
Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach

Abstract
:
1. Introduction
2. Results and Discussion
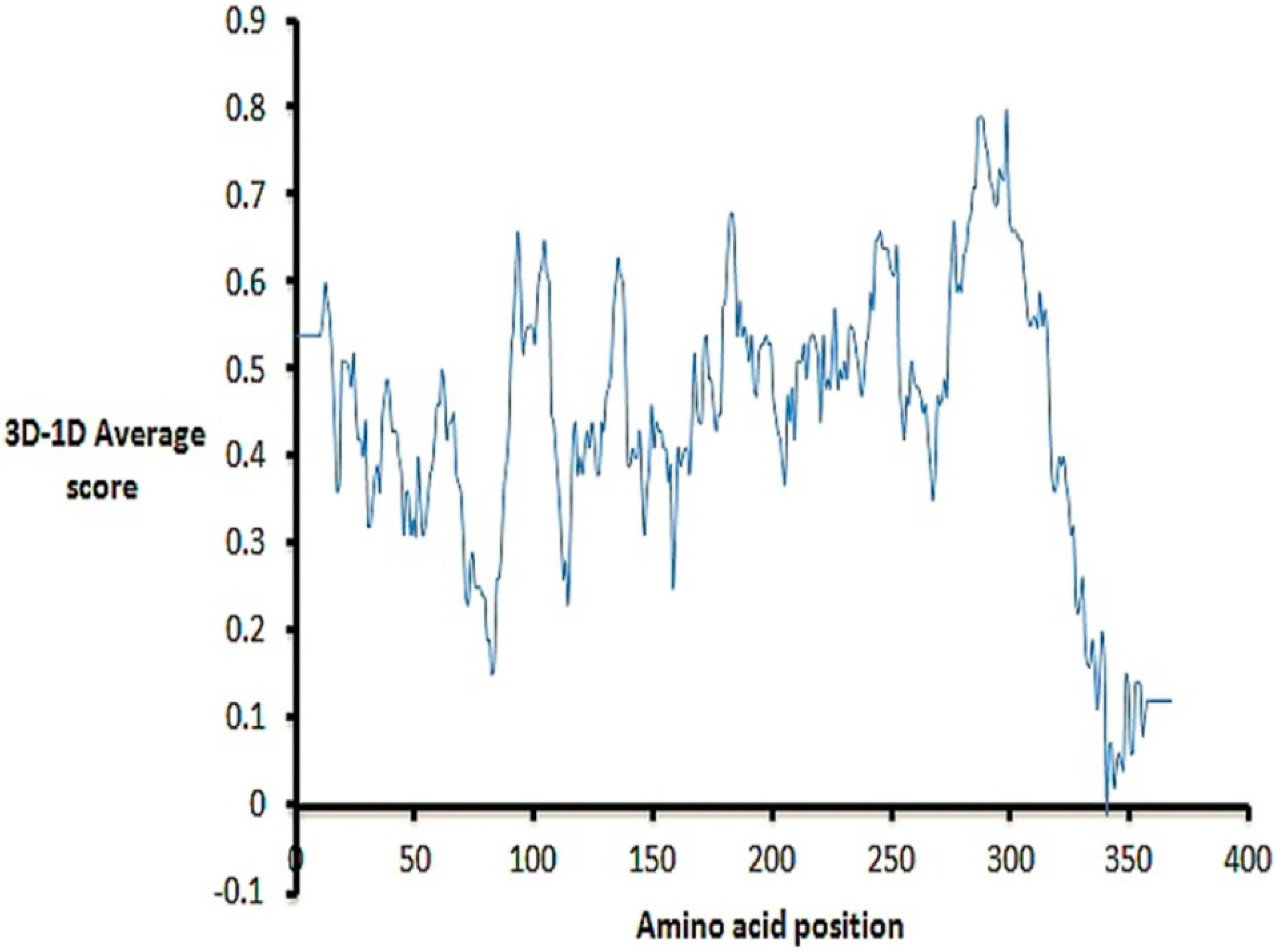
2.1. Model Building and Refinement
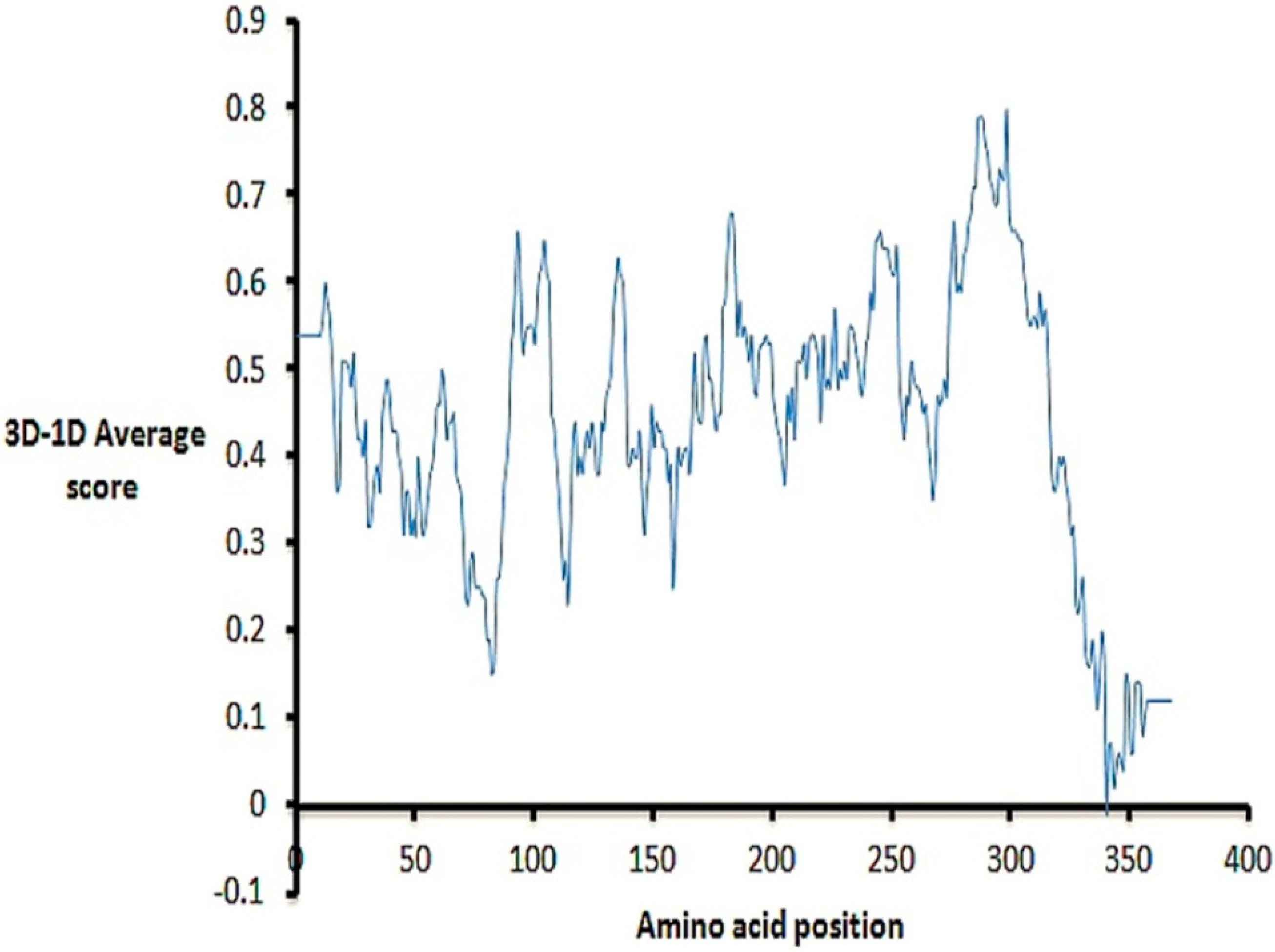
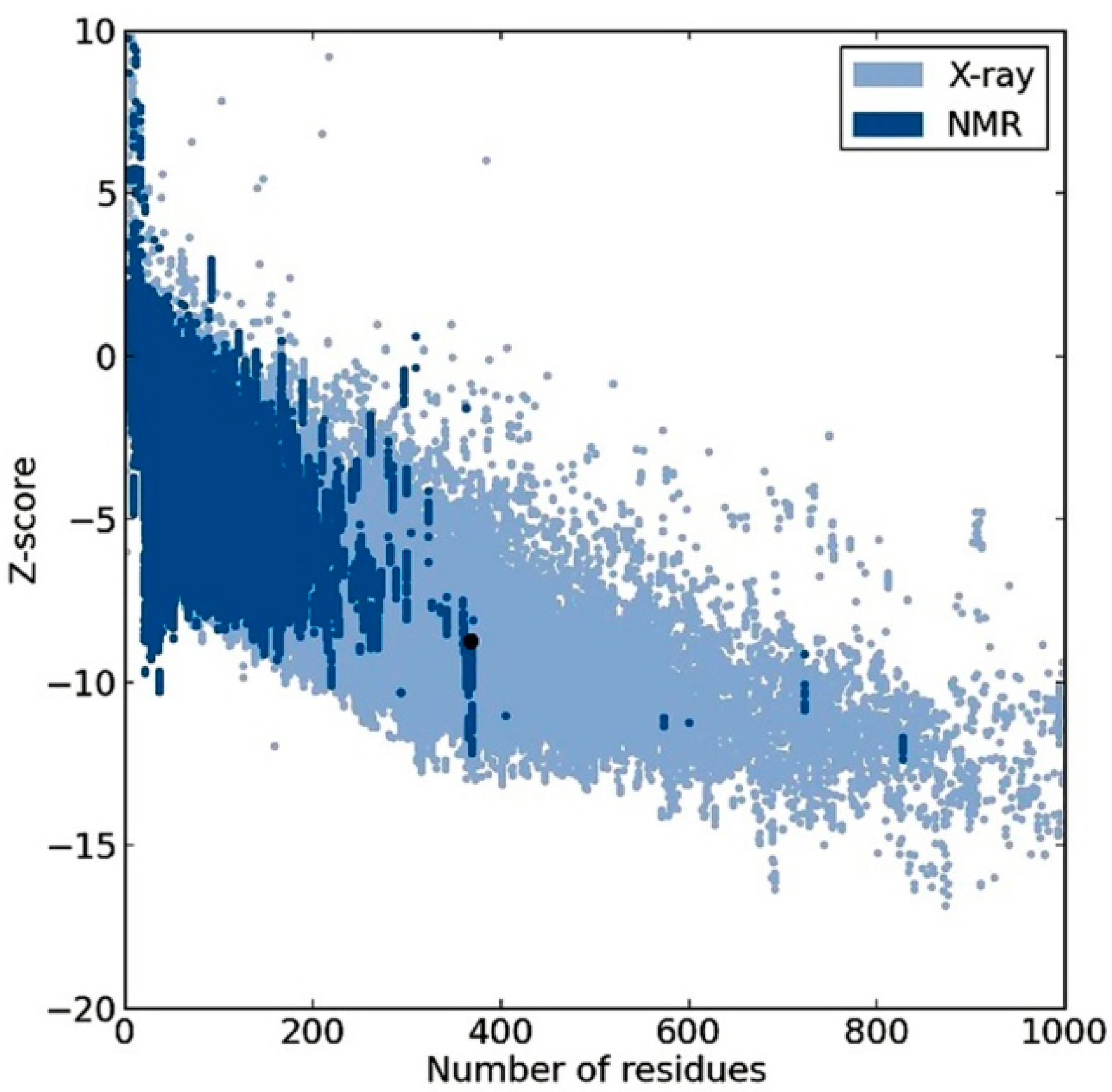
2.2. Model Quality
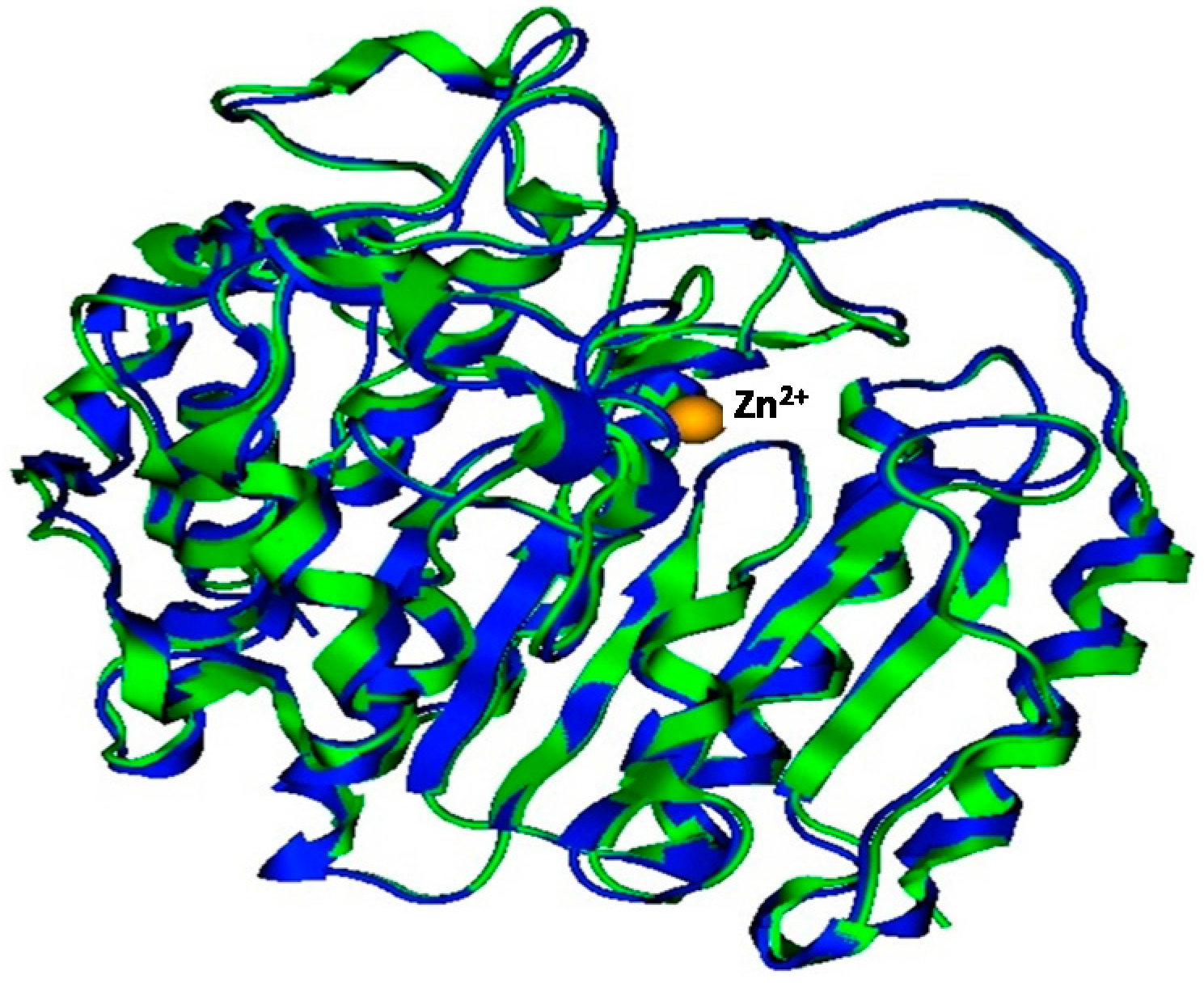
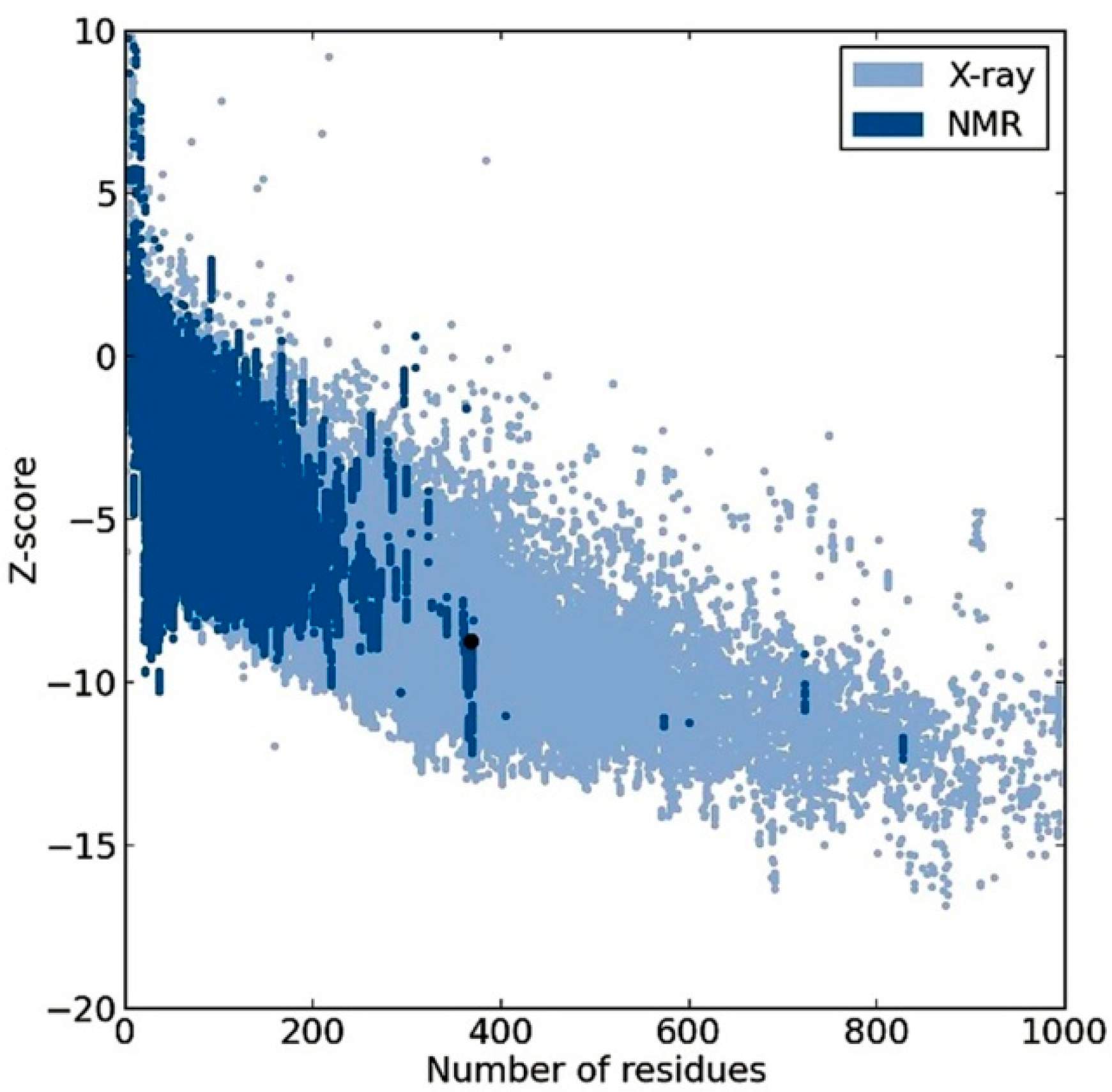
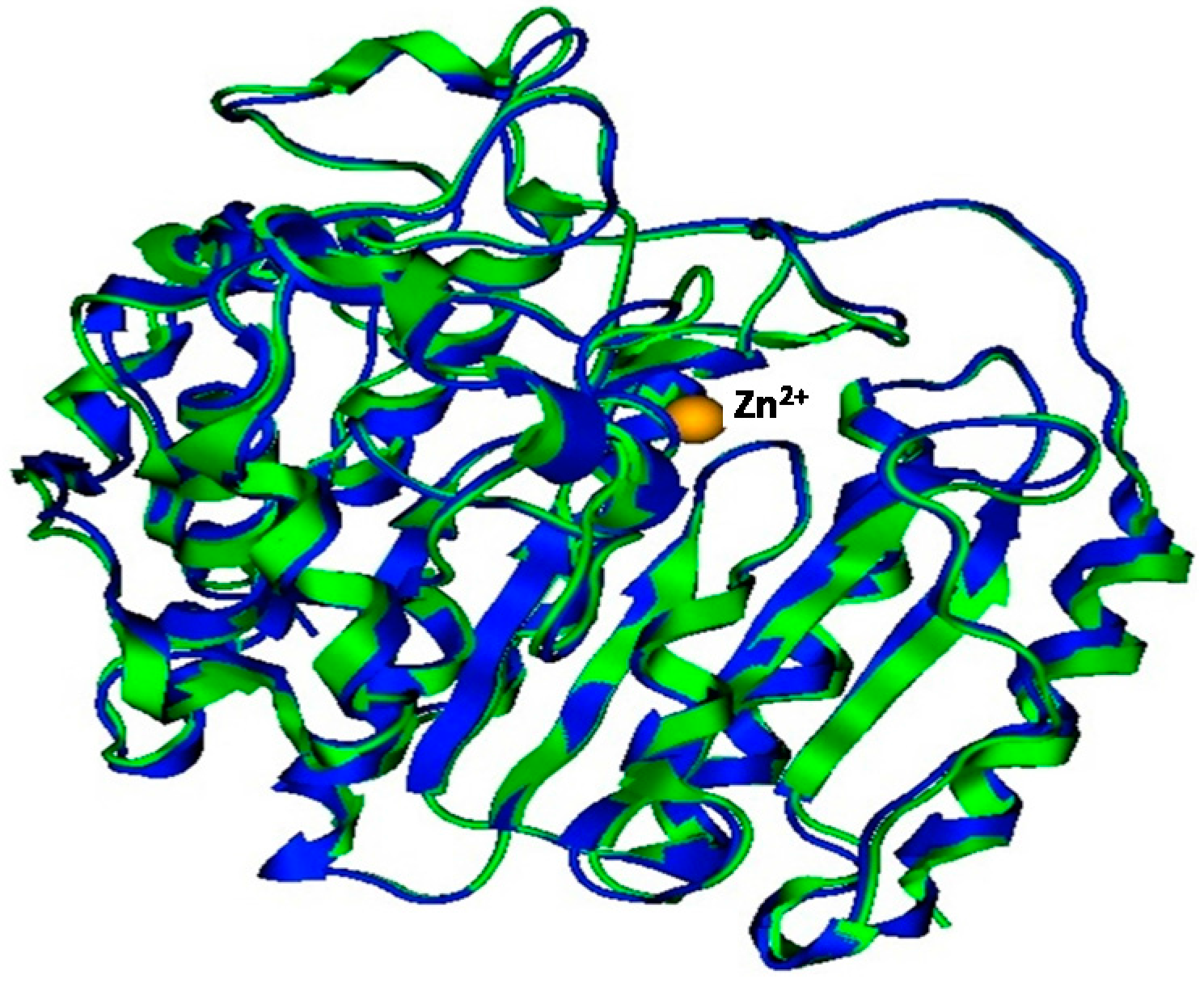
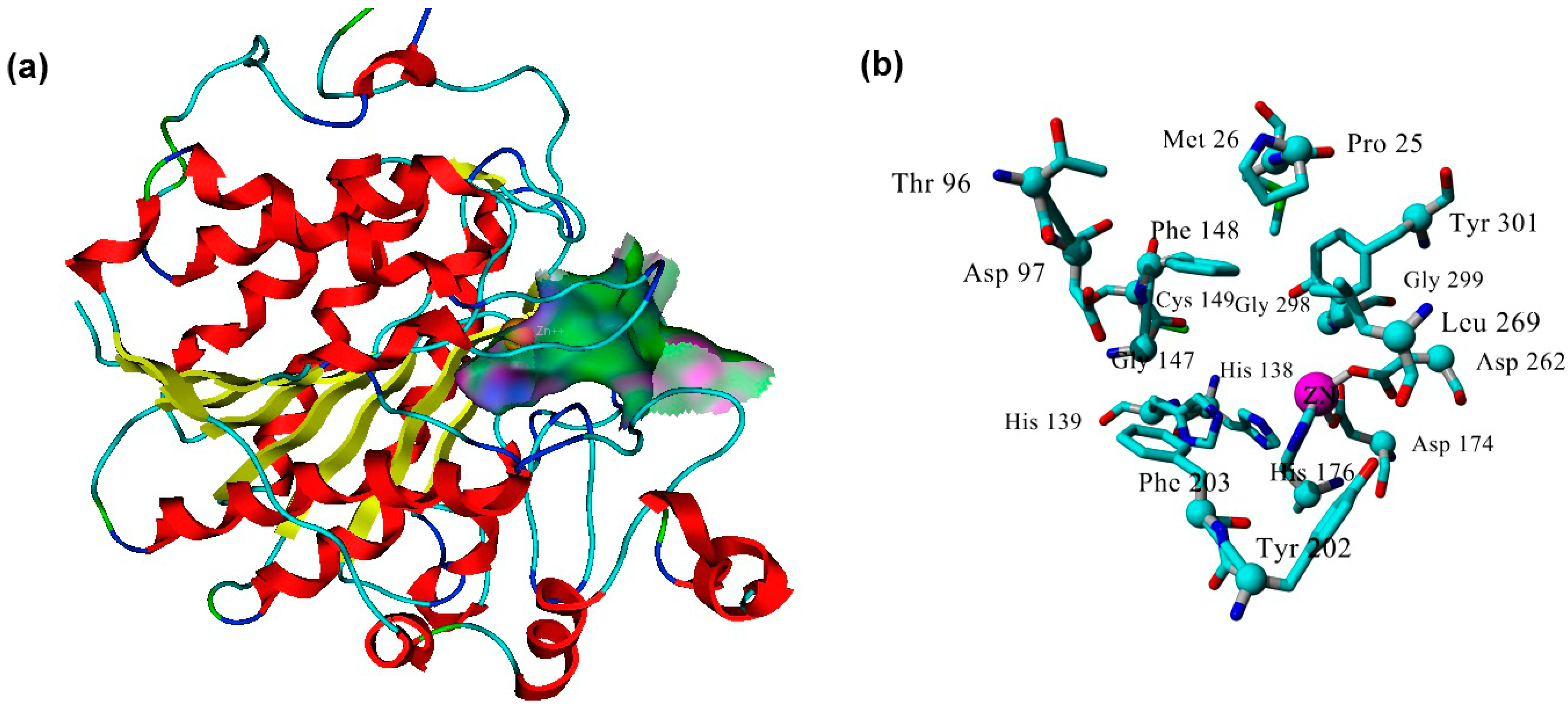
2.3. Plasmodium Falciparum Histone Deacetylase1 Model
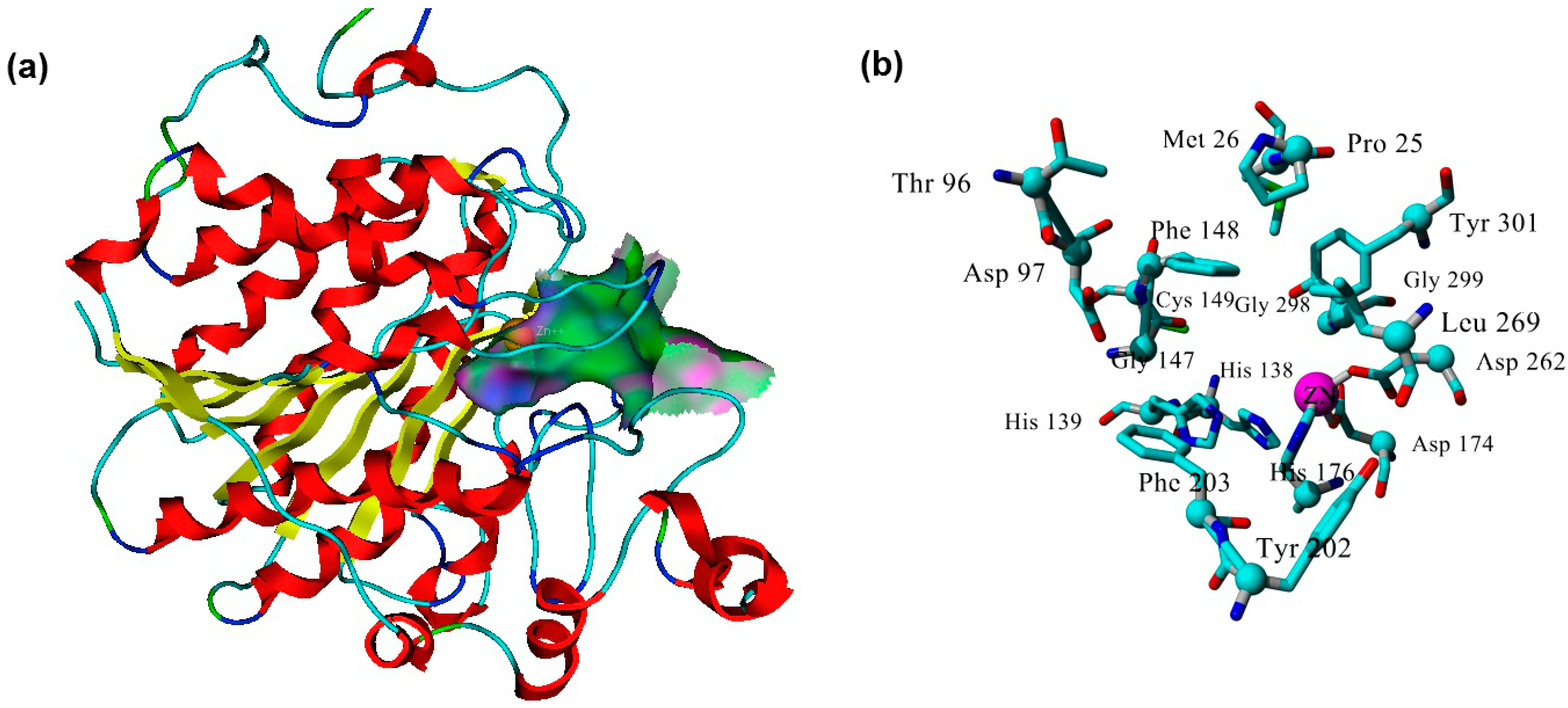
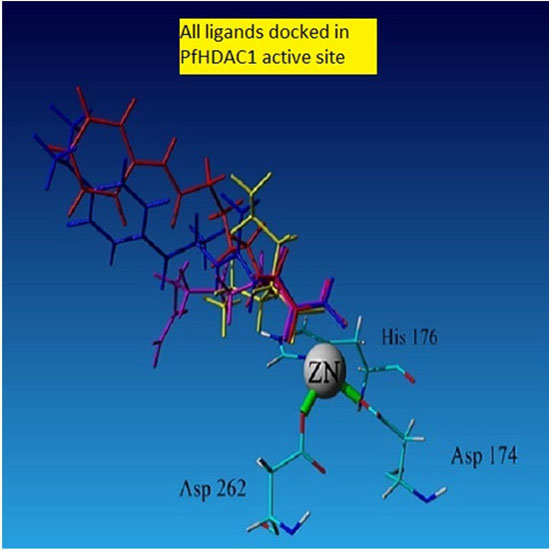
2.4. Docking
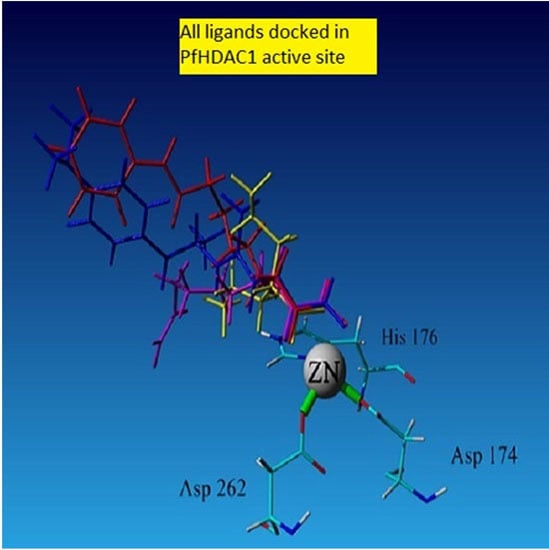
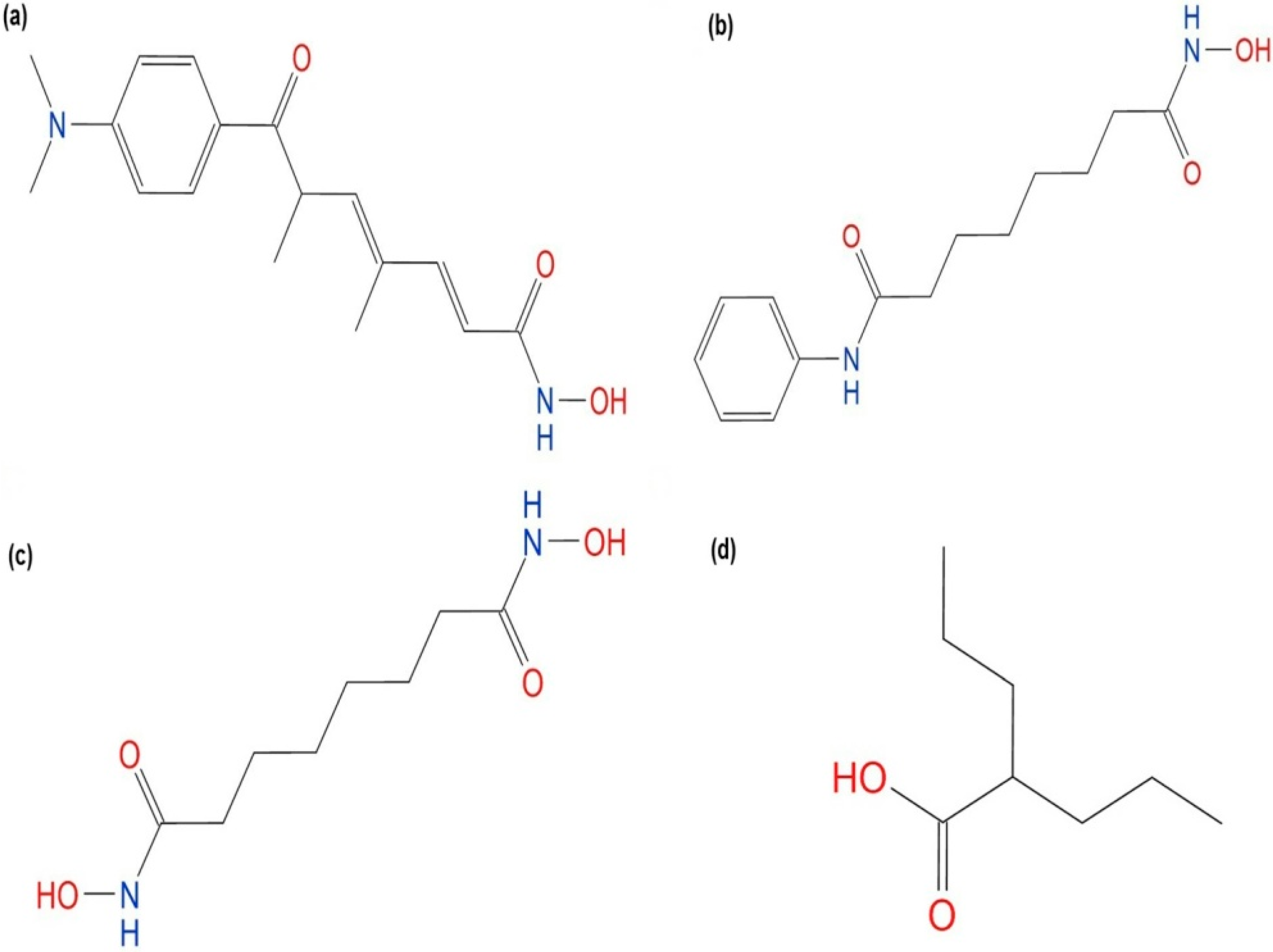
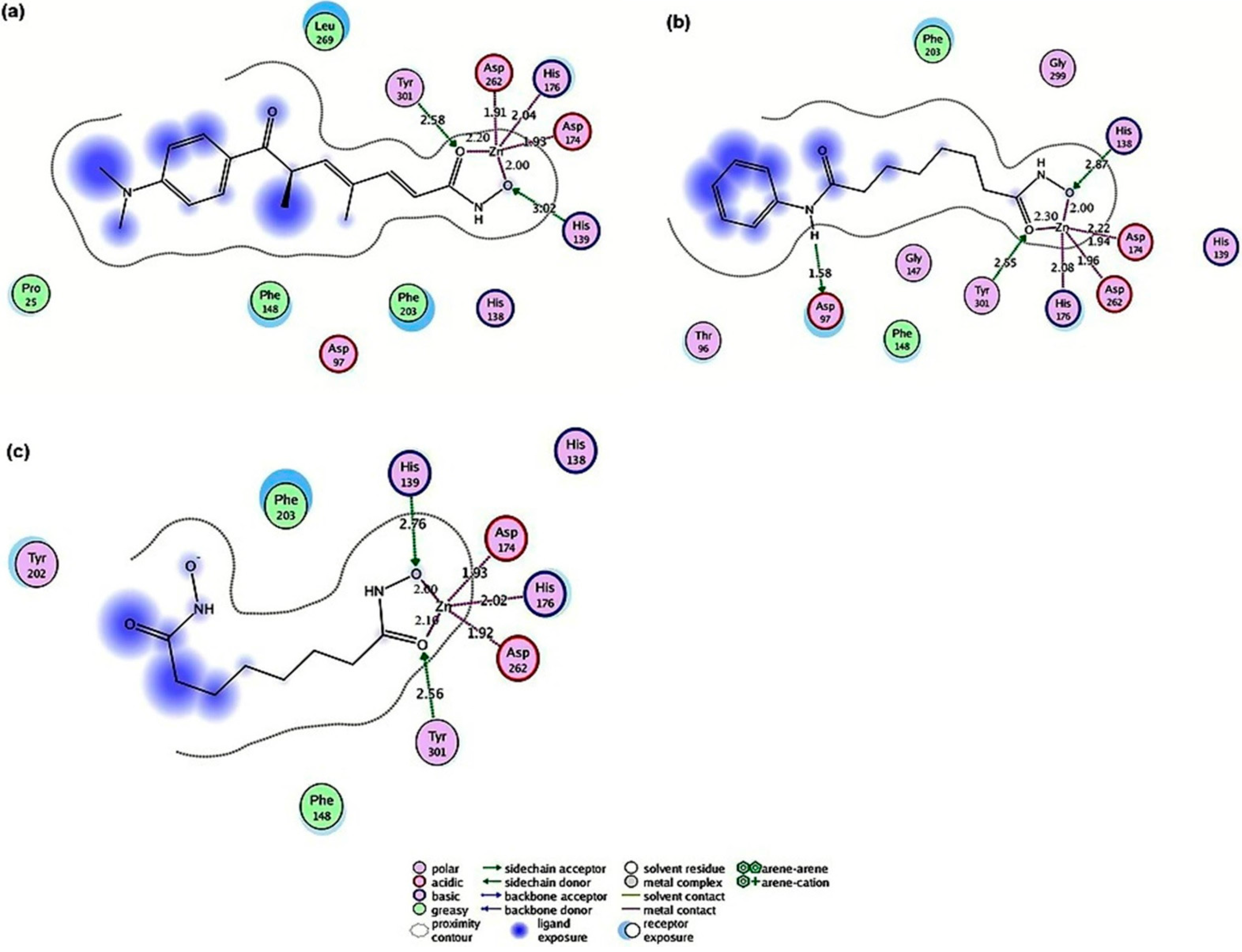
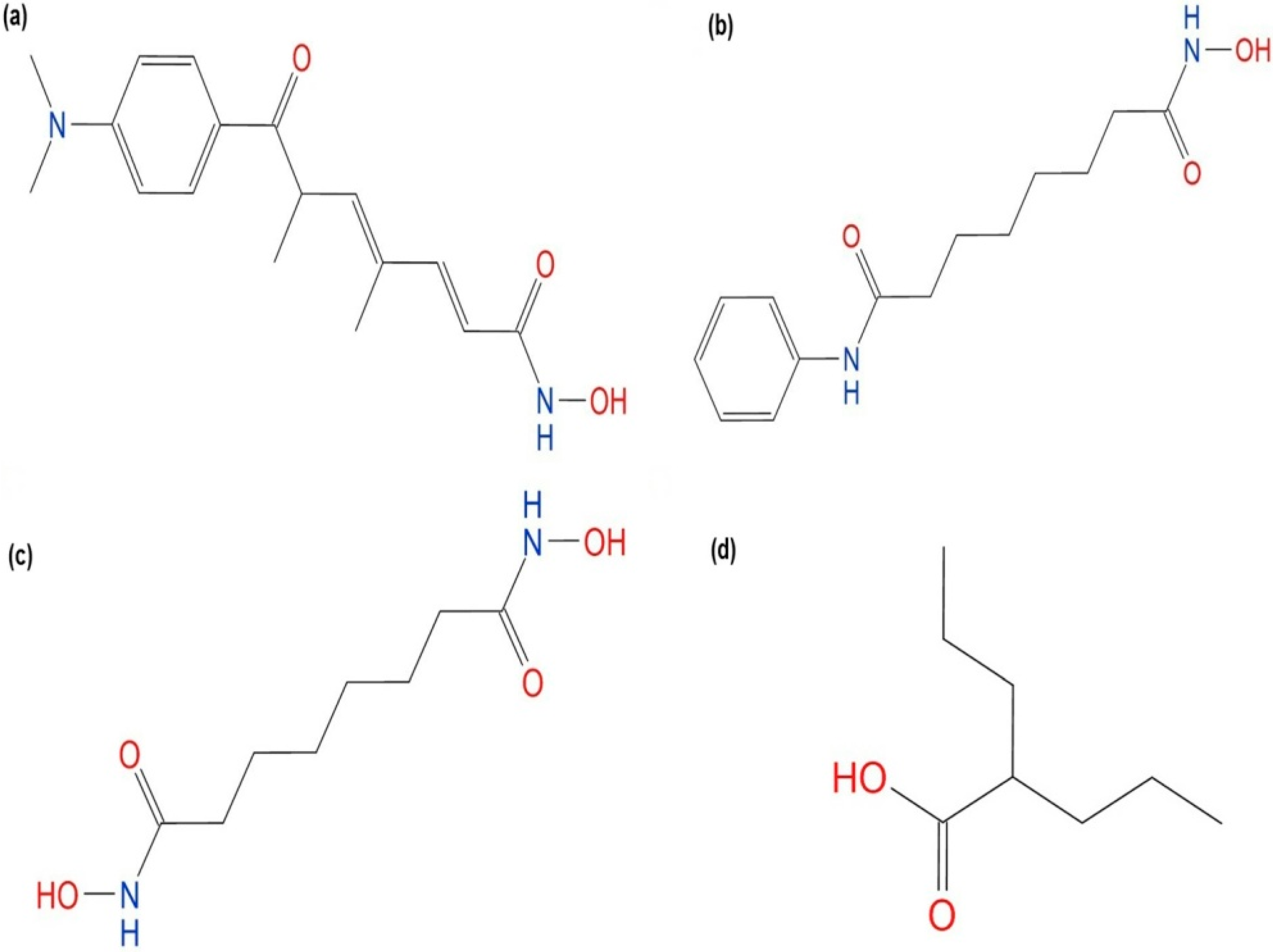
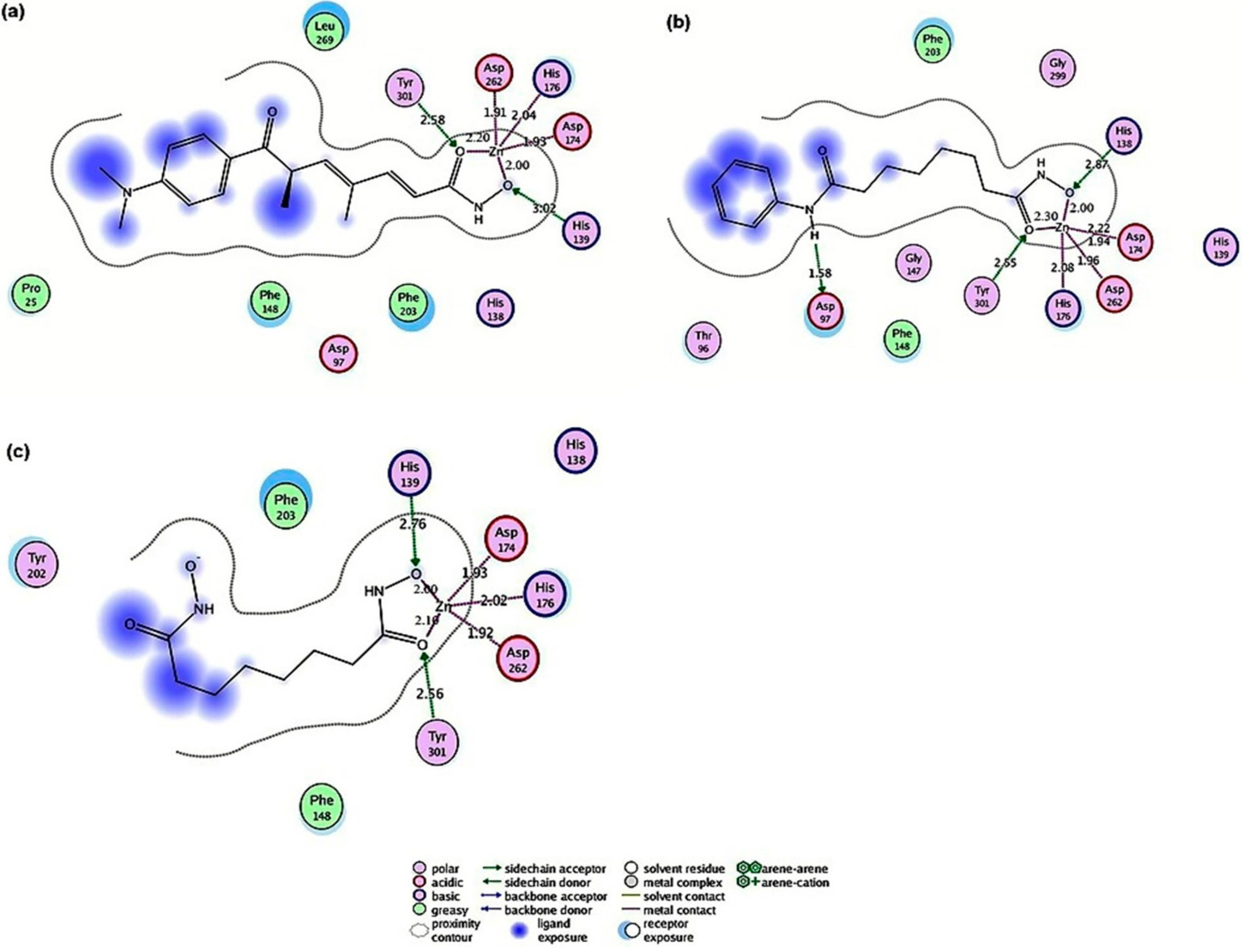
2.4.1. Hydroxamic Acid Inhibitor-PfHDAC1 Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | IC50 (µM) a | Score | Binding Energy (kcal·mol−1) | |
|---|---|---|---|---|
| CQ b Resistant | CQ b Sensitive | |||
| SAHA | 1.78 | 0.94 | −5.15 | 280.81 |
| SBHA | 0.8 | 1.3 | −6.52 | 281.92 |
| TSA | 0.008 | 0.11 | −6.92 | 308.54 |
| Valproic acid | N.A. c | N.A. c | −5.13 | 219.67 |
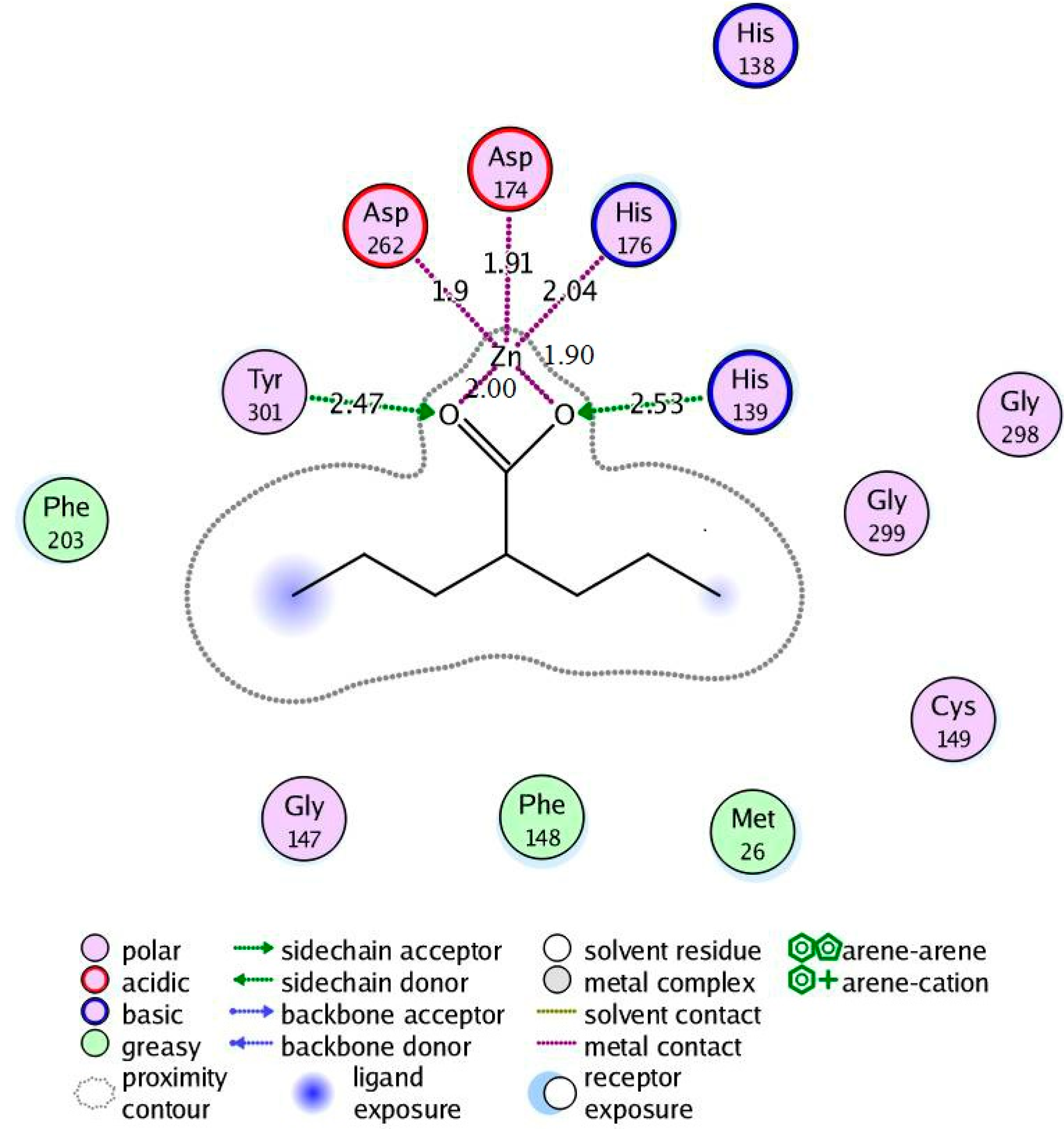
2.4.2. Valproic Acid-PfHDAC1 Complex
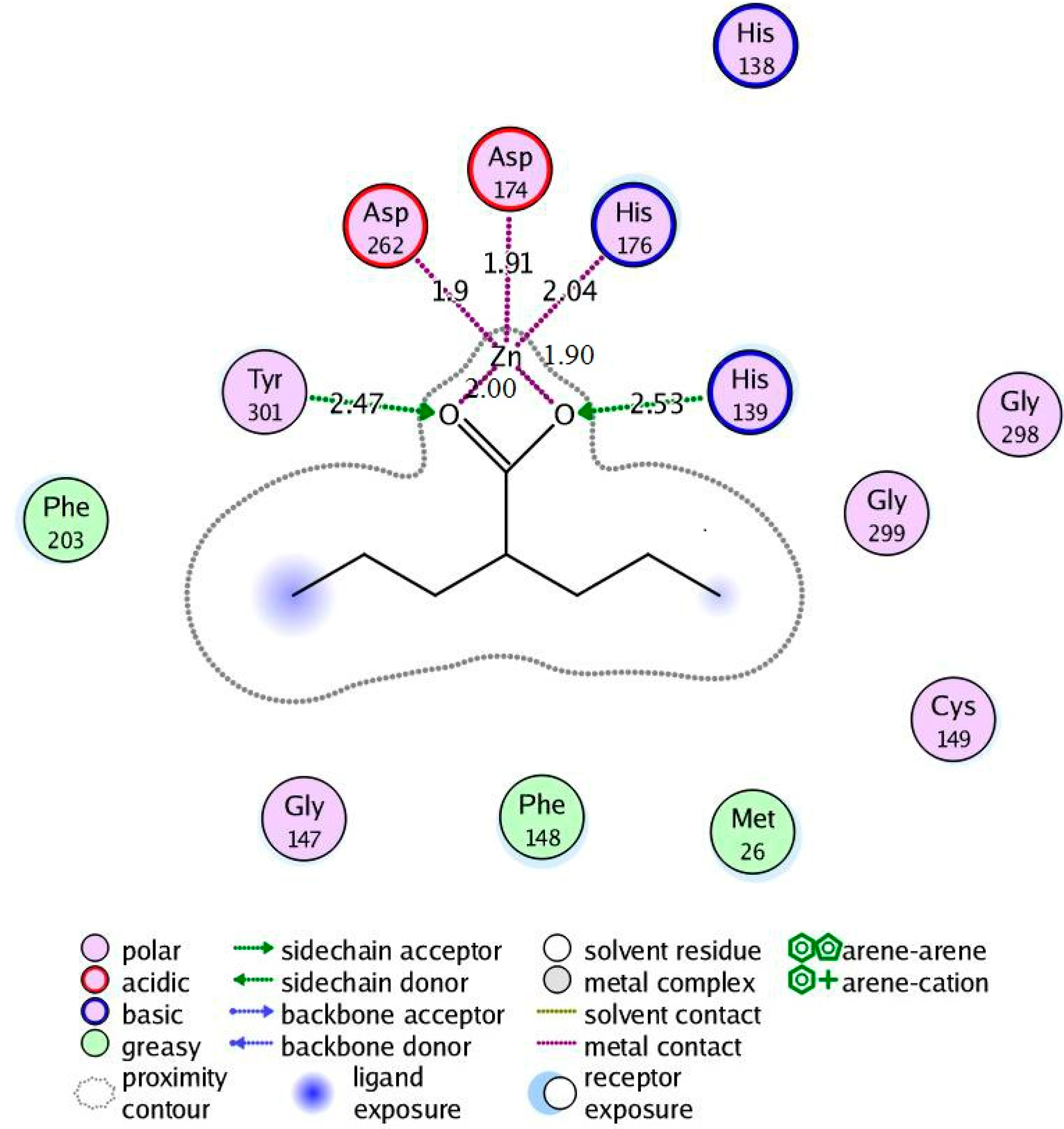
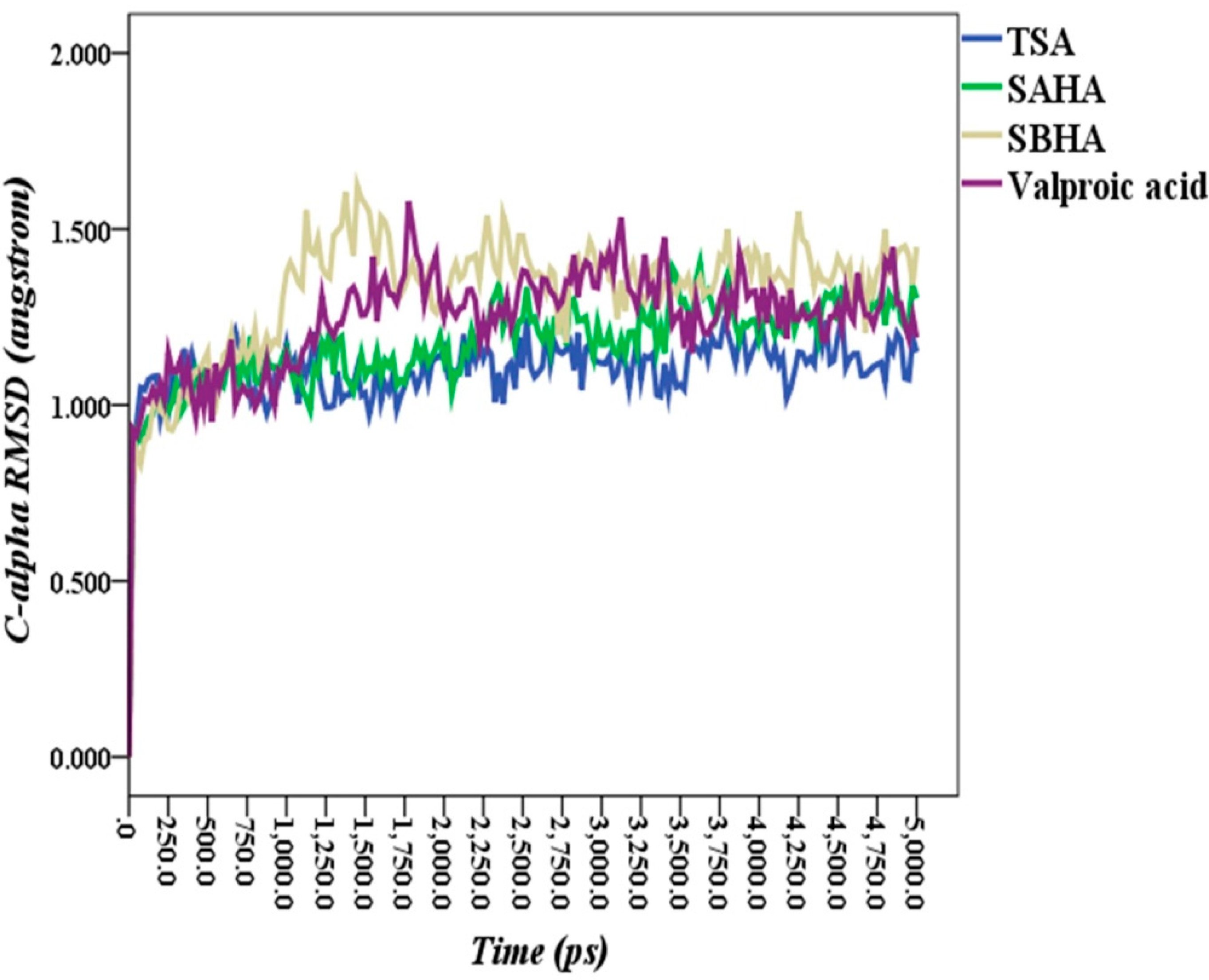
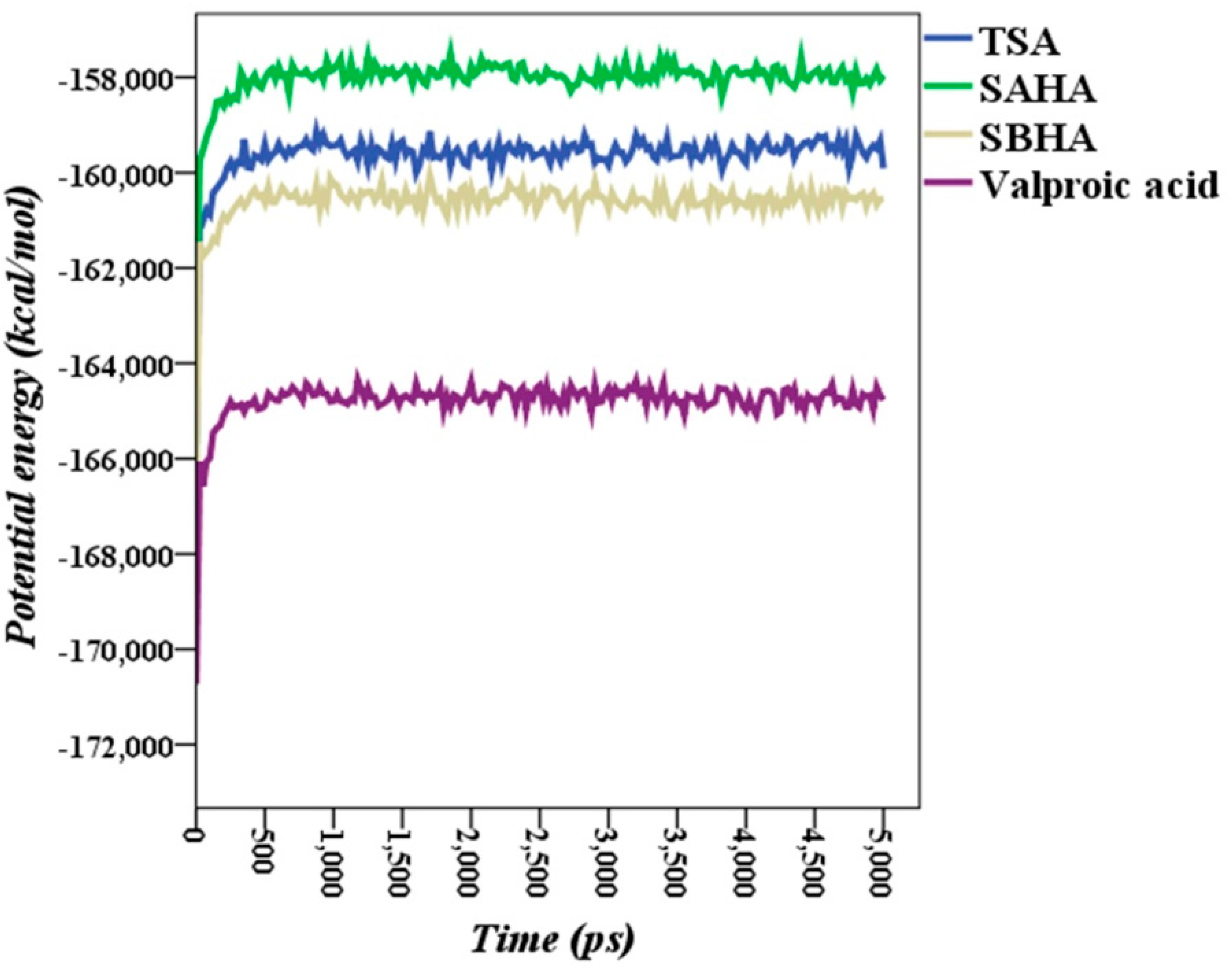
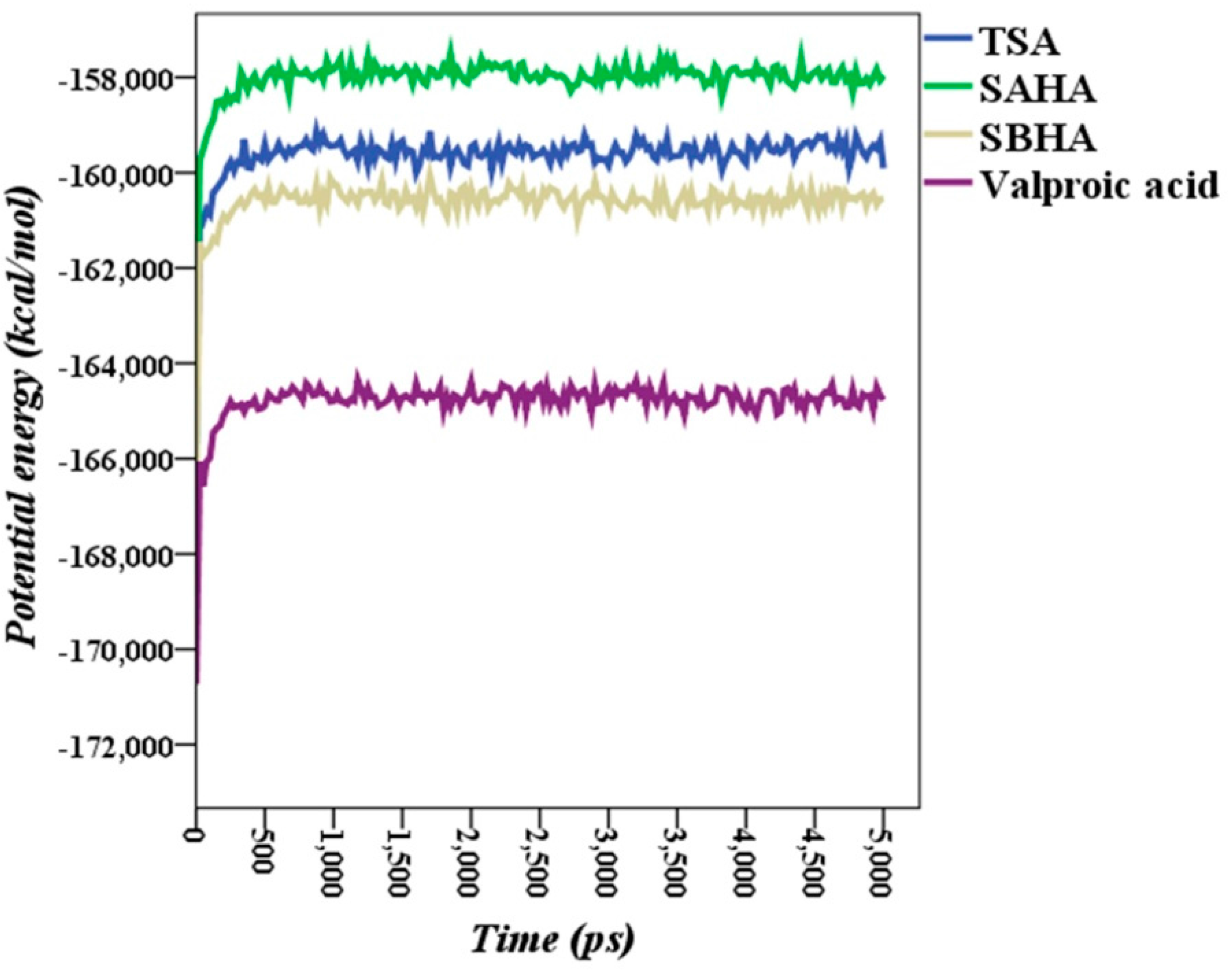
2.5. Molecular Dynamics Simulations
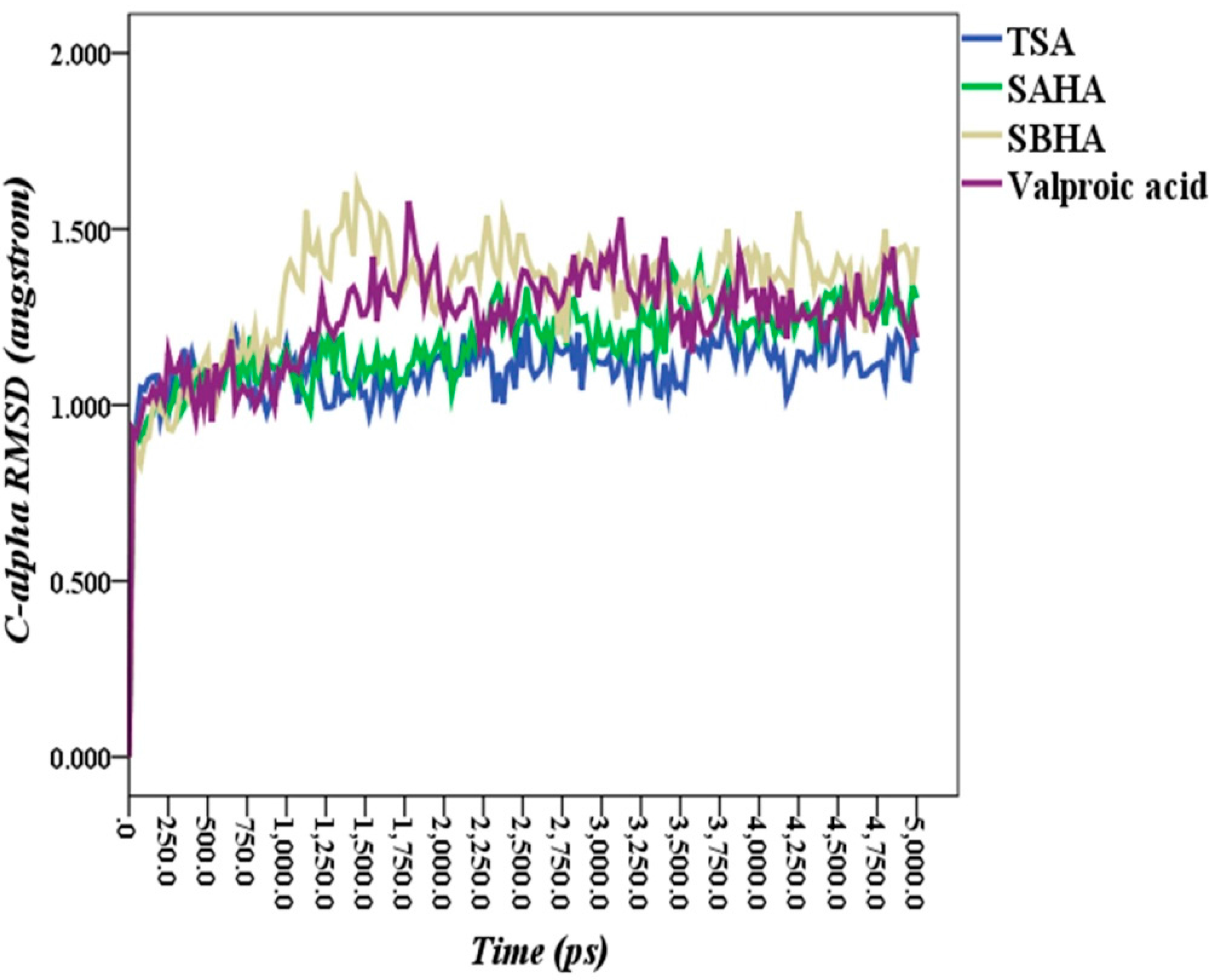
2.6. Theoretical Binding Energies
3. Materials and Methods
3.1. Homology Modeling
3.2. Model Refinement
3.3. Model Quality Validation
3.4. Ligands Preparation
3.5. Docking
3.6. Molecular Dynamics Simulation
3.7. Ligand-Enzyme Complex Theoretical Binding Energy Calculation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Malaria Report 2013. Available online: http://www.who.int/malaria/publications/world_malaria_report_2013/en/ (accessed on 1 June 2014).
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of artemisinin resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M.; Cockell, M.M. The molecular biology of the SIR proteins. Gene 2001, 279, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lin, D.T.; Chiang, P.H.; Goldman, N.D.; Fujioka, H.; Aikawa, M.; Syin, C. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 1999, 99, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Freitas, L.H.; Hernandez-Rivas, R.; Ralph, S.A.; Montiel-Condado, D.; Ruvalcaba-Salazar, O.K.; Rojas-Meza, A.P.; Mâncio-Silva, L.; Leal-Silvestre, R.J.; Gontijo, A.M.; Shorte, S.; et al. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell 2005, 121, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Haque, A.; Jones, M.K. HDAC inhibitors in parasitic diseases. Immunol. Cell Biol. 2012, 90, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Tran, T.N.; Lucke, A.J.; Kahnberg, P.; Le, G.T.; Boyle, G.M.; Gardiner, D.L.; Skinner-Adams, T.S.; Fairlie, D.P. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Sumanadasa, S.D.M.; Goodman, C.D.; Lucke, A.J.; Skinner-Adams, T.; Sahama, I.; Haque, A.; Do, T.A.; McFadden, G.I.; Fairlie, D.P.; Andrews, K.T. Antimalarial activity of the anticancer histone deacetylase inhibitor SB939. Antimicrob. Agents Chemother. 2012, 56, 3849–3856. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.T.; Tran, T.N.; Wheatley, N.C.; Fairlie, D.P. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 2009, 9, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Mazitschek, R.; Coleman, B.; Nguyen, C.; Urgaonkar, S.; Cortese, J.; Barker, R.H.; Greenberg, E.; Tang, W.; Bradner, J.E.; et al. Identification and characterization of small molecule inhibitors of a class I histone deacetylase from Plasmodium falciparum. J. Med. Chem. 2009, 52, 2185–2187. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Jones-Brando, L.; Torrey, E.F.; Yolken, R. Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr. Res. 2003, 62, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wheler, J.J.; Janku, F.; Falchook, G.S.; Jackson, T.L.; Fu, S.; Naing, A.; Tsimberidou, A.M.; Moulder, S.L.; Hong, D.S.; Yang, H.; et al. Phase I study of anti-VEGF monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014, 73, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Tassara, M.; Döhner, K.; Brossart, P.; Held, G.; Götze, K.; Horst, H.-A.; Ringhoffer, M.; Köhne, C.-H.; Kremers, S.; Raghavachar, A.; et al. Valproic acid in combination with all-trans retinoic acid and intensive induction therapy for acute myeloid leukemia in older patients. Blood 2014, 123, 4027–4036. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.F.; Karpenko, M.J.; Liu, Z.; Aimiuwu, J.; Villalona-Calero, M.A.; Chan, K.K.; Grever, M.R.; Otterson, G.A. Phase I study of 5-aza-2'-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 115–121. [Google Scholar] [CrossRef] [PubMed]
- FDA Depakote (divalproex sodium) Tablets for Oral use FDA Approved Labeling Text dated October 7, 2011. NDA 018723/S-037/S-040/S-043/S-045/S-046 Depakote (divalproex sodium) Tablets for Oral use FDA Approved Labeling Text dated 7 October 2011. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/018723s037lbl.pdf (accessed on 3 May 2014).
- The UniProt Consortium. The UniProt consortium activities at the universal protein resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.; Melkus, R.; de Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Pradhan, A.; Shah, F.; Tekwani, B.L.; Avery, M.A. Structural insights into the Plasmodium falciparum histone deacetylase 1 (PfHDAC-1): A novel target for the development of antimalarial therapy. Bioorg. Med. Chem. 2008, 16, 5254–5265. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Adrendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins-Struct. Funct. Genet. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. Verify3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–406. [Google Scholar] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Kannan, S.; Hauser, A.-T.; Moraes Mourão, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef] [PubMed]
- Labute, P.; Santavy, M. Locating Binding Sites in Protein Structures. Available online: http://www.chemcomp.com/journal/sitefind.htm (accessed on 15 January 2015).
- Hassig, C.A. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. USA 1998, 95, 3519–3524. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, D.; Struhl, K. Histone deacetylase activity of Rpd3 is important for transcriptional repression in vivo. Genes Dev. 1998, 12, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Moonsamy, S.; Dash, R.C.; Soliman, M.E.S. Integrated computational tools for identification of CCR5 antagonists as potential HIV-1 entry inhibitors: Homology modeling, virtual screening, molecular dynamics simulations and 3D QSAR analysis. Molecules 2014, 19, 5243–5265. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E. Yet Another Scientific Artificial Reality Application (YASARA). 2004. http://www.yasara.org/.
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). 2008. http://www.chemcomp.com/.
- Wang, D.; Helquist, P.; Wiest, O. Zinc binding in HDAC inhibitors: A DFT study. J. Org. Chem. 2007, 72, 5446–5449. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; de Proft, F.; Loverix, S.; Tourwé, D.; Geerlings, P. Theoretical study revealing the functioning of a novel combination of catalytic motifs in histone deacetylase. Bioorg. Med. Chem. 2005, 13, 3987–3992. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Lu, Z.; Cao, Z.; Zhang, Y. Zinc chelation with hydroxamate in histone deacetylases modulated by water access to the linker binding channel. J. Am. Chem. Soc. 2011, 133, 6110–6113. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, X.; Wu, Y.-D.; Wiest, O. Inhibition and mechanism of HDAC8 revisited. J. Am. Chem. Soc. 2014, 136, 11636–11643. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Edelsbrunner, H. Weighted Alpha Shapes; Department of Computer Science, University of Illinois: Champaign, IL, USA, 1995. [Google Scholar]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pKa prediction with hydrogen bonding network optimization. Methods Mol. Biol. 2012, 819, 405–421. [Google Scholar] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Saenz-Méndez, P.; Elmabsout, A.A.; Sävenstrand, H.; Awadalla, M.K.A.; Strid, Å.; Sirsjö, A.; Eriksson, L.A. Homology models of human all-trans retinoic acid metabolizing enzymes CYP26B1 and CYP26B1 spliced variant. J. Chem. Inf. Model. 2012, 52, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Decroos, C.; Bowman, C.M.; Moser, J.-A.S.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Compromised structure and function of HDAC8 mutants identified in cornelia de lange syndrome spectrum disorders. ACS Chem. Biol. 2014, 9, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbadawi, M.A.A.; Awadalla, M.K.A.; Hamid, M.M.A.; Mohamed, M.A.; Awad, T.A. Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach. Int. J. Mol. Sci. 2015, 16, 3915-3931. https://doi.org/10.3390/ijms16023915
Elbadawi MAA, Awadalla MKA, Hamid MMA, Mohamed MA, Awad TA. Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach. International Journal of Molecular Sciences. 2015; 16(2):3915-3931. https://doi.org/10.3390/ijms16023915
Chicago/Turabian StyleElbadawi, Mohamed A. Abdallah, Mohamed Khalid Alhaj Awadalla, Muzamil Mahdi Abdel Hamid, Magdi Awadalla Mohamed, and Talal Ahmed Awad. 2015. "Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach" International Journal of Molecular Sciences 16, no. 2: 3915-3931. https://doi.org/10.3390/ijms16023915
APA StyleElbadawi, M. A. A., Awadalla, M. K. A., Hamid, M. M. A., Mohamed, M. A., & Awad, T. A. (2015). Valproic Acid as a Potential Inhibitor of Plasmodium falciparum Histone Deacetylase 1 (PfHDAC1): An in Silico Approach. International Journal of Molecular Sciences, 16(2), 3915-3931. https://doi.org/10.3390/ijms16023915