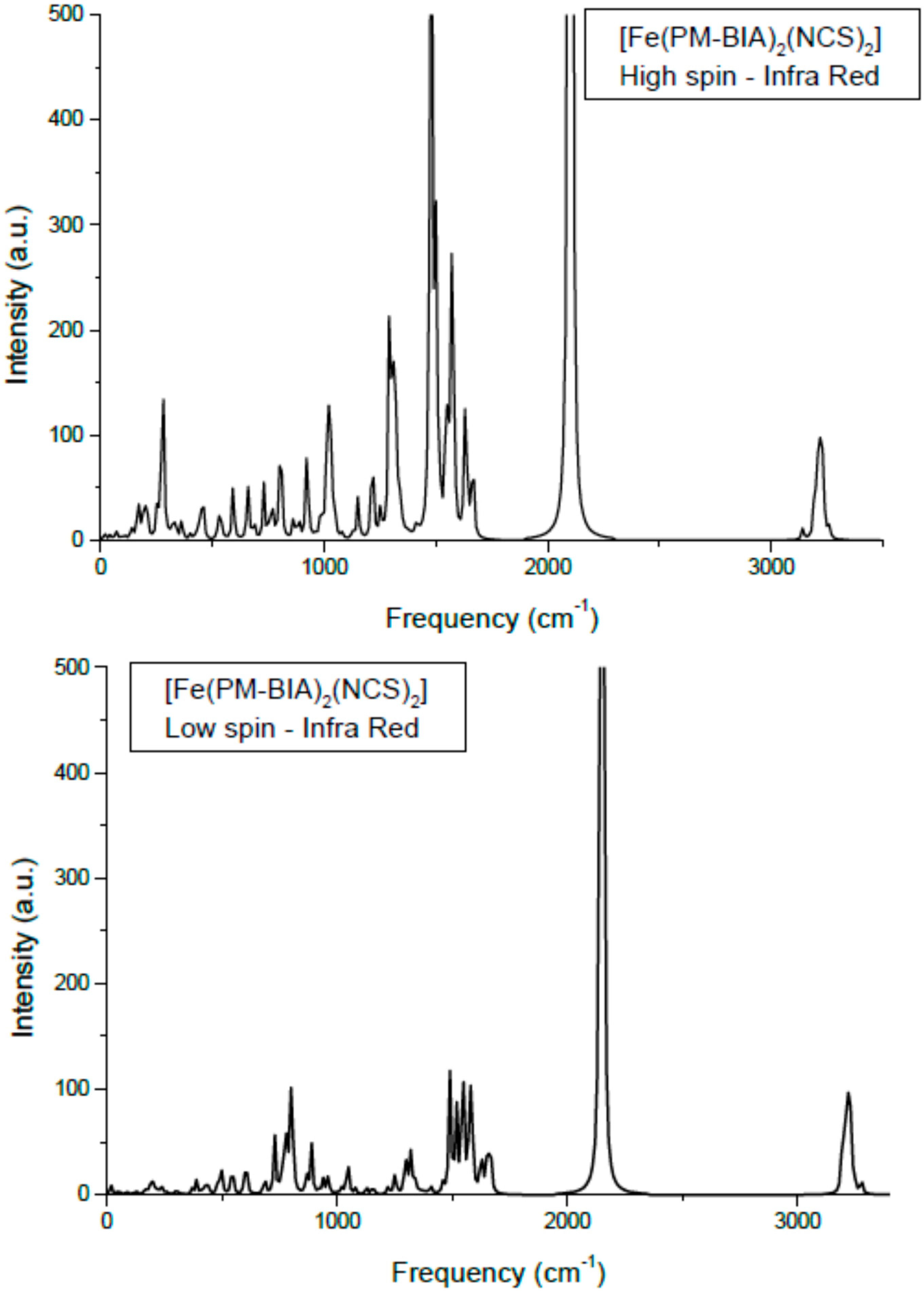
Figure 8.
Calculated Infra Red spectra of [Fe(PM-BiA)2(NCS)2] in the two spin states.
Figure 8.
Calculated Infra Red spectra of [Fe(PM-BiA)2(NCS)2] in the two spin states.
The assignments of the different modes: the lower frequency lying modes are the Fe–N stretching appearing at 220–270 cm−1 (HS) and 340–400 cm−1 (LS). This range corresponds to far-IR. The higher frequency (energy) arises from the average shorter Fe–N in LS (~1.95 Å) versus HS (~2.20 Å) states. Bending modes of NCS, δ(NCS) are found centered around 470 cm−1 and they show a small dependence with spin state. This is also observed for the C–S elongation around 800 cm−1. The intense peaks at ~2000 cm−1 are the usual signature of this class of SCO complexes as they are assigned to C–N symmetric and anti-symmetric stretching in the NCS ligand. They are found here at ~2070 cm−1 for HS and at higher frequency, ~2120 cm−1 for LS.
The calculated frequencies are in good agreement with experiments [
35,
36]. Some differences may appear due to the fact that the calculations were done on a single molecule; subsequently some missing lattice effects might lead to differences. However quantum mechanical calculations were difficult to achieve due to the huge number of atoms for a four formula unit cell, knowing the already large number of atoms in the molecule ~80. Lastly, we mention that Raman-calculated and experimental results showed similarly relative good agreement. Therefore the DFT-based molecular calculations provide a reliable enough description of the two spin states of a SCO complex, specifically the vibration spectroscopic ones and permit-extracting parameters for use in the following steps pertaining to the solid state investigations.
3.4. Semi-Classical Molecular Dynamics
Accounting for intermolecular interactions is necessary for an accurate description of the physcial behavior of an organometallic compound, although such interactions represent a small contribution to the total energy of the chemical system,
i.e., with respect to the contributions arising from the intramolecular forces of the isolated molecules. The intermolecular forces act at the macroscopic level; they mainly result from electrostatic and weak van der Waals interactions. Keeping in mind the difficulties of the purely quantum mechanical DFT approach in evaluating these forces [
31,
35], classical methods like molecular dynamics (MD) ones can be used. In MD methodology the weak van der Waals forces or hydrogen bonding may be modeled within a generalized atom-atom force-field. However this requires the preliminary knowledge of the atomic charges (
qi) and of the potential energy surfaces (PES) deduced from molecular quantum calculations within DFT with its hybrid functionals such as B3LYP [
12,
13]. The results displayed in
Table 2 allow evaluating the force-field parameters. The concomitant combination of DFT-based calculations with molecular dynamics complementarily allows defining an original methodology we call “semi-classical molecular dynamics” (SC-MD).
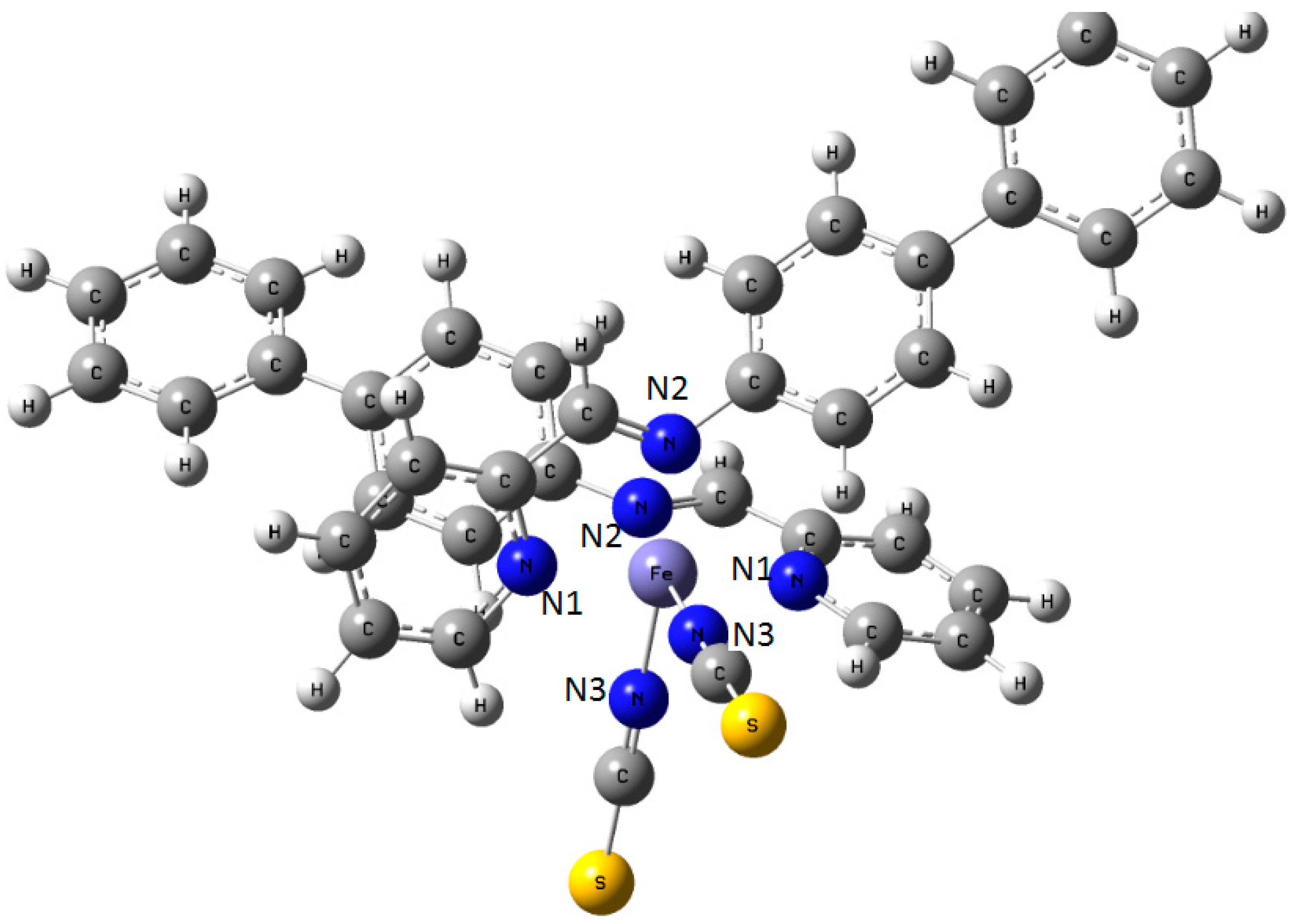
Table 2.
[Fe(PM-BiA)
2(NCS)
2]: Calculated Mulliken charges
q of the atoms forming the
Fe–N6 octahedron (in
e units). Atom labels as in
Figure 9.
Table 2.
[Fe(PM-BiA)2(NCS)2]: Calculated Mulliken charges q of the atoms forming the Fe–N6 octahedron (in e units). Atom labels as in Figure 9.
| Atomic Species | q (LS) | q (HS) |
|---|
| Fe | 1.5099 | 1.0442 |
| N1 | −0.7245 | −0.7662 |
| N2 | −0.7678 | −0.6936 |
| N3 | −0.7567 | −0.7326 |
Practically, in the classical approach the intramolecular force field includes electrostatic forces between atomic charges, two-body stretching elongations, three-body (3 atoms) bending forces, as well as dihedral interactions involving four centers. Using the hybrid B3LYP functional, with effective core potential basis set ECP-LANL2DZ, the resulting optimized molecular structure and point charges allow generating the PES. The curve is then fitted with Morse potential
VFe–N and harmonic
VFe–C–N potentials, by varying alternatively one of the three Fe–N
i distances of the molecule. The Morse expression of short range potentials is written as:
where
Eij is the potential depth, ρ
ij the electronic hardness and
rij the equilibrium distance between two
i and
j species. Following other studies a constant values of ρ
ij = 1.33 Å
−1 was used independently from the nature of
Ni and of the spin state. The three types of nitrogen (
i = 3) are depicted for the molecular entity in
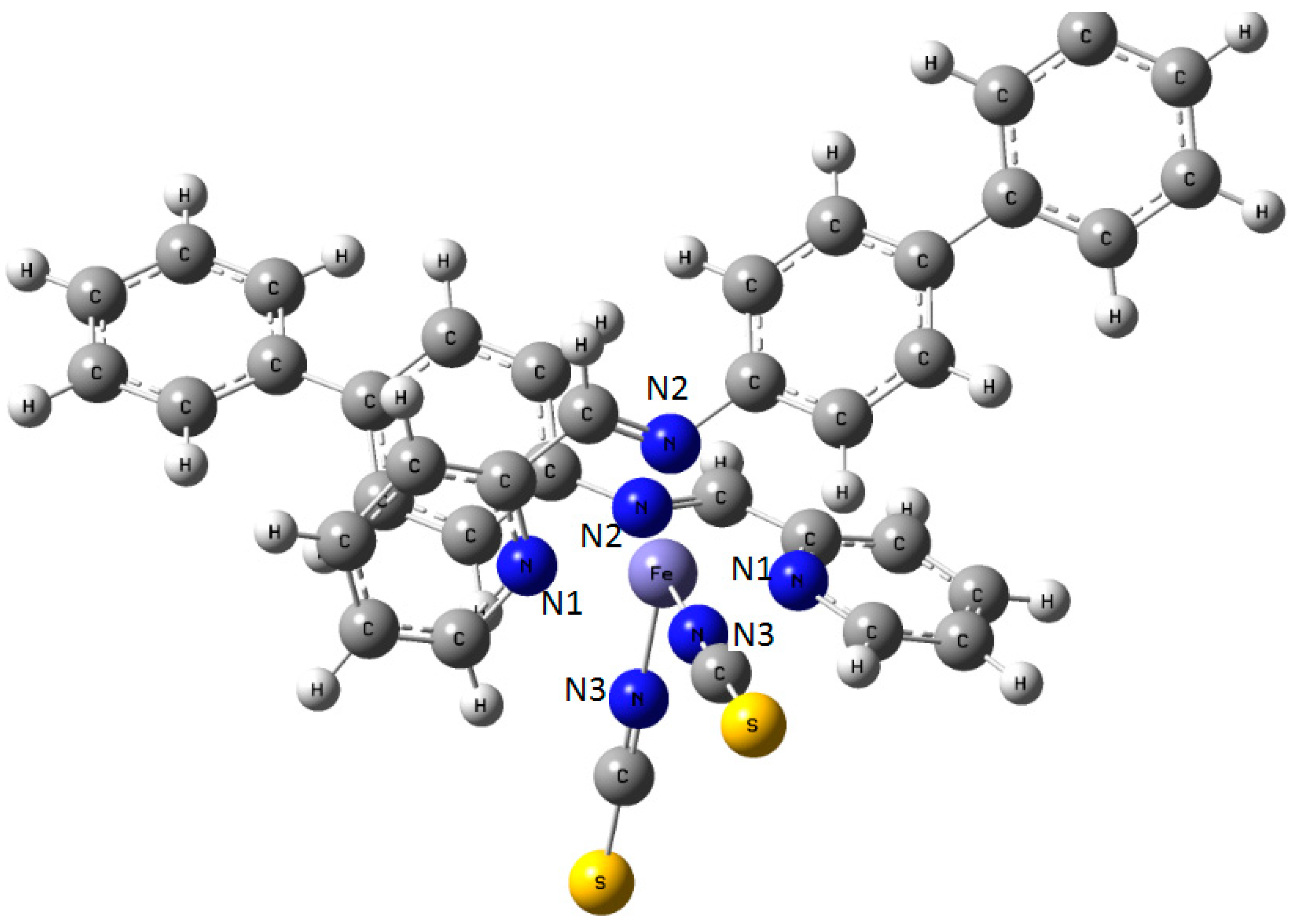
Figure 9. In fact, using a constant value of ρ
ij is generally adopted in classical force field studies even if the coordination polyhedra around a specific atom
i are of different types and strongly distorted [
31].
Figure 9.
Sketch of the molecule of [Fe(PM-BiA)2(NCS)2] SCO compound with the labeling of the three different nitrogen atoms in the coordination sphere of central Fe.
Figure 9.
Sketch of the molecule of [Fe(PM-BiA)2(NCS)2] SCO compound with the labeling of the three different nitrogen atoms in the coordination sphere of central Fe.
In a second step, MD simulations are performed including the intramolecular interactions at the level of the molecule. The parameters are adjusted with respect to the molecular geometry and the spectroscopic properties by using the DL_POLY MD code [
37] which yields a molecular field for the HS and LS spin states.
To finalize, the generalized force fields are applied to the complex in its extended crystal lattice: After a minimization step at ΔT = 1 K for both spin states, MD simulations are performed stepwise up to 300 K; the same protocol is followed for the cooling procedure. Best van der Waals parameters are searched for, in order to reproduce structures both in the LS state (25 and 140 K) and HS state (298 K) [
38]. With these final intermolecular force fields, the complete set of runs allow evaluating respectively
T1/2 ↑ and
T1/2 ↓ and transition enthalpy Δ
H (LS → HS) at
T1/2, the so-called Δ
H1/2.
T1/2 and Δ
H1/2 are related, as shown in a phenomenological thermodynamics view, by modeling the LS-HS domain mixture as a regular solid-solution. This domain mixture arising along the SCO, typical from a first-order phase transition in a crystal, has been observed by means of variable temperature X-ray Diffraction (XRD) [
29].
As a result of the above protocols applied to the [Fe(PM-BiA)
2(NCS)
2] complex, the fitted potentials from quantum
Gaussian09 calculations combined with van der Waals interactions, allow the acquisition of the vibration modes (I.R. and Raman) of the molecule in its two spin states by molecular simulation (
Figure 8). Particularly we found, in agreement with the experiment [
30], that the
Fe–N6 octahedron is more distorted in the high-spin state than in the low-spin one, as illustrated by the distance changes: Δ
dHS = 0.21 Å much larger than Δ
dLS = 0.03 Å as well as for the angles extremes: Δθ
HS = 26.4° > Δθ
LS = 13.7°.
Transferring the obtained fields to the solid structure, the simulations provide the inputs applied to the orthorhombic crystal lattice. Intermolecular interactions show evidence of hydrogen bonding between the NCS ligand at the sulfur end atom with nearest neighbors hydrogen belonging to the aromatic cycles. This feature has been noted as one of the most specific ones of the derived complexes family of general formula [Fe(PM-L)2(NCS)2] where L is an aromatic ligand. The evolution of the structure with temperature shows that the LS → HS transition occurs at T1/2 ↑ = 120 K which is 50 K lower than the experimental value. The change of the transition enthalpy, ΔMDH1/2 = 14 ± 2 kJ/mol corresponds to the transition temperature T1/2 = 150 K. Experimental measurements give ΔHLS → HS ~11 kJ/mol. This allows casting confidence on the calculation procedure. We note however that the departure from the experimental value is likely due to the drawbacks of using a semi-classical model in view of the probable prevalence of quantum effects below 100 K.
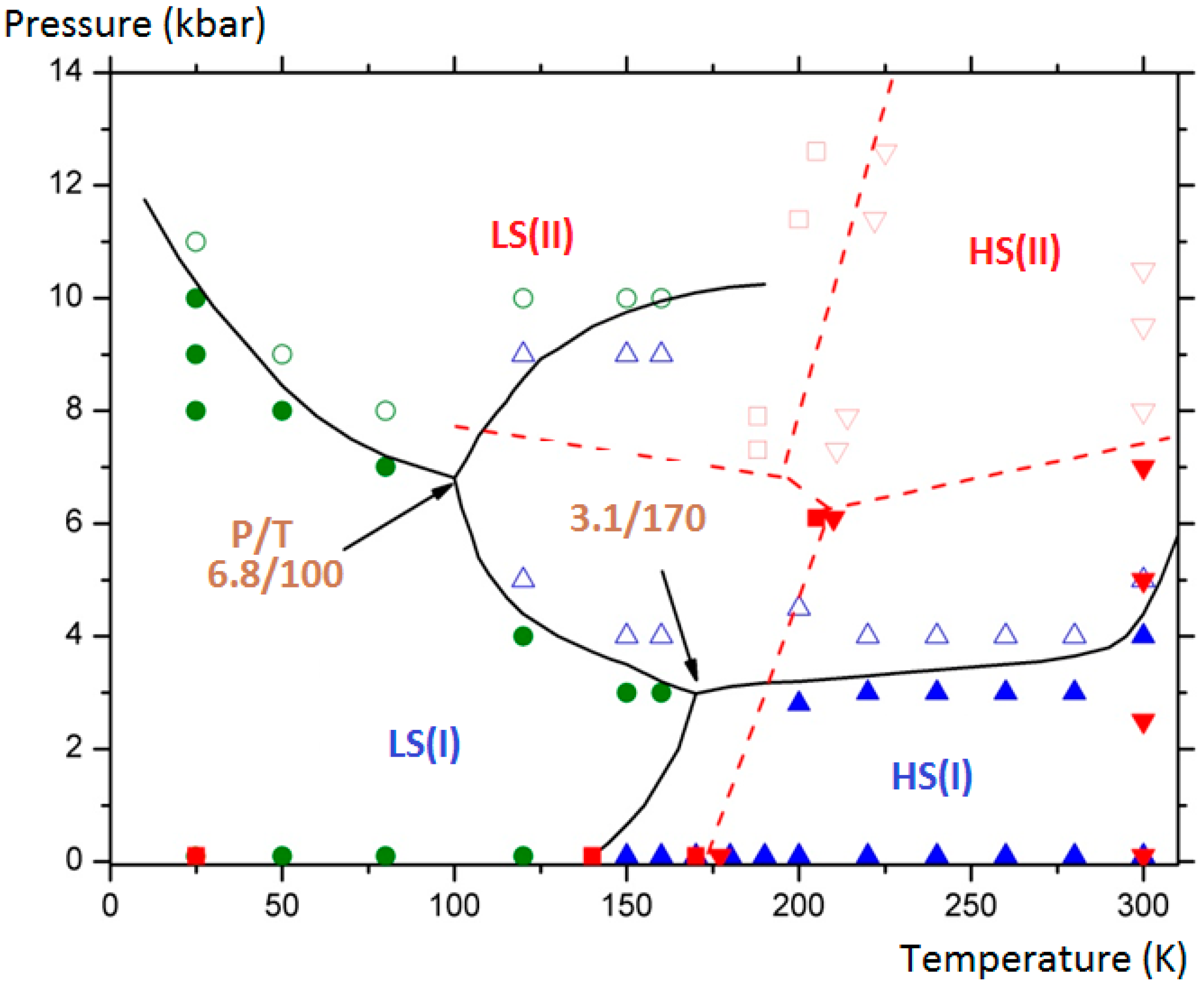
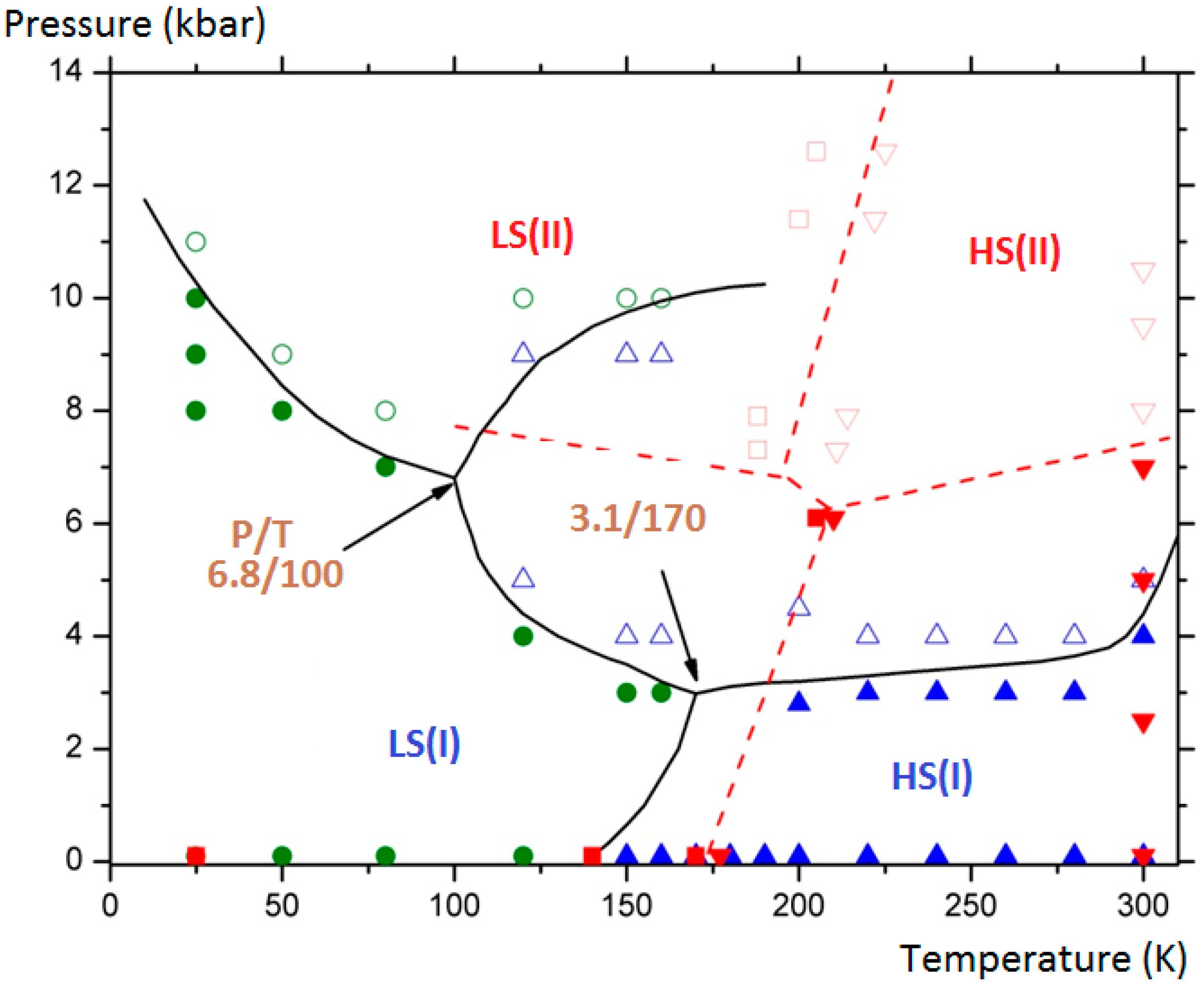
The final aim of the calculations was to assess and complete the experimental (P, T) phase diagram of the compound by showing the domains of existence of the monoclinic polymorph (II) in its two spin states (LS/HS). Furthermore the delimitation of the different domains allowed generating two triple points. The results are depicted in
Figure 10 where the connected lines are drawn as a guide for the eye: for LS (I), HS (I) and HS (II) equilibrium: at T = 170 K and P = 3.1 kbar; for LS (I), HS (I) and HS (II) equilibrium, at T = 100 K and P = 6.8 kbar.
Figure 10.
[Fe(PM-BiA)
2(NCS)
2]: (P, T) phase diagram showing four solid phases: LS (I): filled green circles; HS (I): filled blue triangles; LS (II): empty green circles; HS (II): empty blue triangles). The domains delimitated by straight curves generate two triple points. Experimental points from [
30] are presented for comparison (LS (I): filled red squares; HS (I): filled red down triangles; LS (II): empty red square; HS (II): empty red up triangles).
Figure 10.
[Fe(PM-BiA)
2(NCS)
2]: (P, T) phase diagram showing four solid phases: LS (I): filled green circles; HS (I): filled blue triangles; LS (II): empty green circles; HS (II): empty blue triangles). The domains delimitated by straight curves generate two triple points. Experimental points from [
30] are presented for comparison (LS (I): filled red squares; HS (I): filled red down triangles; LS (II): empty red square; HS (II): empty red up triangles).
It needs to be stressed that subsequent experimental investigations at low temperature under high pressure by means of neutron diffraction have confirmed the predicted phase diagram (
Figure 10). Indeed the possible HS (I) to LS (II) and HS (II) to LS (I) switching by applying high pressure at variable temperature was demonstrated. Experimental difficulties to extract accurate structural data as well as an accurate determination of the effective sample temperature have prevented covering the whole (P, T) phase diagram. The molecular simulations, once validated by some experimental points as performed here, appear to be a predominant tool to investigate the full phase diagrams. We also note that the behavior of this sample is probably even more complicated than described here since diffuse reflectance investigations under high pressure have revealed that a third phase could be obtained at moderated pressure. The localization of this new phase is not clearly assigned either to the surface solely or to the whole bulk material. Comparative investigations of surface and volume behaviors are still a challenge in both the multiscale experimental and theoretical approaches.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}