
Transcriptome Profiling and Identification of Transcription Factors in Ramie (Boehmeria nivea L. Gaud) in Response to PEG Treatment, Using Illumina Paired-End Sequencing Technology

 ,
,
Abstract
:1. Introduction
2. Results
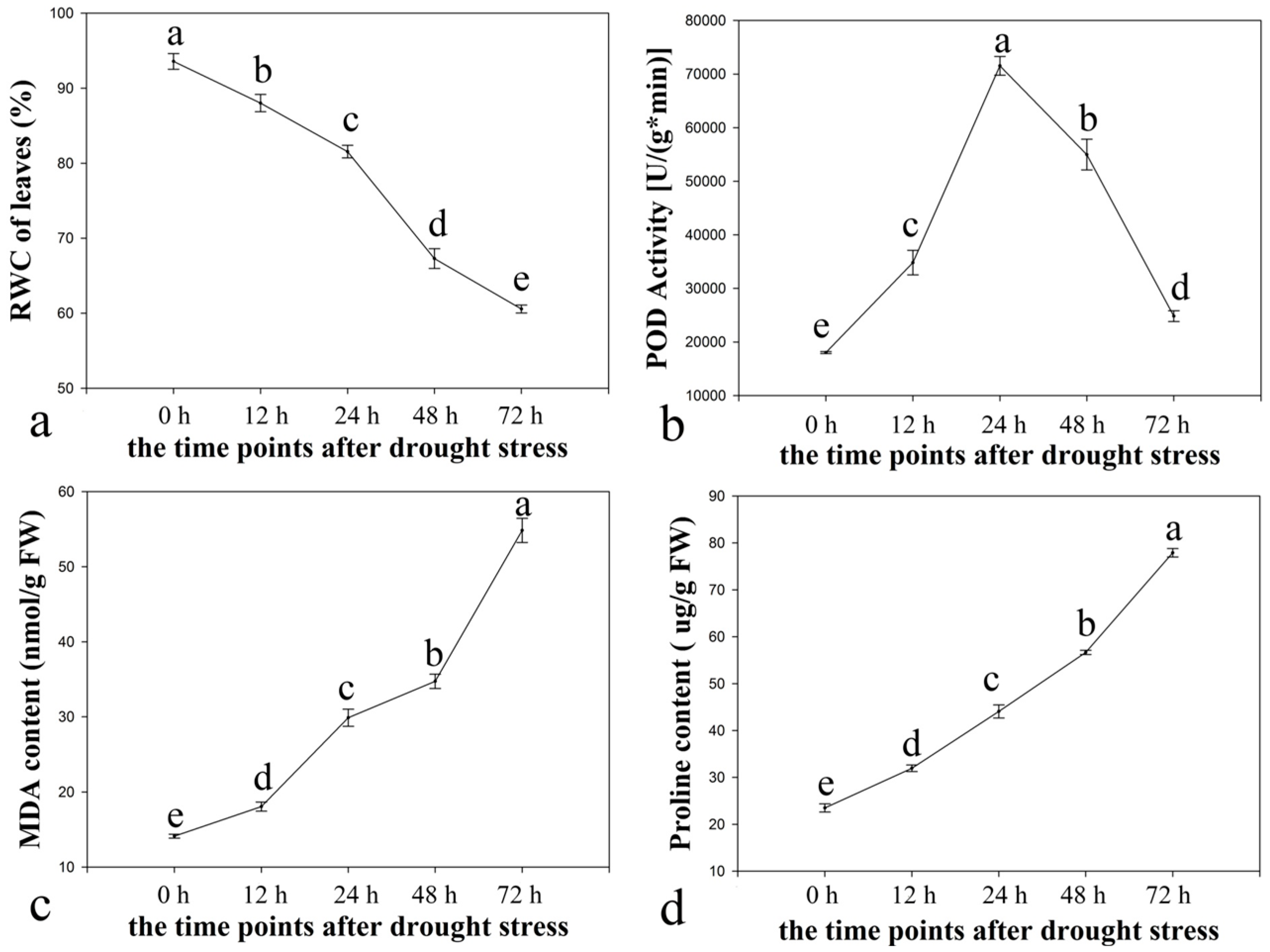
2.1. Physiological Responses of Ramie to Drought Stress
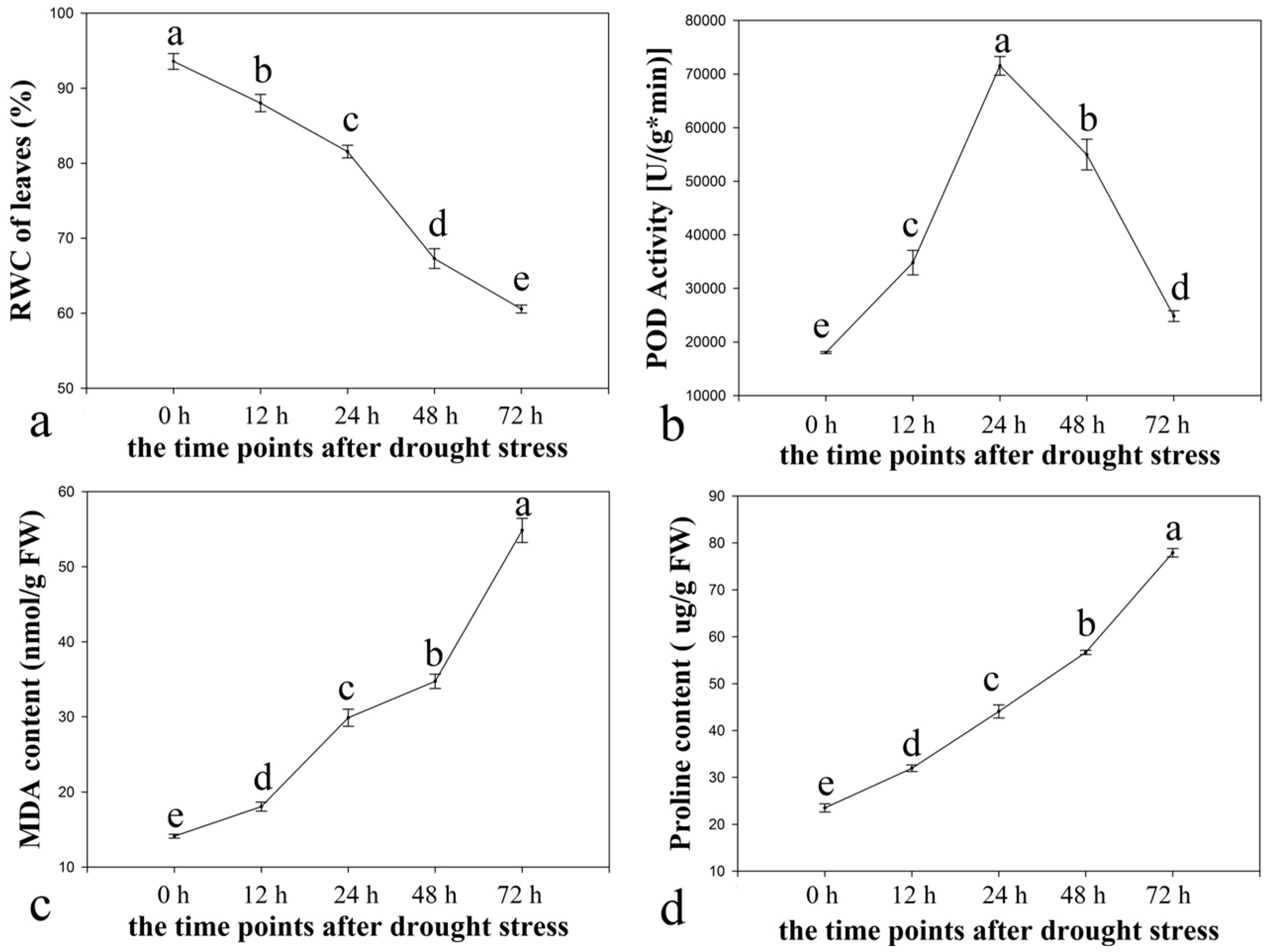
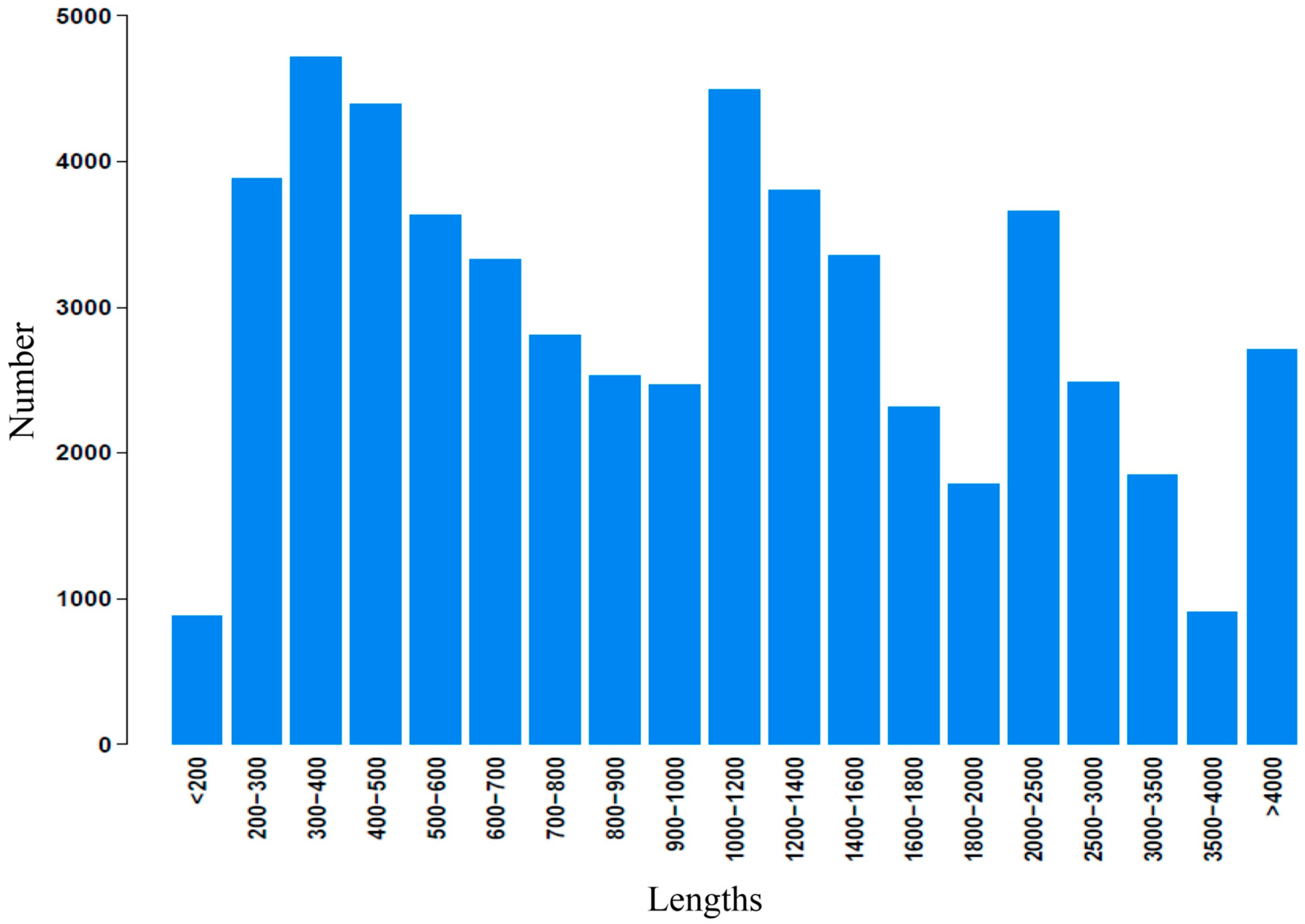
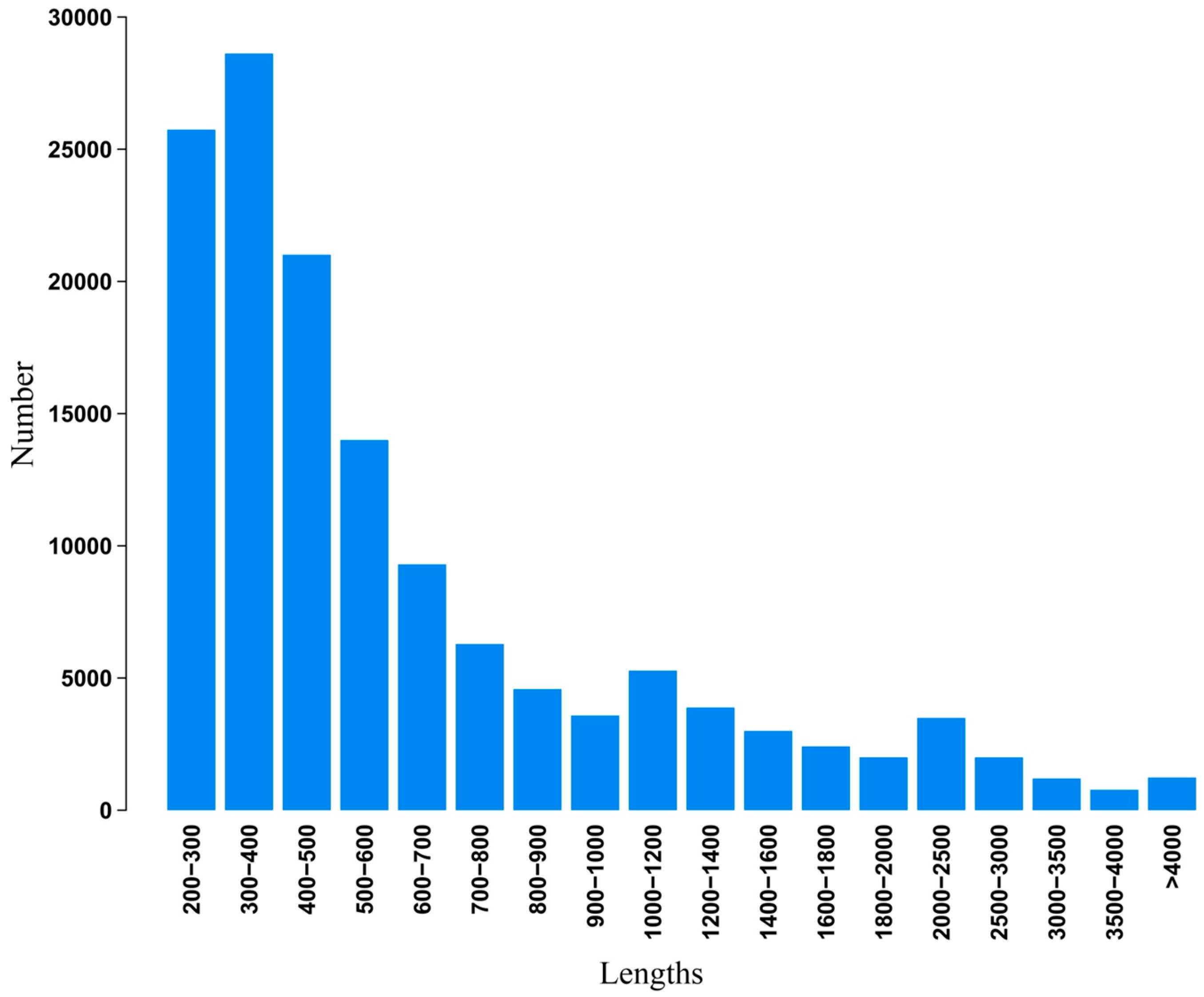
2.2. Illumina Paired-End Sequencing, Reads Assembly, and Annotation
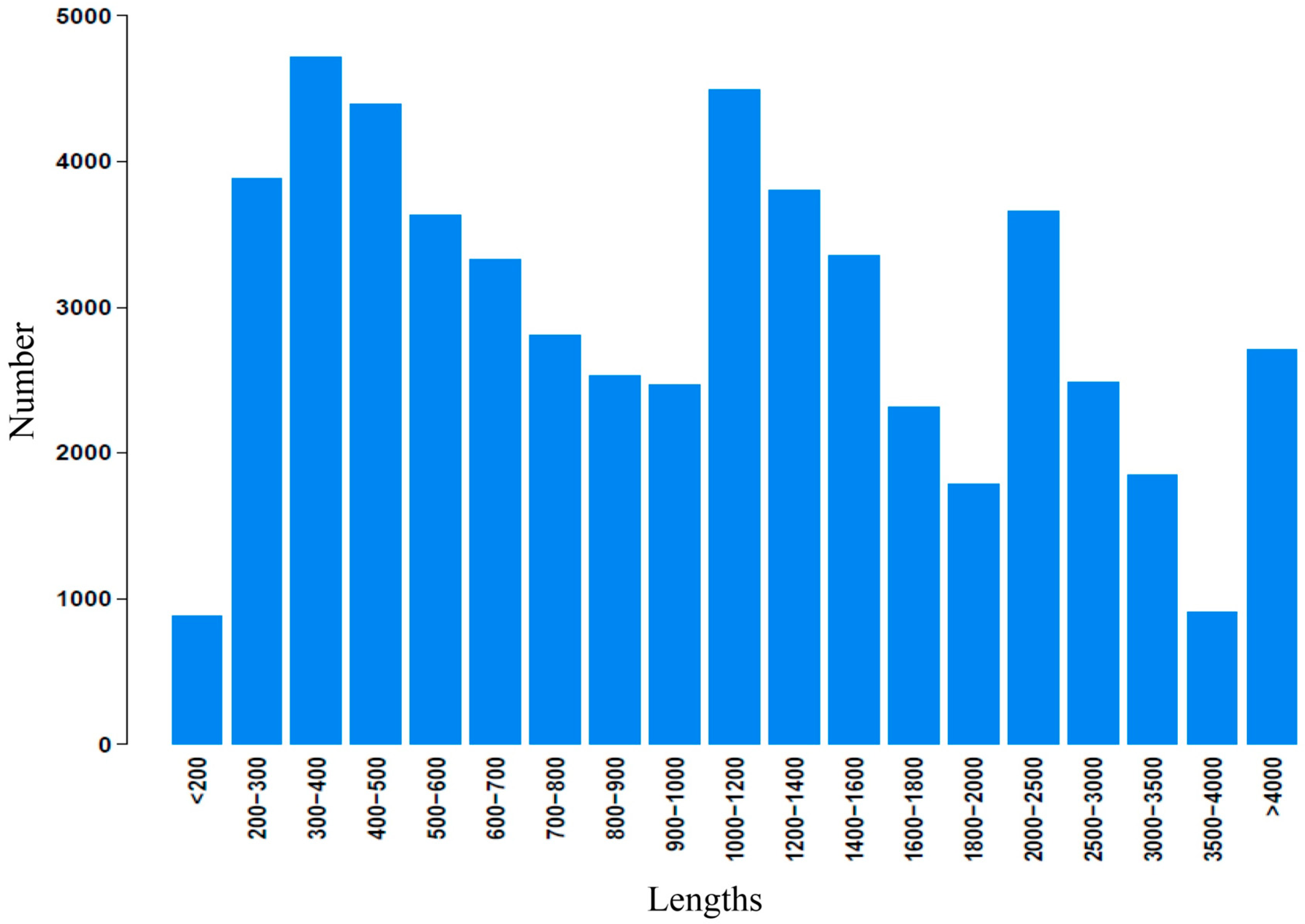
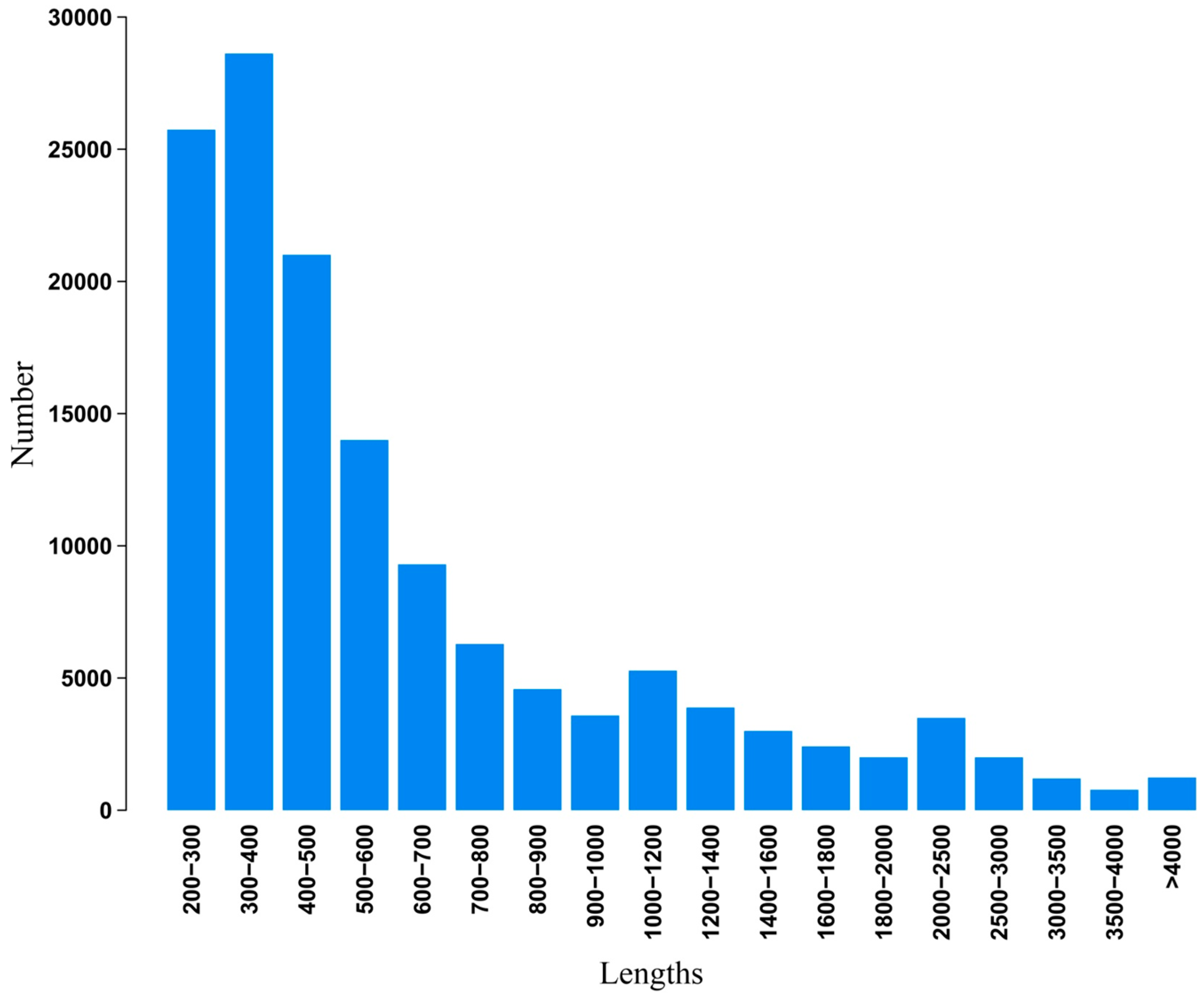
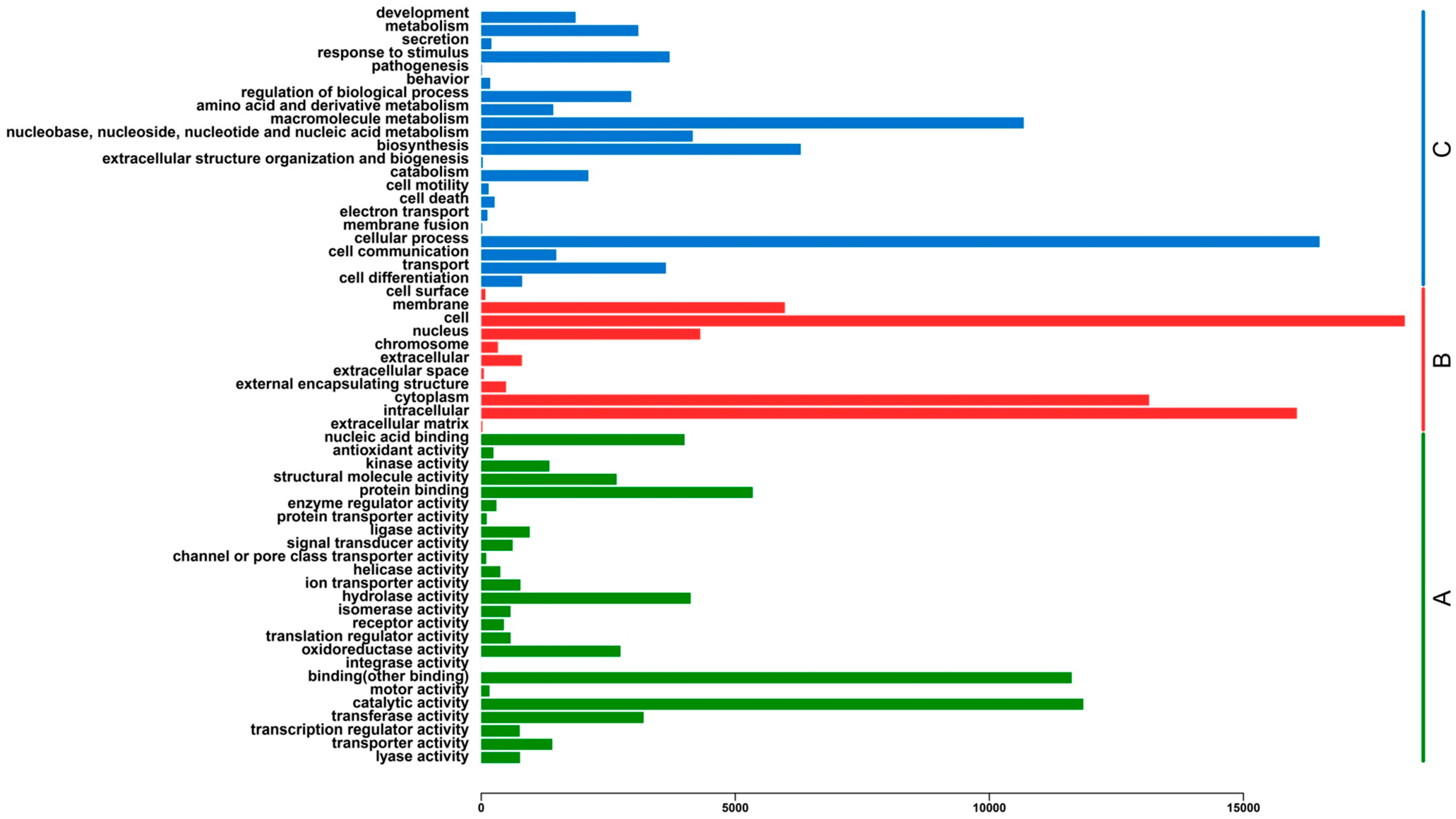
2.3. Functional Classification and Metabolic Pathway Assignment
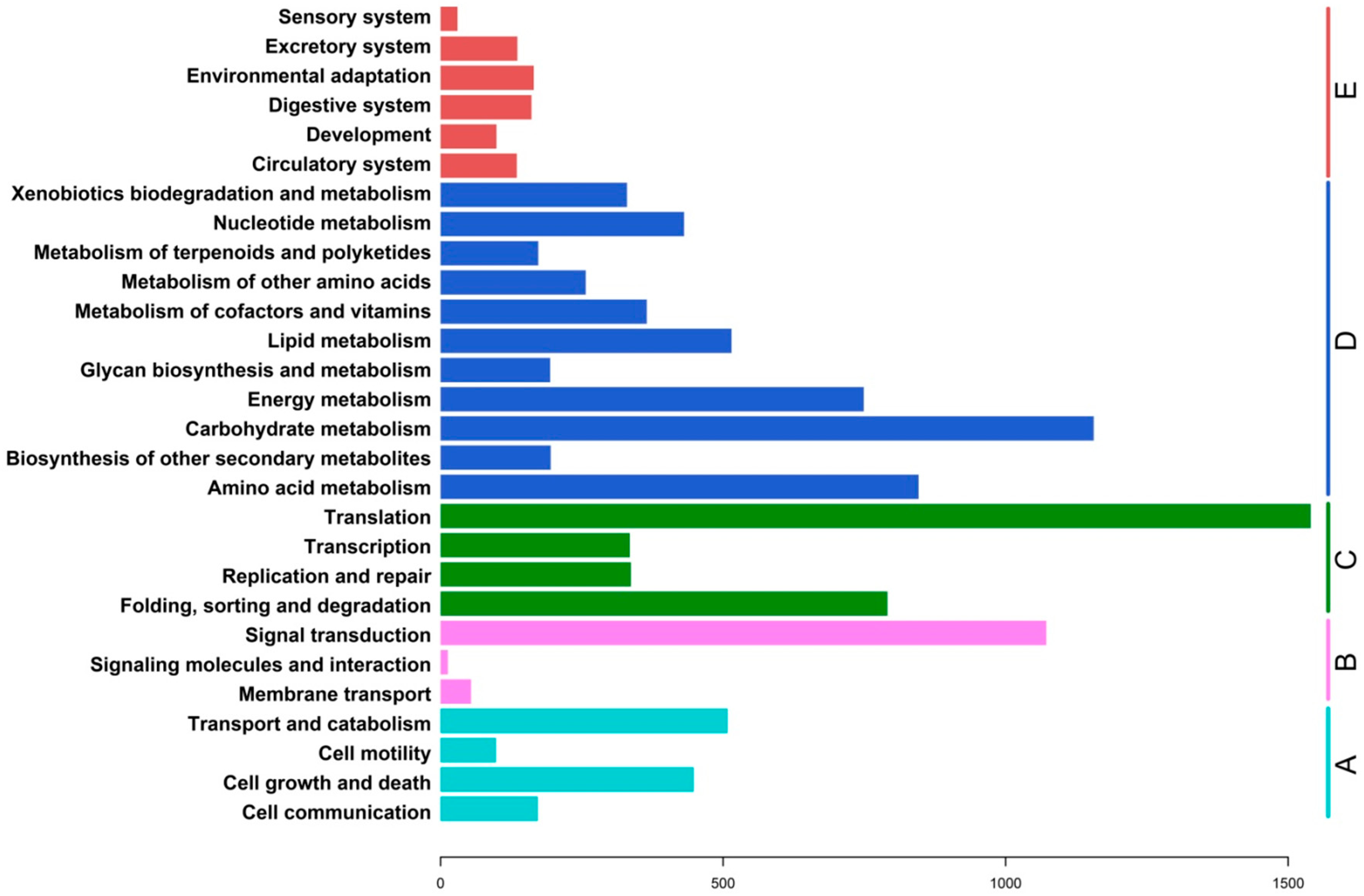
2.4. Analysis of Differential Gene Expression

{kind=link}
{kind=link}
{kind=link}
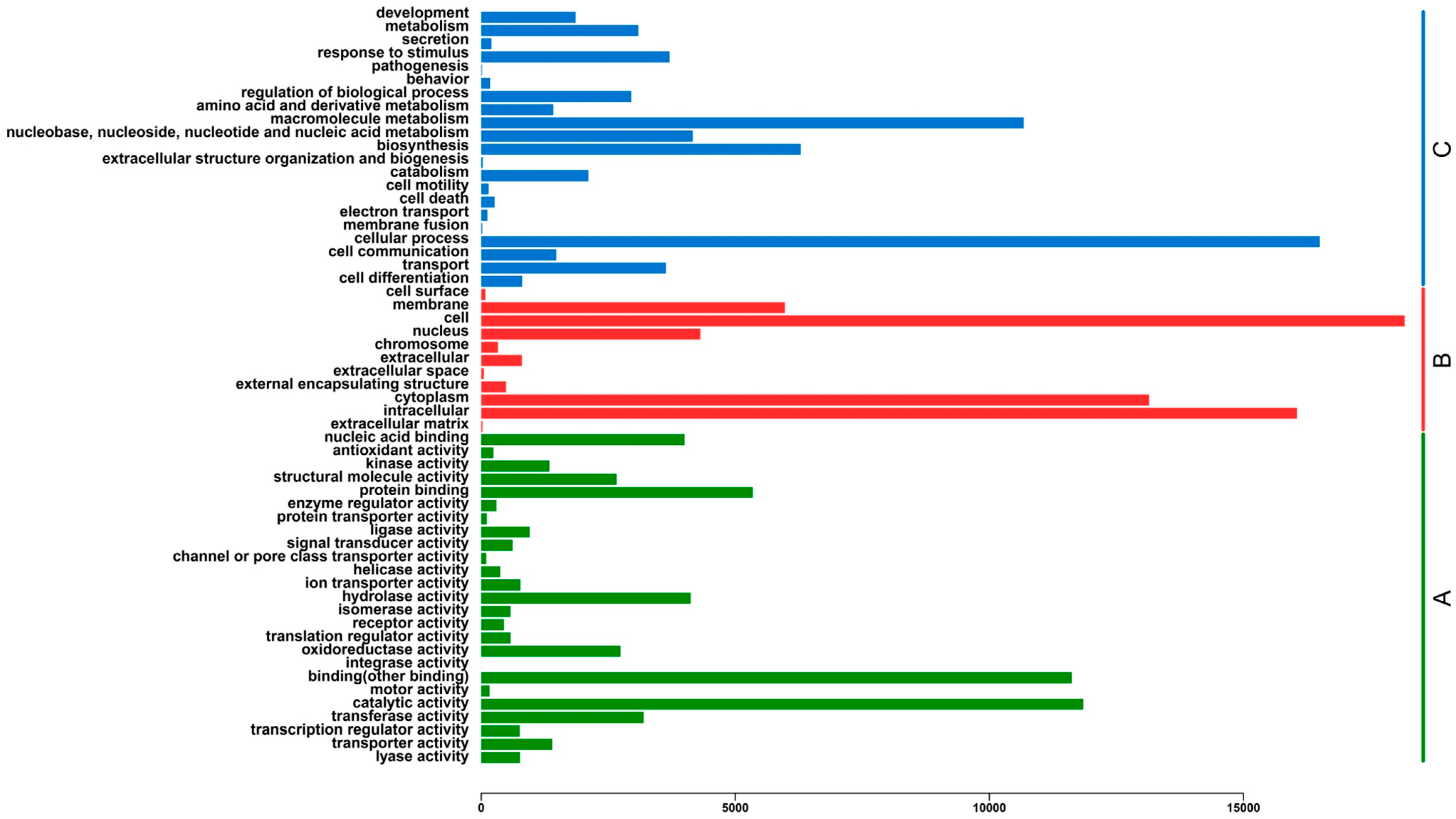{kind=link}
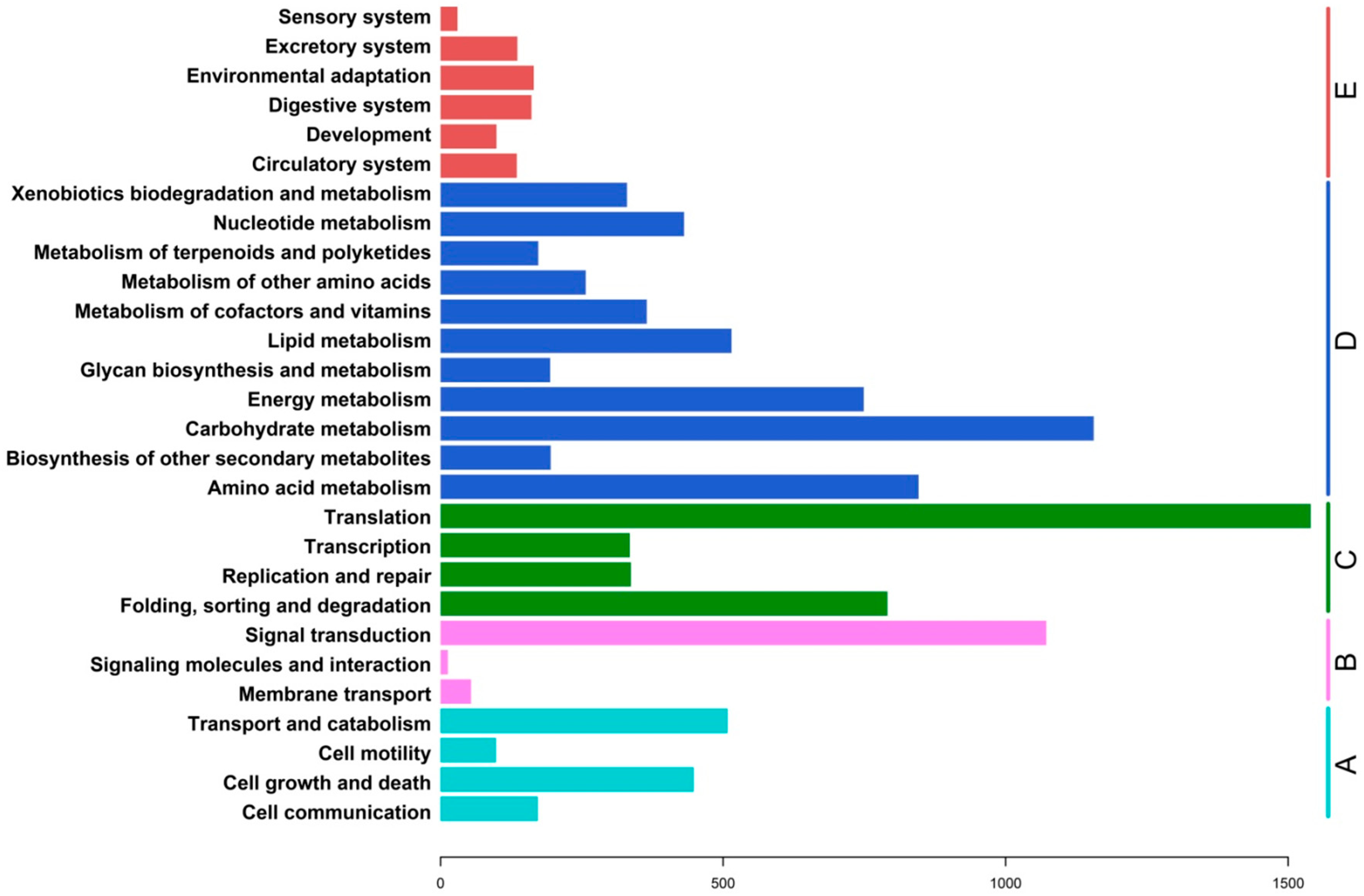{kind=link}
{kind=link}
| Sample | Up-Regulated | Down-Regulated |
|---|---|---|
| L1–L2 | 1450 | 2631 |
| L2–L3 | 2281 | 3120 |
| R1–R2 | 3872 | 1913 |
| R2–R3 | 2090 | 4590 |
2.5. Identification of Drought-Responsive TFs
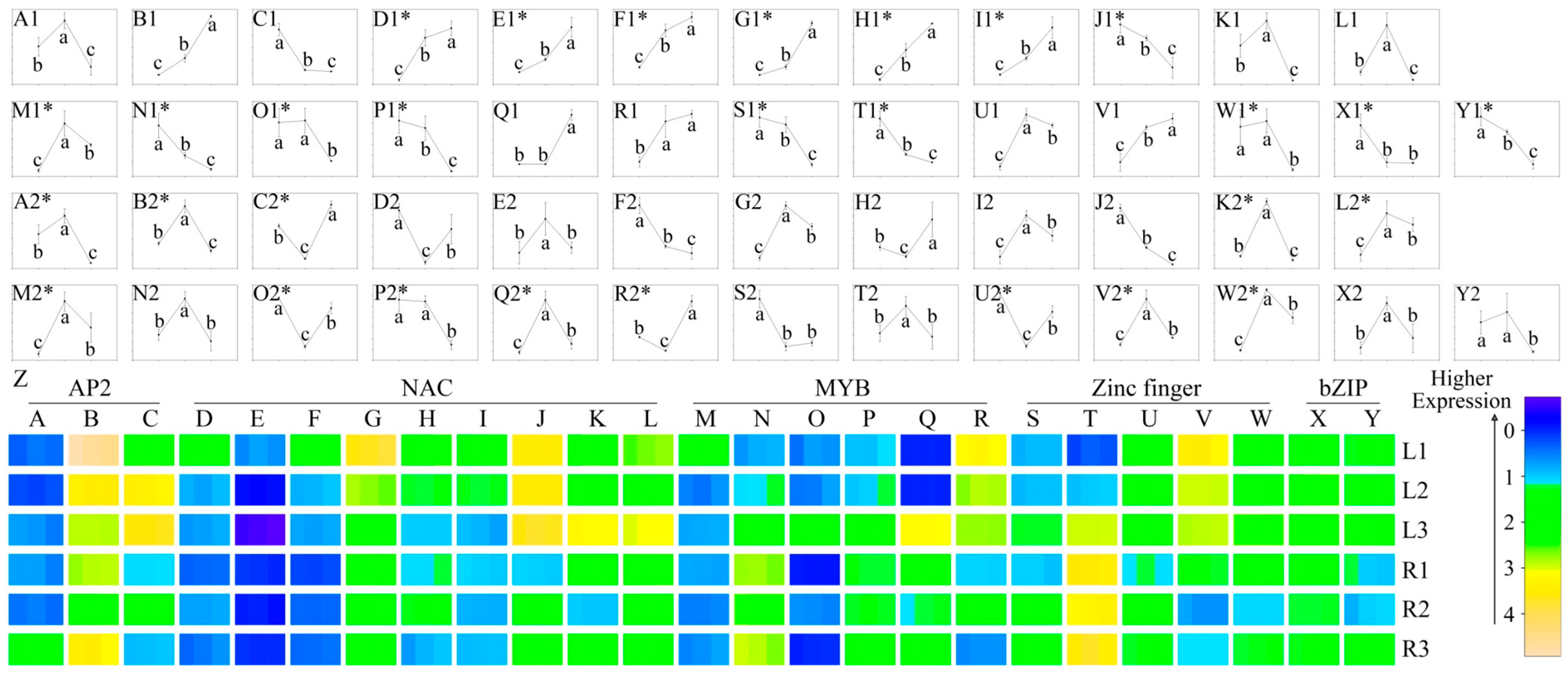
| Samples | All-up | All-down | Up–down | Down–up |
|---|---|---|---|---|
| Leaves | 379 | 637 | 348 | 98 |
| Roots | 28 | 57 | 3048 | 675 |
3. Discussion
3.1. Characterization of the Ramie Transcriptome
3.2. Physiological Traits Changed under PEG Treatment
3.3. The Five Main Families of TFs Responding to Drought Stress in Ramie
4. Material and Methods
4.1. Plant Materials Preparation, RNA Extraction, and cDNA Library Construction
4.2. Sequence Assembly, Annotation, and GO Terms/KEGG Pathways Construction
4.3. Differential Expression Redundancy and Enrichment Analyses
4.4. Real-Time Quantitative RT-PCR
4.5. Estimations of the RWC, the MDA, the Proline Content, and POD
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular responses to dehydration and low temperature: Differences and cross-talk between two stress signaling pathways. Curr. Opin. Plant Biol. 2000, 3, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Meng, F.R.; Zhang, C.Y.; Zhang, N.; Sun, M.S.; Ren, J.P.; Niu, H.B.; Wang, X.; Yin, J. Comparative analysis of water stress-responsive transcriptomes in drought-susceptible and -tolerant wheat (triticum aestivum L.). J. Plant Biol. 2012, 55, 349–360. [Google Scholar] [CrossRef]
- Pastori, G.M.; Foyer, C.H. Common components, networks, and pathways of cross-tolerance to stress. The central role of “redox” and abscisic acid-mediated controls. Plant Physiol. 2002, 129, 460–468. [Google Scholar]
- Dong, Y.P.; Fan, G.Q.; Zhao, Z.L.; Deng, M.J. Transcriptome expression profiling in response to drought stress in paulownia australis. Int. J. Mol. Sci. 2014, 15, 4583–4607. [Google Scholar] [CrossRef] [PubMed]
- Li, M.R.; Li, Y.; Li, H.Q.; Wu, G.J. Overexpression of AtNHX5 improves tolerance to both salt and drought stress in Broussonetia papyrifera (L.) Vent. Tree Physiol. 2011, 31, 349–357. [Google Scholar] [CrossRef]
- Liu, G.Z.; Li, X.L.; Jin, S.X.; Liu, X.Y.; Zhu, L.F.; Nie, Y.C.; Zhang, X.L. Overexpression of rice NAC Gene SNAC1 improves drought and salt tolerance by enhancing root development and reducing transpiration rate in transgenic cotton. PLoS One 2014, 9, e86895. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006, 11, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.L.; Jin, X.H.; Yang, Y.; Lin, Z.W.; Zhang, Y.L. The regulatory role of riboflavin in the drought tolerance of tobacco plants depends on ROS production. Plant Growth Regul. 2014, 72, 269–277. [Google Scholar] [CrossRef]
- Urano, K.; Maruyama, K.; Ogata, Y.; Morishita, Y.; Takeda, M.; Sakurai, N.; Suzuki, H.; Saito, K.; Shibata, D.; Kobayashi, M.; et al. Characterization of the ABA-regulated global responses to dehydration in Arabidopsis by metabolomics. Plant J. 2009, 57, 1065–1078. [Google Scholar]
- Rabbani, M.A.; Maruyama, K.; Abe, H.; Khan, M.A.; Katsura, K.; Ito, Y.; Yoshiwara, K.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Monitoring expression profiles of rice genes under cold, drought, and high-salinity stresses and abscisic acid application using cDNA microarray and RNA get-blot analyses. Plant Physiol. 2003, 133, 1755–1767. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Zhu, S.Y.; Tang, Q.M.; Chen, P.; Yu, Y.T.; Tang, S.W. De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L. Gaud). BMC Genomics 2013, 14. [Google Scholar] [CrossRef]
- Liu, T.M.; Zhu, S.Y.; Tang, Q.M.; Yu, Y.T.; Tang, S.W. Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud). BMC Plant Biol. 2013, 13. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, 355–360. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, M.; Luo, Q.F.; Wang, J.; Li, H.G. De novo transcriptome analysis of Liriodendron chinense petals and leaves by Illumina sequencing. Gene 2014, 534, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.B.; Fu, H.T.; Zhou, Q.; Sun, S.M.; Jiang, S.F.; Xiong, Y.W.; Gong, Y.S.; Qiao, H.; Zhang, W.Y. Transcriptome analysis of androgenic gland for discovery of novel genes from the oriental river prawn, Macrobrachium nipponense, using Illumina Hiseq 2000. PLoS One 2013, 8, e76840. [Google Scholar] [CrossRef] [PubMed]
- Verslues, P.E.; Agarwal, M.; Katiyar-Agarwal, S.; Zhu, J.; Zhu, J.K. Methods and concepts in quantifying resistance to drought, salt and freezing, abiotic stresses that affect plant water status. Plant J. 2006, 45, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M. Biotechnological approach of improving plant salt tolerance using antioxidants as markers. Biotechnol. Adv. 2009, 27, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Shigeoka, S. Understanding oxidative stress and antioxidant functions to enhance photosynthesis. Plant Physiol. 2011, 155, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.M.; Cui, X.L.; Chen, Y.; Gu, C.S.; Miao, H.B.; Gao, H.S.; Chen, F.D.; Liu, Z.L.; Guan, Z.Y.; Fang, W.M. CgDREBa transgenic chrysanthemum confers drought and salinity tolerance. Environ. Exp. Bot. 2011, 74, 255–260. [Google Scholar] [CrossRef]
- Tamura, T.; Hara, K.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Osmotic stress tolerance of transgenic tobacco expressing a gene encoding a membrane-located receptor-like protein from tobacco plants. Plant Physiol. 2003, 131, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Schroeder, J.I. Calcium-activated K+ channels and calcium-induced calcium release by slow vacuolar ion channels in guard cell vacuoles implicated in the control of stomatal closure. Plant Cell 1994, 6, 669–683. [Google Scholar] [PubMed]
- Klein, M.; Geisler, M.; Suh, S.J.; Kolukisaoglu, H.U.; Azevedo, L.; Plaza, S.; Curtis, M.D.; Richter, A.; Weder, B.; Schulz, B.; et al. Disruption of AtMRP4, a guard cell plasma membrane ABCC-type ABC transporter, leads to deregulation of stomatal opening and increased drought susceptibility. Plant J. 2004, 39, 219–236. [Google Scholar]
- Bartels, D. Targeting detoxification pathways: An efficient approach to obtain plants with multiple stress tolerance? Trends Plant Sci. 2001, 6, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in Arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 2011, 124, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.B.; Foley, R.C.; Onate-Sanchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Aida, M.; Ishida, T.; Fukaki, H.; Fujisawa, H.; Tasaka, M. Genes involved in organ separation in Arabidopsis: An analysis of the cup-shaped cotyledon mutant. Plant Cell 1997, 9, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.N.; Ernst, H.A.; Lo Leggio, L.; Skriver, K. NAC transcription factors: Structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.Y.; Jiang, H.L.; Li, C.B.; Zhai, Q.Z.; Zhang, J.Y.; Wu, X.Q.; Sun, J.Q.; Xie, Q.; Li, C.Y. Role of the Arabidopsis thaliana NAC transcription factors ANAC019 and ANAC055 in regulating jasmonic acid-signaled defense responses. Cell Res. 2008, 18, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.S.P.; Nakashima, K.; Sakuma, Y.; Simpson, S.D.; Fujita, Y.; Maruyama, K.; Fujita, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 2004, 16, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Dai, M.Q.; Yao, J.L.; Xiao, B.Z.; Li, X.H.; Zhang, Q.F.; Xiong, L.Z. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.H.; Do Choi, Y.; Kim, M.; Reuzeau, C.; Kim, J.K. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.C.; Zhao, G.Y.; Xia, C.; Jia, J.Z.; Liu, X.; Kong, X.Y. A wheat R2R3-MYB gene, TaMYB30-B, improves drought stress tolerance in transgenic Arabidopsis. J. Exp. Bot. 2012, 63, 5873–5885. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, E.; Sala, T.; Calvi, D.; Gusmaroli, G.; Tonelli, C. Over-expression of the Arabidopsis AtMYB41 gene alters cell expansion and leaf surface permeability. Plant J. 2008, 53, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Lippold, F.; Sanchez, D.H.; Musialak, M.; Schlereth, A.; Scheible, W.R.; Hincha, D.K.; Udvardi, M.K. AtMyb41 regulates transcriptional and metabolic responses to osmotic stress in Arabidopsis. Plant Physiol. 2009, 149, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, E.; Galbiati, M.; Vavasseur, A.; Conti, L.; Sala, T.; Vuylsteke, M.; Leonhardt, N.; Dellaporta, S.L.; Tonelli, C. A guard-cell-specific MYB transcription factor regulates stomatal movements and plant drought tolerance. Curr. Biol. 2005, 15, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Su, L.T.; Li, J.W.; Liu, D.Q.; Zhai, Y.; Zhang, H.J.; Li, X.W.; Zhang, Q.L.; Wang, Y.; Wang, Q.Y. A novel MYB transcription factor, GmMYBJ1, from soybean confers drought and cold tolerance in Arabidopsis thaliana. Gene 2014, 538, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Seo, J.S.; Han, S.W.; Koo, Y.J.; Kim, C.H.; Song, S.I.; Nahm, B.H.; Choi, Y.D.; Cheong, J.J. Overexpression of AtMYB44 enhances stomatal closure to confer abiotic stress tolerance in transgenic Arabidopsis. Plant Physiol. 2008, 146, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.B.; Nadeau, J.A.; Lucas, J.; Lee, E.K.; Nakagawa, T.; Zhao, L.; Geisler, M.; Sack, F.D. The Arabidopsis R2R3 MYB proteins FOUR LIPS and MYB88 restrict divisions late in the stomatal cell lineage. Plant Cell 2005, 17, 2754–2767. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Lee, E.; Lucas, J.R.; Morohashi, K.; Li, D.; Murray, J.A.; Sack, F.D.; Grotewold, E. Regulation of cell proliferation in the stomatal lineage by the Arabidopsis MYB FOUR LIPS via direct targeting of core cell cycle genes. Plant Cell 2010, 22, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Rahaie, M.; Xue, G.P.; Naghavi, M.R.; Alizadeh, H.; Schenk, P.M. A MYB gene from wheat (Triticum aestivum L.) is up-regulated during salt and drought stresses and differentially regulated between salt-tolerant and sensitive genotypes. Plant Cell Rep. 2010, 29, 835–844. [Google Scholar]
- Luo, X.; Bai, X.; Zhu, D.; Li, Y.; Ji, W.; Cai, H.; Wu, J.; Liu, B.H.; Zhu, Y.M. GsZFP1, a new Cys2/His2-type zinc-finger protein, is a positive regulator of plant tolerance to cold and drought stress. Planta 2012, 235, 1141–1155. [Google Scholar] [CrossRef] [PubMed]
- Ben Saad, R.; Zouari, N.; Ben Ramdhan, W.; Azaza, J.; Meynard, D.; Guiderdoni, E.; Hassairi, A. Improved drought and salt stress tolerance in transgenic tobacco overexpressing a novel A20/AN1 zinc-finger “AlSAP” gene isolated from the halophyte grass Aeluropus littoralis. Plant Mol. Biol. 2010, 72, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.L.; Xu, K.D.; Zhong, M.; Pan, Y.Z.; Jiang, B.B.; Liu, G.L.; Jia, Y.; Zhang, H.Q. Overexpression of a novel chrysanthemum Cys2/His2-type zinc finger protein gene DgZFP3 confers drought tolerance in tobacco. Biotechnol. Lett. 2013, 35, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Y.; Chao, D.Y.; Gao, J.P.; Zhu, M.Z.; Shi, M.; Lin, H.X. A previously unknown zinc finger protein, DST, regulates drought and salt tolerance in rice via stomatal aperture control. Genes Dev. 2009, 23, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.T.; Wu, Y.B.; Wang, X.P. bZIP transcription factor OsbZIP52/RISBZ5: A potential negative regulator of cold and drought stress response in rice. Planta 2012, 235, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.S.; Liu, J.H.; Chen, X.J. Overexpression of PtrABF gene, a bZIP transcription factor isolated from Poncirus trifoliata, enhances dehydration and drought tolerance in tobacco via scavenging ROS and modulating expression of stress-responsive genes. BMC Plant Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta 2012, 1819, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Broeckling, C.D.; Blancaflor, E.B.; Sledge, M.K.; Sumner, L.W.; Wang, Z.Y. Overexpression of WXP1, a putative Medicago truncatula AP2 domain-containing transcription factor gene, increases cuticular wax accumulation and enhances drought tolerance in transgenic alfalfa (Medicago sativa). Plant J. 2005, 42, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Song, C.P.; Agarwal, M.; Ohta, M.; Guo, Y.; Halfter, U.; Wang, P.; Zhu, J.K. Role of an Arabidopsis AP2/EREBP-type transcriptional repressor in abscisic acid and drought stress responses. Plant Cell 2005, 17, 2384–2396. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Wang, B.; Liu, L.J.; Jiang, H.; Chen, J.; Ye, S.T.; Chen, L.Y.; Guo, P.A.; Huang, X.; Peng, D.X. Agrobacterium-mediated genetic transformation and regeneration of transgenic plants using leaf midribs as explants in ramie (Boehmeria nivea (L.) Gaud). Mol. Biol. Rep. 2014, 41, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar]
- Chen, Z.Z.; Xue, C.H.; Zhu, S.; Zhou, F.F.; Ling, X.F.B.; Liu, G.P.; Chen, L.B. GoPipe: Streamlined Gene Ontology annotation for batch anonymous sequences with statistics. Prog. Biochem. Biophys. 2005, 32, 187–190. [Google Scholar]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, X.; Chen, J.; Zhang, J.; Liao, Y.; Dai, L.; Wang, B.; Liu, L.; Peng, D. Transcriptome Profiling and Identification of Transcription Factors in Ramie (Boehmeria nivea L. Gaud) in Response to PEG Treatment, Using Illumina Paired-End Sequencing Technology. Int. J. Mol. Sci. 2015, 16, 3493-3511. https://doi.org/10.3390/ijms16023493
An X, Chen J, Zhang J, Liao Y, Dai L, Wang B, Liu L, Peng D. Transcriptome Profiling and Identification of Transcription Factors in Ramie (Boehmeria nivea L. Gaud) in Response to PEG Treatment, Using Illumina Paired-End Sequencing Technology. International Journal of Molecular Sciences. 2015; 16(2):3493-3511. https://doi.org/10.3390/ijms16023493
Chicago/Turabian StyleAn, Xia, Jie Chen, Jingyu Zhang, Yiwen Liao, Lunjin Dai, Bo Wang, Lijun Liu, and Dingxiang Peng. 2015. "Transcriptome Profiling and Identification of Transcription Factors in Ramie (Boehmeria nivea L. Gaud) in Response to PEG Treatment, Using Illumina Paired-End Sequencing Technology" International Journal of Molecular Sciences 16, no. 2: 3493-3511. https://doi.org/10.3390/ijms16023493
APA StyleAn, X., Chen, J., Zhang, J., Liao, Y., Dai, L., Wang, B., Liu, L., & Peng, D. (2015). Transcriptome Profiling and Identification of Transcription Factors in Ramie (Boehmeria nivea L. Gaud) in Response to PEG Treatment, Using Illumina Paired-End Sequencing Technology. International Journal of Molecular Sciences, 16(2), 3493-3511. https://doi.org/10.3390/ijms16023493