
Cyclic Nucleotide Signalling in Kidney Fibrosis


Abstract
:1. Renal Fibrosis and Involved Signalling Pathways
1.1. Fibrotic Kidney Diseases
1.2. Renal Fibrotic Models

{kind=link}
{kind=link}
| Renal Fibrotic Disease | Causes | Profibrotic Signalling Pathways | Actual or Clinically Tested Treatments | Literature |
|---|---|---|---|---|
| Diabetic nephropathy | Hyperglykaemia; Hypertension | DM I, II; RAAS; JAK/STAT eNOS-dysfunction TGFβ | RAAS blockade Pirfenidone | [16,21] |
| Glomerulo-sclerosis (e.g., FSGS) | DN, Hypertension; Nephrotic syndrome; (FSGS) | e.g., DM I, II; RAAS; suPAR (FSGS) | RAAS blockade, Pirfenidone, FSGS: Glucocorticoids, Cytostatics, ACTH, Rituximab | [25,26,29,30,43] |
| Lupus nephritis | Autoimmune antibodies; Expansion of inflammatory cells [44] | TWEAK/Fn14 | Steroids, Belimumab, Cytostatics: Azathioprin, Cyclophosphamide Hydroxychloroquine | [35,36,37,45] |
2. Cyclic Nucleotide Signalling Pathways and Their Potential as Therapeutic Options in Renal Fibrosis
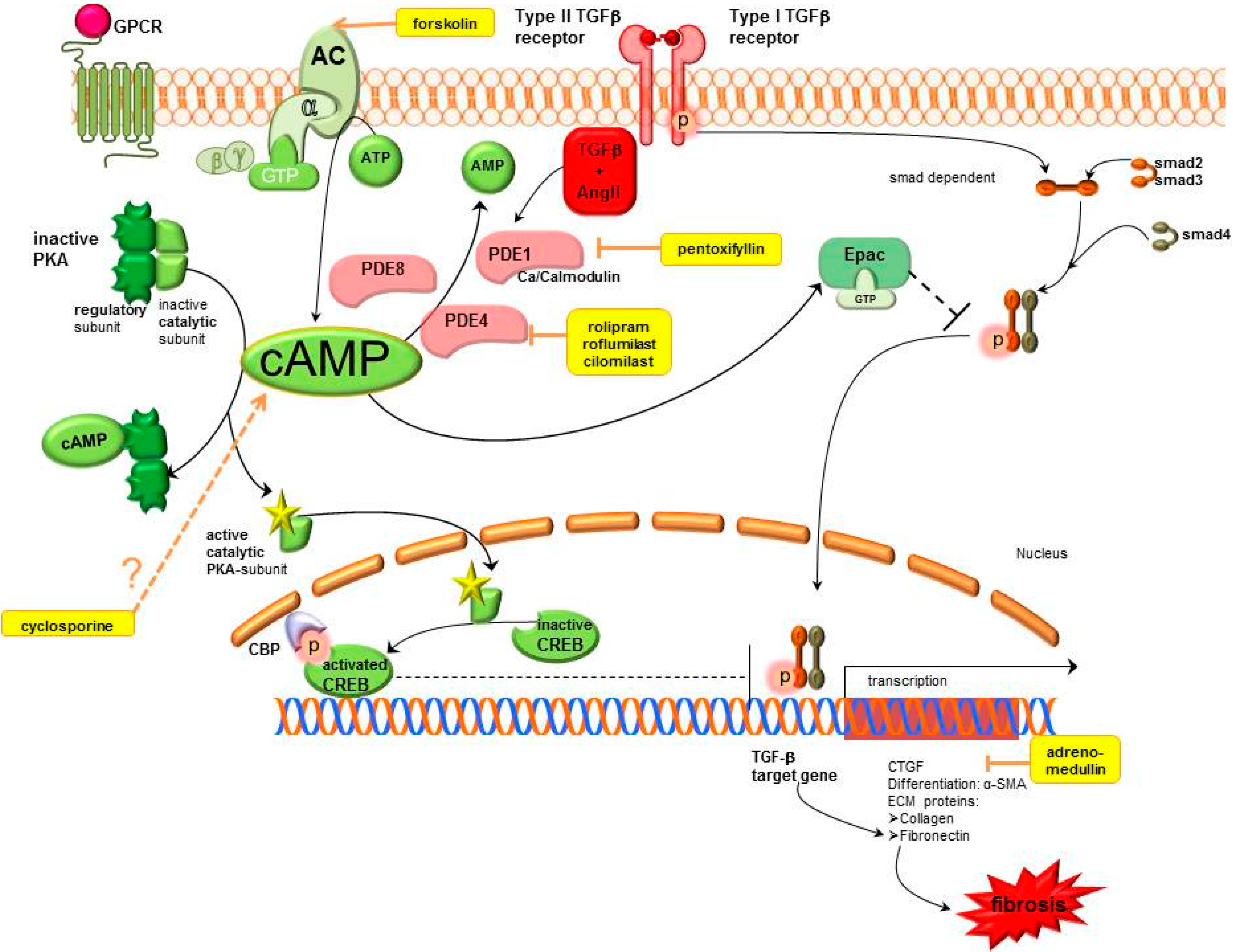
2.1. Cyclic Adenosine Monophosphate (cAMP) Pathway
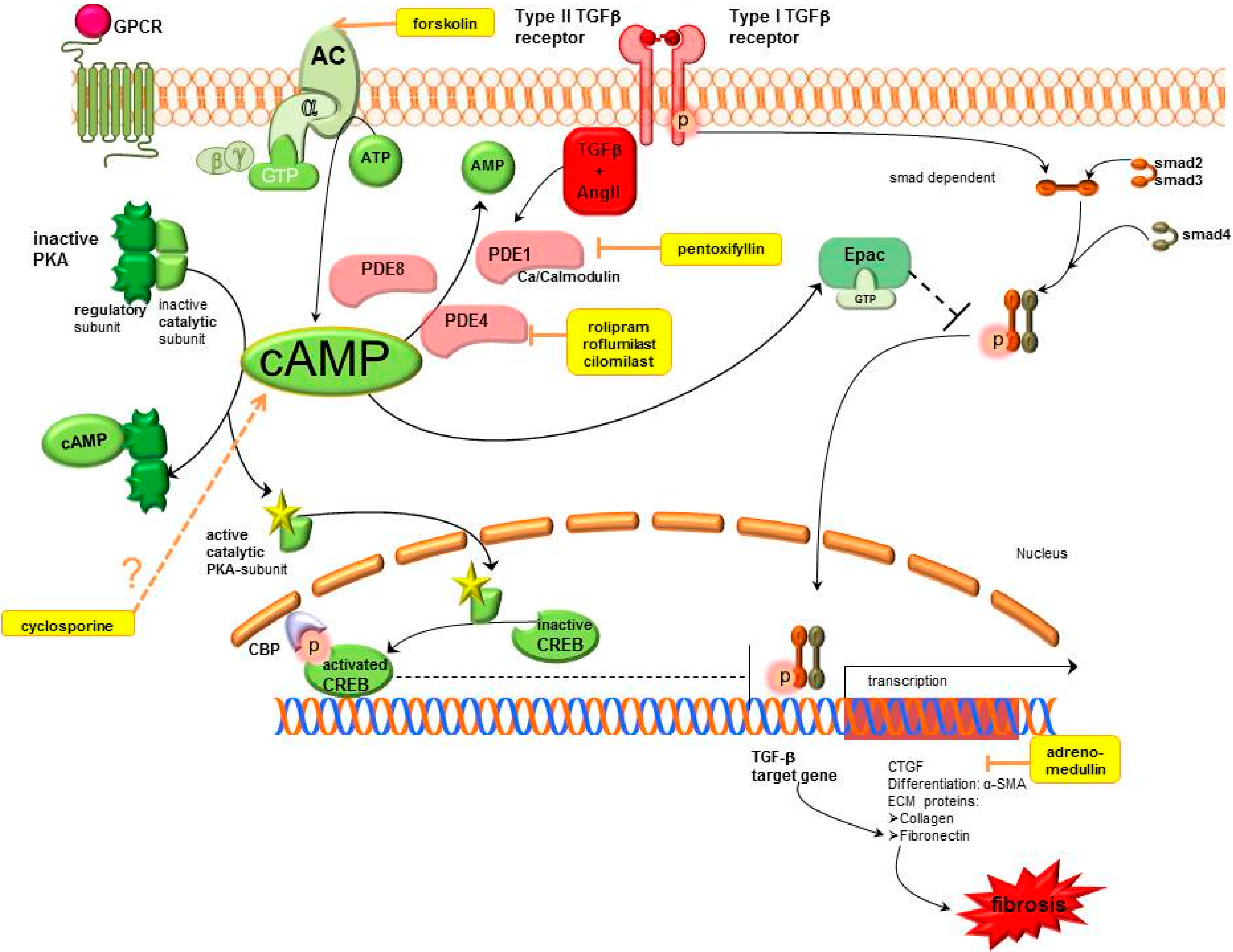
2.1.1. cAMP Modulation
Adenylyl Cyclase (AC)
Phosphodiesterases (PDEs).
2.1.2. cAMP Effectors
Protein Kinase A-cAMP Response Element Binding (PKA-CREB)
Exchange Protein Directly Activated by cAMP (Epac)
2.1.3. Further cAMP Influencing Systems
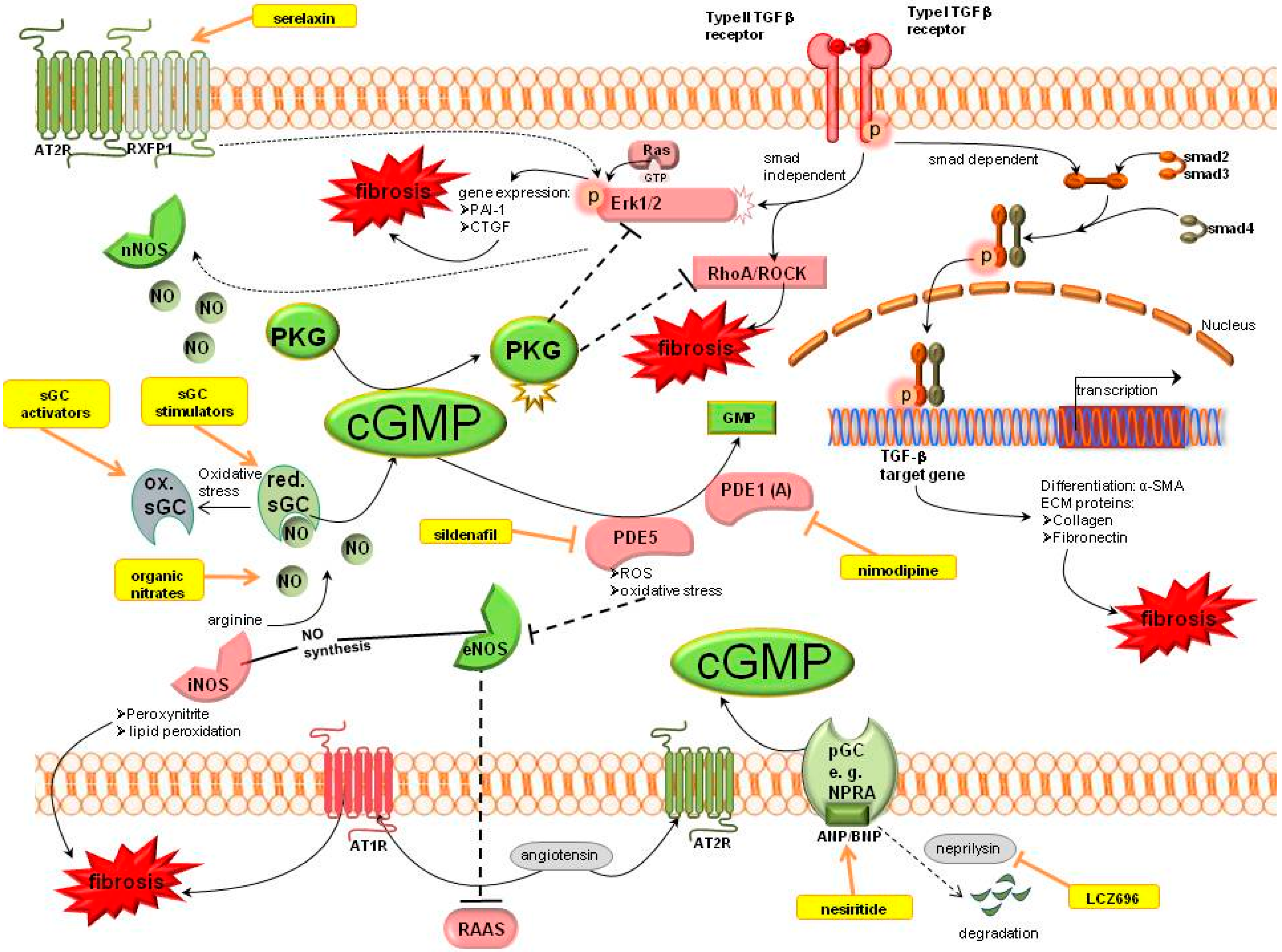
2.2. Cyclic Guanosine Monophosphate (cGMP) Pathway
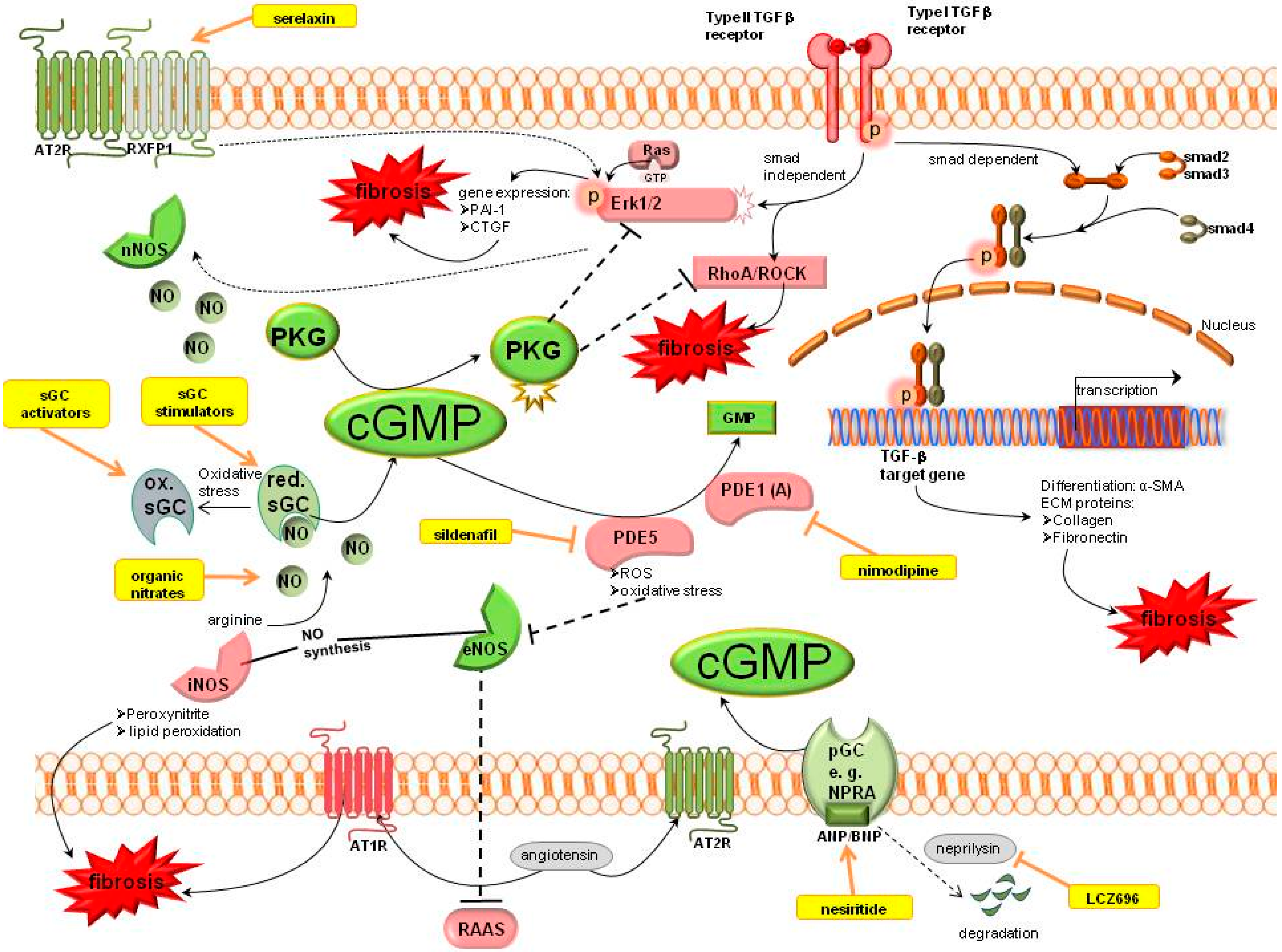
2.2.1. cGMP Modulation
Organic Nitrates
Nitric Oxide Synthase
Soluble Guanylyl Cyclase (sGC)
PDE Inhibitors
Relaxin
Natriuretic Peptides
2.2.2. cGMP Effectors
cGMP Dependent Protein Kinases (PKG)
2.2.3. Further cGMP Influencing Systems
Renin Angiotensin Aldosterone System (RAAS)
Kallikrein
All-Trans-Retinoic Acid/Sodium Butyrate
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Eddy, A.A. The origin of scar-forming kidney myofibroblasts. Nat. Med. 2013, 19, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Honda, E.; Park, A.-M.; Yoshida, K.; Tabuchi, M.; Munakata, H. Myofibroblasts: Biochemical and proteomic approaches to fibrosis. Tohoku J. Exp. Med. 2013, 230, 67–73. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2014. [CrossRef]
- Lin, S.L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am. J. Pathol. 2008, 173, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Campanholle, G.; Ligresti, G.; Gharib, S.A.; Duffield, J.S. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C591–C603. [Google Scholar] [CrossRef] [PubMed]
- Wakino, S.; Kanda, T.; Hayashi, K. Rho/Rho kinase as a potential target for the treatment of renal disease. Drug News Perspect. 2005, 18, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Vernon, M.A.; Mylonas, K.J.; Hughes, J. Macrophages and renal fibrosis. Semin. Nephrol. 2010, 30, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Matavelli, L.C.; Siragy, H.M. Reduction of aldosterone production improves renal oxidative stress and fibrosis in diabetic rats. J. Cardiovasc. Pharmacol. 2013, 61, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Iwano, M.; Higgins, D.F.; Yamaguchi, Y.; Nakatani, K.; Harada, K.; Kubo, A.; Akai, Y.; Rankin, E.B.; Neilson, E.G.; et al. Stable expression of HIF-1α in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Renal Physiol. 2008, 295, F1023–F1029. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Wauquier, F.; Eid, A.A.; Roman, L.J.; Ghosh-Choudhury, G.; Khazim, K.; Block, K.; Gorin, Y. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: Role of mitochondrial reactive oxygen species. J. Biol. Chem. 2013, 288, 28668–28686. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Mendoza, F.A.; Jimenez, S.A. Strategies for anti-fibrotic therapies. Biochim. Biophys. Acta 2013, 1832, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Liu, H.; Ni, H.F.; Lv, L.L.; Zhang, M.H.; Zhang, A.H.; Tang, R.N.; Chen, P.S.; Liu, B.C. Improved mitochondrial function underlies the protective effect of pirfenidone against tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS One 2013, 8, e83593. [Google Scholar] [CrossRef] [PubMed]
- RamachandraRao, S.P.; Zhu, Y.; Ravasi, T.; McGowan, T.A.; Toh, I.; Dunn, S.R.; Okada, S.; Shaw, M.A.; Sharma, K. Pirfenidone is renoprotective in diabetic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krämer, S.; Loof, T.; Martini, S.; Kron, S.; Kawachi, H.; Shimizu, F.; Neumayer, H.; Peters, H. Enhancing cGMP in experimental progressive renal fibrosis: Soluble guanylate cyclase stimulation vs. phosphodiesterase inhibition. Am. J. Physiol. Renal Physiol. 2006, 290, F167–F176. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.; Conway, B.R. Recent advances in animal models of diabetic nephropathy. Nephron. Exp Nephrol. 2014, 126, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Hodgin, J.B.; Nair, V.; Zhang, H.; Randolph, A.; Harris, R.C.; Nelson, R.G.; Weil, E.J.; Cavalcoli, J.D.; Patel, J.M.; Brosius, F.C., III; et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes 2013, 62, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C., III. New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocr. Metab. Disord. 2008, 9, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Heerspink, H.J.; de Zeeuw, D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat. Rev. Nephrol. 2014, 10, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Komers, R.; Oyama, T.T.; Beard, D.R.; Tikellis, C.; Xu, B.; Lotspeich, D.F.; Anderson, S. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int. 2011, 79, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ott, I.M.; Alter, M.L.; von Websky, K.; Kretschmer, A.; Tsuprykov, O.; Sharkovska, Y.; Krause-Relle, K.; Raila, J.; Henze, A.; Stasch, J.P.; et al. Effects of stimulation of soluble guanylate cyclase on diabetic nephropathy in diabetic eNOS knockout mice on top of angiotensin II receptor blockade. PLoS One 2012, 7, e42623. [Google Scholar] [CrossRef] [PubMed]
- Braun, N.; Schweisfurth, A.; Lohofener, C.; Lange, C.; Grundemann, C.; Kundt, G.; Grone, H.J. Epidemiology of glomerulonephritis in Northern Germany. Int. Urol. Nephrol. 2011, 43, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B.; Appel, A.S. New diagnostic tests and new therapies for glomerular diseases. Blood Purif. 2013, 35, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Graziani, G. Current and emerging treatments for idiopathic focal and segmental glomerulosclerosis in adults. Expert. Rev. Clin. Immunol. 2013, 9, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Shankland, S.J.; Pollak, M.R. A suPAR circulating factor causes kidney disease. Nat. Med. 2011, 17, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Hogan, J.; Bomback, A.S.; Mehta, K.; Canetta, P.A.; Rao, M.K.; Appel, G.B.; Radhakrishnan, J.; Lafayette, R.A. Treatment of idiopathic FSGS with adrenocorticotropic hormone gel. Clin. J. Am. Soc. Nephrol. 2013, 8, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Kerschbaum, J.; Fernandez-Fresnedo, G.; Hoxha, E.; Kurschat, C.E.; Busch, M.; Bruchfeld, A.; Mayer, G.; Rudnicki, M. Rituximab treatment for relapsing minimal change disease and focal segmental glomerulosclerosis: A systematic review. Am. J. Nephrol. 2014, 39, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Cyclosporine protects glomeruli from FSGS factor via an increase in glomerular cAMP. Transplantation 1996, 62, 1916–1920. [Google Scholar] [CrossRef] [PubMed]
- Balow, J.E.; Austin, H.A., III. Renal disease in systemic lupus erythematosus. Rheum. Dis. Clin. N. Am. 1988, 14, 117–133. [Google Scholar]
- Michaelson, J.S.; Wisniacki, N.; Burkly, L.C.; Putterman, C. Role of TWEAK in lupus nephritis: A bench-to-bedside review. J. Autoimmun. 2012, 39, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Crampton, S.P.; Morawski, P.A.; Bolland, S. Linking susceptibility genes and pathogenesis mechanisms using mouse models of systemic lupus erythematosus. Dis. Model. Mech. 2014, 7, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma. Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Runkel, L.; Stacey, J. Lupus clinical development: Will belimumab’s approval catalyse a new paradigm for SLE drug development? Expert Opin. Biol. Ther. 2014, 14, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Donadio, J.V., Jr.; Glassock, R.J. Immunosuppressive drug therapy in lupus nephritis. Am. J. Kidney Dis. 1993, 21, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin. Arthritis. Rheum. 1993, 23, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Huang, Y.; Yang, L.; Chen, T.; Xu, J.; Epstein, P.N. Uninephrectomy of diabetic OVE26 mice greatly accelerates albuminuria, fibrosis, inflammatory cell infiltration and changes in gene expression. Nephron. Exp. Nephrol. 2011, 119, e21–e32. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.R.; Rennie, J.; Bailey, M.A.; Dunbar, D.R.; Manning, J.R.; Bellamy, C.O.; Hughes, J.; Mullins, J.J. Hyperglycemia and renin-dependent hypertension synergize to model diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A.; Lopez-Guisa, J.M.; Okamura, D.M.; Yamaguchi, I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr. Nephrol. 2012, 27, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Komers, R. Rho kinase inhibition in diabetic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 551–559. [Google Scholar] [PubMed]
- Wakeland, E.K.; Liu, K.; Graham, R.R.; Behrens, T.W. Delineating the genetic basis of systemic lupus erythematosus. Immunity 2001, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I.; Kang, H.I. Mechanism of action of antimalarial drugs: Inhibition of antigen processing and presentation. Lupus 1993, 2, S9–S12. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Schmidt, P.M.; Stasch, J.P. NO- and haem-independent soluble guanylate cyclase activators. Handb. Exp. Pharmacol. 2009, 191, 309–339. [Google Scholar] [PubMed]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Renal Physiol. 2000, 279, F400–F416. [Google Scholar] [PubMed]
- Swaney, J.S.; Roth, D.M.; Olson, E.R.; Naugle, J.E.; Meszaros, J.G.; Insel, P.A. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. USA 2005, 102, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Dunkern, T.R.; Torsten, R.; Feurstein, D.; Rossi, G.A.; Sabatini, F.; Hatzelmann, A. Inhibition of TGFβ induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur. J. Pharmacol. 2007, 572, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Vatner, S.F.; Yan, L.; Ishikawa, Y.; Vatner, D.E.; Sadoshima, J. Adenylyl cyclase type 5 disruption prolongs longevity and protects the heart against stress. Circ. J. 2009, 73, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, C.; Rajamani, K.; Vadivel, E. Coleus forskohlii—A comprehensive review on morphology, phytochemistry and pharmacological aspects. J. Med. Plants Res. 2010, 4, 278–285. [Google Scholar]
- Dubey, R.K.; Rosselli, M.; Gillespie, D.G.; Mi, Z.; Jackson, E.K. Extracellular 3',5'-cAMP-adenosine pathway inhibits glomerular mesangial cell growth. J. Pharmacol. Exp. Ther. 2010, 333, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kong, Q.; Kone, B.C. CREB trans-activation of disruptor of telomeric silencing-1 mediates forskolin inhibition of CTGF transcription in mesangial cells. Am. J. Physiol. Renal Physiol. 2010, 298, F617–F624. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.J.; Park, K.M.; Choi, H.E.; Chung, K.S.; Lim, H.J.; Park, H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008, 20, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Turko, I.V.; Corbin, J.D. Cyclic nucleotide phosphodiesterases: Relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 1–52. [Google Scholar] [PubMed]
- Sonnenburg, W.K.; Seger, D.; Beavo, J.A. Molecular cloning of a cDNA encoding the “61 kDa” calmodulin-stimulated cyclic nucleotide phosphodiesterase. Tissue-specific expression of structurally related isoforms. J. Biol. Chem. 1993, 268, 645–652. [Google Scholar] [PubMed]
- Snyder, P.B.; Florio, V.A.; Ferguson, K.; Loughney, K. Isolation, expression and analysis of splice variants of a human Ca2+/calmodulin-stimulated phosphodiesterase (PDE1A). Cell Signal. 1999, 11, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Cai, Y.; Oikawa, M.; Thomas, T.; Dostmann, W.R.; Zaccolo, M.; Fujiwara, K.; Yan, C. Cyclic nucleotide phosphodiesterase 1A: A key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res. Cardiol. 2011, 106, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Dousa, T.P. Cyclic-3',5'-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney. Kidney Int. 1999, 55, 29–62. [Google Scholar] [CrossRef] [PubMed]
- Souness, J.E.; Aldous, D.; Sargent, C. Immunosuppressive and anti-inflammatory effects of cyclic AMP phosphodiesterase (PDE) type 4 inhibitors. Immunopharmacology 2000, 47, 127–162. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.; Grande, J.P.; Chini, E.N.; Dousa, T.P. Compartmentalization of cAMP signaling in mesangial cells by phosphodiesterase isozymes PDE3 and PDE4. Regulation of superoxidation and mitogenesis. J. Biol. Chem. 1997, 272, 9854–9859. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, T.; Liu, X.; Wen, F.Q.; Zhu, Y.K.; Wang, H.; Kim, H.J.; Takizawa, H.; Cieslinski, L.B.; Barnette, M.S.; Rennard, S.I. PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am. J. Respir. Cell Mol. Biol. 2002, 26, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Kohyama, T.; Liu, X.; Zhu, Y.K.; Wen, F.Q.; Wang, H.J.; Fang, Q.; Kobayashi, T.; Rennard, S.I. Phosphodiesterase 4 inhibitor cilomilast inhibits fibroblast-mediated collagen gel degradation induced by tumor necrosis factor-α and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 2002, 27, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Togo, S.; Liu, X.; Wang, X.; Sugiura, H.; Kamio, K.; Kawasaki, S.; Kobayashi, T.; Ertl, R.F.; Ahn, Y.; Holz, O.; et al. PDE4 inhibitors roflumilast and rolipram augment PGE2 inhibition of TGFβ1-stimulated fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L959–L969. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Shankland, S.J.; Grande, J.P.; Walker, H.J.; Johnson, R.J.; Dousa, T.P. Suppression of mesangial proliferative glomerulonephritis development in rats by inhibitors of cAMP phosphodiesterase isozymes types III and IV. J. Clin. Investig. 1996, 98, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Tam, F.W.; Smith, J.; Agarwal, S.; Karkar, A.M.; Morel, D.; Thompson, E.M.; Pusey, C.D. Type IV phosphodiesterase inhibitor is effective in prevention and treatment of experimental crescentic glomerulonephritis. Nephron 2000, 84, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Tikoo, K.; Lodea, S.; Karpe, P.A.; Kumar, S. Calorie restriction mimicking effects of roflumilast prevents diabetic nephropathy. Biochem. Biophys. Res. Commun. 2014, 450, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Calverley, P.M.; Rabe, K.F.; Goehring, U.M.; Kristiansen, S.; Fabbri, L.M.; Martinez, F.J. Roflumilast in symptomatic chronic obstructive pulmonary disease: Two randomised clinical trials. Lancet 2009, 374, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, C.; Kase, Y.; Koseki, J.; Hasegawa, Y.; Shindo, S.; Maruyama, H.; Takeda, S.; Takeda, H.; Hattori, T. Effects of TJN-598, a new selective phosphodiesterase type IV inhibitor on anti-Thy1 nephritis in rats. Clin. Exp. Nephrol. 2011, 15, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Monneaux, F.; Yougbare, I.; Gazi, L.; Bourguignon, J.J.; Muller, S.; Lugnier, C. Disease progression in MRL/lpr lupus-prone mice is reduced by NCS 613, a specific cyclic nucleotide phosphodiesterase type 4 (PDE4) inhibitor. PLoS One 2012, 7, e28899. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Aroonsakool, N.; Yokoyama, U.; Patel, H.H.; Insel, P.A. Increase in cellular cyclic AMP concentrations reverses the profibrogenic phenotype of cardiac myofibroblasts: A novel therapeutic approach for cardiac fibrosis. Mol. Pharmacol. 2013, 84, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Clissold, S.P. Pentoxifylline—A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 1987, 34, 50–97. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Chien, C.T.; Hu-Tsai, M.I.; Wu, K.D.; Tsai, C.C.; Wu, M.S.; Tsai, T.J. Pentoxifylline attenuates experimental mesangial proliferative glomerulonephritis. Kidney Int. 1999, 56, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chen, Y.M.; Chien, C.T.; Chiang, W.C.; Tsai, C.C.; Tsai, T.J. Pentoxifylline attenuated the renal disease progression in rats with remnant kidney. J. Am. Soc. Nephrol. 2002, 13, 2916–2929. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chen, R.H.; Chen, Y.M.; Chiang, W.C.; Lai, C.F.; Wu, K.D.; Tsai, T.J. Pentoxifylline attenuates tubulointerstitial fibrosis by blocking Smad3/4-activated transcription and profibrogenic effects of connective tissue growth factor. J. Am. Soc. Nephrol. 2005, 16, 2702–2713. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, M.; Noorafshan, A.; Farrokhi, A. Effects of pentoxifylline on renal structure after urethral obstruction in rat: A stereological study. Cent. Eur. J. Urol. 2011, 64, 30–33. [Google Scholar] [CrossRef]
- Yagmurlu, A.; Boleken, M.E.; Ertoy, D.; Ozsan, M.; Gokcora, I.H.; Dindar, H. Preventive effect of pentoxifylline on renal scarring in rat model of pyelonephritis. Urology 2003, 61, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Cooper, M.E. Diabetic nephropathy: Renoprotective effects of pentoxifylline in the PREDIAN trial. Nat. Rev. Nephrol. 2014, 10, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Schiller, M.; Verrecchia, F.; Mauviel, A. Cyclic adenosine 3',5'-monophosphate-elevating agents inhibit transforming growth factor-β-induced SMAD3/4-dependent transcription via a protein kinase A-dependent mechanism. Oncogene 2003, 22, 8881–8890. [Google Scholar] [CrossRef] [PubMed]
- Mehra, A.; Wrana, J.L. TGFβ and the Smad signal transduction pathway. Biochem. Cell Biol. 2002, 80, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGFβ signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Suzuki, S.; Jin, M.; Jiao, Q.; Watanabe, M.; Otsu, K.; Iwasaki, S.; et al. Prostaglandin E2-activated Epac promotes neointimal formation of the rat ductus arteriosus by a process distinct from that of cAMP-dependent protein kinase A. J. Biol. Chem. 2008, 283, 28702–28709. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Stokman, G.; Qin, Y.; Booij, T.H.; Ramaiahgari, S.; Lacombe, M.; Dolman, M.E.; van Dorenmalen, K.M.; Teske, G.J.; Florquin, S.; Schwede, F.; et al. Epac-Rap signaling reduces oxidative stress in the tubular epithelium. J. Am. Soc. Nephrol. 2014, 25, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Dendooven, A.; Ishola, D.A., Jr.; Nguyen, T.Q.; van der Giezen, D.M.; Kok, R.J.; Goldschmeding, R.; Joles, J.A. Oxidative stress in obstructive nephropathy. Int. J. Exp. Pathol. 2011, 92, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Nagae, T.; Mori, K.; Mukoyama, M.; Kasahara, M.; Yokoi, H.; Suganami, T.; Sawai, K.; Yoshioka, T.; Koshikawa, M.; Saito, Y.; et al. Adrenomedullin inhibits connective tissue growth factor expression, extracellular signal-regulated kinase activation and renal fibrosis. Kidney Int. 2008, 74, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Kato, K.; Zhang, J.J.; Dobrzynski, E.; Wang, C.; Agata, J.; Chao, L. Human adrenomedullin gene delivery protects against cardiovascular remodeling and renal injury. Peptides 2001, 22, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Yoshida, H.; Chao, L.; Chao, J. Human adrenomedullin gene delivery protects against cardiac hypertrophy, fibrosis, and renal damage in hypertensive dahl salt-sensitive rats. Hum. Gene Ther. 2000, 11, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Schlossmann, J.; Schinner, E. cGMP becomes a drug target. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 243–252. [Google Scholar] [CrossRef]
- Peters, H.; Border, W.A.; Noble, N.A. Tandem antifibrotic actions of l-arginine supplementation and low protein diet during the repair phase of experimental glomerulonephritis. Kidney Int. 2000, 57, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Daig, U.; Martini, S.; Ruckert, M.; Schaper, F.; Liefeldt, L.; Kramer, S.; Neumayer, H.H. NO mediates antifibrotic actions of l-arginine supplementation following induction of anti-thy1 glomerulonephritis. Kidney Int. 2003, 64, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Yamaleyeva, L.M.; Lindsey, S.H.; Varagic, J.; Zhang, L.L.; Gallagher, P.E.; Chen, A.F.; Chappell, M.C. Amelioration of renal injury and oxidative stress by the nNOS inhibitor L-VNIO in the salt-sensitive mRen2.Lewis congenic rat. J. Cardiovasc. Pharmacol. 2012, 59, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ezzidi, I.; Mtiraoui, N.; Mohamed, M.B.; Mahjoub, T.; Kacem, M.; Almawi, W.Y. Association of endothelial nitric oxide synthase Glu298Asp, 4b/a, and −786T>C gene variants with diabetic nephropathy. J. Diabetes Complicat. 2008, 22, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Gheorghiade, M.; Marti, C.N.; Sabbah, H.N.; Roessig, L.; Greene, S.J.; Bohm, M.; Burnett, J.C.; Campia, U.; Cleland, J.G.; Collins, S.P.; et al. Soluble guanylate cyclase: A potential therapeutic target for heart failure. Heart Fail. Rev. 2013, 18, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Ni, Z.; Oveisi, F.; Liang, K.; Pandian, R. Enhanced nitric oxide inactivation and protein nitration by reactive oxygen species in renal insufficiency. Hypertension 2002, 39, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.P.; Kuo, M.S.; Wu, B.N.; Chai, C.Y.; Huang, H.T.; Chung, P.W.; Chen, I.J. NO-releasing xanthine KMUP-1 bonded by simvastatin attenuates bleomycin-induced lung inflammation and delayed fibrosis. Pulm Pharmacol. Ther. 2014, 27, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Knorr, A.; Hirth-Dietrich, C.; Alonso-Alija, C.; Harter, M.; Hahn, M.; Keim, Y.; Wunder, F.; Stasch, J.P. Nitric oxide-independent activation of soluble guanylate cyclase by BAY 60–2770 in experimental liver fibrosis. Arzneimittelforschung 2008, 58, 71–80. [Google Scholar] [PubMed]
- Kashiwagi, M.; Shinozaki, M.; Hirakata, H.; Tamaki, K.; Hirano, T.; Tokumoto, M.; Goto, H.; Okuda, S.; Fujishima, M. Locally activated renin-angiotensin system associated with TGFβ1 as a major factor for renal injury induced by chronic inhibition of nitric oxide synthase in rats. J. Am. Soc. Nephrol. 2000, 11, 616–624. [Google Scholar] [PubMed]
- Forbes, M.S.; Thornhill, B.A.; Park, M.H.; Chevalier, R.L. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am. J. Pathol. 2007, 170, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol. Lett. 2003, 140, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.H.; Lee, T.W.; Ihm, C.G.; Lee, S.H.; Moon, J.Y.; Lim, S.J. Effects of sildenafil on oxidative and inflammatory injuries of the kidney in streptozotocin-induced diabetic rats. Am. J. Nephrol. 2009, 29, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Tsuruda, T.; Kato, J.; Imamura, T.; Asada, Y.; Stasch, J.P.; Kitamura, K.; Eto, T. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens. Res. 2009, 32, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Keil, A.; Blom, I.E.; Goldschmeding, R.; Rupprecht, H.D. Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int. 2002, 62, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Hofmann, F.; Stasch, J.P. Handbook of Experimental Pharmacology 191. cGMP: Generators, effectors and therapeutic implications. Pref. Handb. Exp. Pharmacol. 2009, 191, v–vi. [Google Scholar]
- Nossaman, B.; Pankey, E.; Kadowitz, P. Stimulators and activators of soluble guanylate cyclase: Review and potential therapeutic indications. Crit. Care Res. Pract. 2012, 2012, 290805. [Google Scholar] [PubMed]
- Deruelle, P.; Balasubramaniam, V.; Kunig, A.M.; Seedorf, G.J.; Markham, N.E.; Abman, S.H. BAY 41–2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Neonatology 2006, 90, 135–144. [Google Scholar]
- Dumitrascu, R.; Weissmann, N.; Ghofrani, H.A.; Dony, E.; Beuerlein, K.; Schmidt, H.; Stasch, J.P.; Gnoth, M.J.; Seeger, W.; Grimminger, F.; et al. Activation of soluble guanylate cyclase reverses experimental pulmonary hypertension and vascular remodeling. Circulation 2006, 113, 286–295. [Google Scholar] [CrossRef]
- Masuyama, H.; Tsuruda, T.; Kato, J.; Imamura, T.; Asada, Y.; Stasch, J.P.; Kitamura, K.; Eto, T. Soluble guanylate cyclase stimulation on cardiovascular remodeling in angiotensin II-induced hypertensive rats. Hypertension 2006, 48, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef]
- Friebe, A.; Mullershausen, F.; Smolenski, A.; Walter, U.; Schultz, G.; Koesling, D. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol. Pharmacol. 1998, 54, 962–967. [Google Scholar]
- Hwang, T.L.; Wu, C.C.; Guh, J.H.; Teng, C.M. Potentiation of tumor necrosis factor-α expression by YC-1 in alveolar macrophages through a cyclic GMP-independent pathway. Biochem. Pharmacol. 2003, 66, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Feelisch, M.; Kotsonis, P.; Siebe, J.; Clement, B.; Schmidt, H.H. The soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3,-a] quinoxalin-1-one is a nonselective heme protein inhibitor of nitric oxide synthase and other cytochrome P-450 enzymes involved in nitric oxide donor bioactivation. Mol. Pharmacol. 1999, 56, 243–253. [Google Scholar] [PubMed]
- Ko, F.N.; Wu, C.C.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1, a novel activator of platelet guanylate cyclase. Blood 1994, 84, 4226–4233. [Google Scholar] [PubMed]
- Wu, C.C.; Ko, F.N.; Kuo, S.C.; Lee, F.Y.; Teng, C.M. YC-1 inhibited human platelet aggregation through NO-independent activation of soluble guanylate cyclase. Br. J. Pharmacol. 1995, 116, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.W.; Kim, S.Y.; Lee, J.H.; Lee, Y.S. YC-1 attenuates hypoxia-induced pulmonary arterial hypertension in mice. Pulm Pharmacol. Ther. 2011, 24, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Schinner, E.; Schramm, A.; Kees, F.; Hofmann, F.; Schlossmann, J. The cyclic GMP-dependent protein kinase Iα suppresses kidney fibrosis. Kidney Int. 2013, 84, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kramer, S.; Loof, T.; Martini, S.; Kron, S.; Kawachi, H.; Shimizu, F.; Neumayer, H.H.; Peters, H. Stimulation of soluble guanylate cyclase slows progression in anti-thy1-induced chronic glomerulosclerosis. Kidney Int. 2005, 68, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, B.; Daniel, C.; Wagner, A.; Stasch, J.P.; Hugo, C. Stimulation of soluble guanylyl cyclase inhibits mesangial cell proliferation and matrix accumulation in experimental glomerulonephritis. Am. J. Physiol Renal Physiol. 2005, 288, F685–F693. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Dembowsky, K.; Perzborn, E.; Stahl, E.; Schramm, M. Cardiovascular actions of a novel NO-independent guanylyl cyclase stimulator, BAY 41-8543: In vivo studies. Br. J. Pharmacol. 2002, 135, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Wang-Rosenke, Y.; Mika, A.; Khadzhynov, D.; Loof, T.; Neumayer, H.H.; Peters, H. Stimulation of soluble guanylate cyclase improves renal recovery after relief of unilateral ureteral obstruction. J. Urol. 2011, 186, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Hobbs, A.J. NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb. Exp. Pharmacol. 2009, 191, 277–308. [Google Scholar] [PubMed]
- Conole, D.; Scott, L.J. Riociguat: First global approval. Drugs 2013, 73, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Geschka, S.; Kretschmer, A.; Sharkovska, Y.; Evgenov, O.V.; Lawrenz, B.; Hucke, A.; Hocher, B.; Stasch, J.P. Soluble guanylate cyclase stimulation prevents fibrotic tissue remodeling and improves survival in salt-sensitive Dahl rats. PLoS One 2011, 6, e21853. [Google Scholar] [CrossRef] [PubMed]
- Sharkovska, Y.; Kalk, P.; Lawrenz, B.; Godes, M.; Hoffmann, L.S.; Wellkisch, K.; Geschka, S.; Relle, K.; Hocher, B.; Stasch, J.P. Nitric oxide-independent stimulation of soluble guanylate cyclase reduces organ damage in experimental low-renin and high-renin models. J. Hypertens. 2010, 28, 1666–1675. [Google Scholar] [CrossRef]
- Kalk, P.; Godes, M.; Relle, K.; Rothkegel, C.; Hucke, A.; Stasch, J.P.; Hocher, B. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br. J. Pharmacol. 2006, 148, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Benz, K.; Orth, S.R.; Simonaviciene, A.; Linz, W.; Schindler, U.; Rutten, H.; Amann, K. Blood pressure-independent effect of long-term treatment with the soluble heme-independent guanylyl cyclase activator HMR1766 on progression in a model of noninflammatory chronic renal damage. Kidney Blood Press Res. 2007, 30, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, E.; Semigran, M.J.; Nieminen, M.S.; Gheorghiade, M.; Agrawal, R.; Mitrovic, V.; Mebazaa, A. Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur. Heart J. 2013, 34, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yougbare, I.; Keravis, T.; Abusnina, A.; Decossas, M.; Schall, N.; Muller, S.; Lugnier, C. Cyclic GMP catabolism up-regulation in MRL/lpr lupus-prone mice is associated with organ remodeling. Biochim. Biophys. Acta 2014, 1842, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Maimaitiyiming, H.; Qi, X.; Norman, H.; Zhou, Q.; Wang, X.; Fu, J.; Wang, S. Increasing cGMP-dependent protein kinase activity attenuates unilateral ureteral obstruction-induced renal fibrosis. Am. J. Physiol. Renal Physiol. 2014, 306, F996–F1007. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Iturbe, B.; Ferrebuz, A.; Vanegas, V.; Quiroz, Y.; Espinoza, F.; Pons, H.; Vaziri, N.D. Early treatment with cGMP phosphodiesterase inhibitor ameliorates progression of renal damage. Kidney Int. 2005, 68, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Tapia, E.; Sanchez-Lozada, L.G.; Soto, V.; Manrique, A.M.; Ortiz-Vega, K.M.; Santamaria, J.; Medina-Campos, O.N.; Cristobal, M.; Avila-Casado, C.; Pedraza-Chaverri, J.; et al. Sildenafil treatment prevents glomerular hypertension and hyperfiltration in rats with renal ablation. Kidney Blood Press Res. 2012, 35, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.H.; Kim, I.J.; Joo, S.Y.; Kim, E.Y.; Kim, C.S.; Choi, J.S.; Ma, S.K.; Kim, S.H.; Lee, J.U.; Kim, S.W. Renoprotective effects of sildenafil in DOCA-salt hypertensive rats. Kidney Blood Press Res. 2012, 36, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Muzaffar, S.; Shukla, N.; Srivastava, A.; Angelini, G.D.; Jeremy, J.Y. Sildenafil citrate and sildenafil nitrate (NCX 911) are potent inhibitors of superoxide formation and gp91phox expression in porcine pulmonary artery endothelial cells. Br. J. Pharmacol. 2005, 146, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Guzeloglu, M.; Yalcinkaya, F.; Atmaca, S.; Bagriyanik, A.; Oktar, S.; Yuksel, O.; Fansa, I.; Hazan, E. The beneficial effects of tadalafil on renal ischemia-reperfusion injury in rats. Urol. Int. 2011, 86, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Sohotnik, R.; Nativ, O.; Abbasi, A.; Awad, H.; Frajewicki, V.; Bishara, B.; Sukhotnik, I.; Armaly, Z.; Aronson, D.; Heyman, S.N.; et al. Phosphodiesterase-5 inhibition attenuates early renal ischemia-reperfusion-induced acute kidney injury: Assessment by quantitative measurement of urinary NGAL and KIM-1. Am. J. Physiol. Renal Physiol. 2013, 304, F1099–F1104. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.Y.; Nakabayashi, K.; Nishi, S.; Kumagai, J.; Kudo, M.; Sherwood, O.D.; Hsueh, A.J. Activation of orphan receptors by the hormone relaxin. Science 2002, 295, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Bathgate, R.A.; Halls, M.L.; van der Westhuizen, E.T.; Callander, G.E.; Kocan, M.; Summers, R.J. International Union of Pharmacology LVII: Recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol. Rev. 2006, 58, 7–31. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.G. Relaxin and its role in the development and treatment of fibrosis. Transl. Res. 2009, 154, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Heeg, M.H.; Koziolek, M.J.; Vasko, R.; Schaefer, L.; Sharma, K.; Muller, G.A.; Strutz, F. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005, 68, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, I.; Hewitson, T.D.; Halls, M.L.; Summers, R.J.; Mathai, M.L.; Bathgate, R.A.; Tregear, G.W.; Samuel, C.S. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009, 23, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Masterson, R.; Hewitson, T.D.; Kelynack, K.; Martic, M.; Parry, L.; Bathgate, R.; Darby, I.; Becker, G. Relaxin down-regulates renal fibroblast function and promotes matrix remodelling in vitro. Nephrol. Dial. Transplant. 2004, 19, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.; Chew, E.G.; Zhao, C.; Bathgate, R.A.; Hewitson, T.D.; Samuel, C.S. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: The additional involvement of iNOS. PLoS One 2012, 7, e42714. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.; Kocan, M.; Bosnyak, S.; Sarwar, M.; Wigg, B.; Jones, E.S.; Widdop, R.E.; Summers, R.J.; Bathgate, R.A.; Hewitson, T.D.; et al. Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int. 2014, 86, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Gai, Y.; Yang, N.; Lu, B.; Samuel, C.S.; Thannickal, V.J.; Zhou, Y. Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am. J. Pathol. 2011, 179, 2751–2765. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kumagai, H.; Suzuki, A.; Kobayashi, N.; Ohkawa, S.; Odamaki, M.; Kohsaka, T.; Yamamoto, T.; Ikegaya, N. Relaxin ameliorates salt-sensitive hypertension and renal fibrosis. Nephrol. Dial. Transplant. 2012, 27, 2190–2197. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Mookerjee, I.; Masterson, R.; Zhao, C.; Tregear, G.W.; Becker, G.J.; Samuel, C.S. Endogenous relaxin is a naturally occurring modulator of experimental renal tubulointerstitial fibrosis. Endocrinology 2007, 148, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Zhao, C.; Wigg, B.; Lee, S.W.; Simpson, E.R.; Boon, W.C.; Samuel, C.S. Relaxin and castration in male mice protect from, but testosterone exacerbates, age-related cardiac and renal fibrosis, whereas estrogens are an independent determinant of organ size. Endocrinology 2012, 153, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.S.; Zhao, C.; Bond, C.P.; Hewitson, T.D.; Amento, E.P.; Summers, R.J. Relaxin-1-deficient mice develop an age-related progression of renal fibrosis. Kidney Int. 2004, 65, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.A.; Welford, A.; Harris, A. Relaxin improves renal function and histology in aging Munich Wistar rats. J. Am. Soc. Nephrol. 2006, 17, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.A.; Sarkar, P.; Rennke, H.; Unemori, E.; Kalluri, R.; Sukhatme, V.P. Relaxin increases ubiquitin-dependent degradation of fibronectin in vitro and ameliorates renal fibrosis in vivo. Am. J. Physiol. Renal Physiol. 2003, 285, F59–F67. [Google Scholar] [PubMed]
- Sasser, J.M.; Molnar, M.; Baylis, C. Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not Nω-nitro-l-arginine methyl ester hypertensive rats. Hypertension 2011, 58, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Garber, S.L.; Mirochnik, Y.; Brecklin, C.S.; Unemori, E.N.; Singh, A.K.; Slobodskoy, L.; Grove, B.H.; Arruda, J.A.; Dunea, G. Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int. 2001, 59, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Garber, S.L.; Mirochnik, Y.; Brecklin, C.; Slobodskoy, L.; Arruda, J.A.; Dunea, G. Effect of relaxin in two models of renal mass reduction. Am. J. Nephrol. 2003, 23, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Lekgabe, E.D.; Kiriazis, H.; Zhao, C.; Xu, Q.; Moore, X.L.; Su, Y.; Bathgate, R.A.; Du, X.J.; Samuel, C.S. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 2005, 46, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Haase, N.; Rugor, J.; Przybyl, L.; Qadri, F.; Muller, D.N.; Dechend, R. Relaxin does not improve angiotensin II-induced target-organ damage. PLoS One 2014, 9, e93743. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.E.; Samuel, C.S.; Kelly, D.J.; Zhang, Y.; Becker, G.J.; Hewitson, T.D. The anti-fibrotic hormone relaxin is not reno-protective, despite being active, in an experimental model of type 1 diabetes. Protein Pept. Lett. 2013, 20, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Pini, A.; Shemesh, R.; Samuel, C.S.; Bathgate, R.A.; Zauberman, A.; Hermesh, C.; Wool, A.; Bani, D.; Rotman, G. Prevention of bleomycin-induced pulmonary fibrosis by a novel antifibrotic peptide with relaxin-like activity. J. Pharmacol. Exp. Ther. 2010, 335, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Sager, G. Cyclic GMP transporters. Neurochem. Int. 2004, 45, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, Y.; Li, X.; Zhang, J.; Xu, D. C-type natriuretic peptide ameliorates ischemia/reperfusion-induced acute kidney injury by inhibiting apoptosis and oxidative stress in rats. Life Sci. 2014, 117, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Oparil, S.; Novak, L.; Cao, X.; Shi, W.; Lucas, J.; Chen, Y.F. ANP signaling inhibits TGFβ-induced Smad2 and Smad3 nuclear translocation and extracellular matrix expression in rat pulmonary arterial smooth muscle cells. J. Appl. Physiol. 2007, 102, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.F. Atrial natriuretic peptide inhibits transforming growth factor β-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Au, E.; Krazit, S.T.; Pandey, K.N. Targeted disruption of guanylyl cyclase-A/natriuretic peptide receptor-A gene provokes renal fibrosis and remodeling in null mutant mice: Role of proinflammatory cytokines. Endocrinology 2010, 151, 5841–5850. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Periyasamy, R.; Das, S.; Neerukonda, S.; Mani, I.; Pandey, K.N. All-trans retinoic acid and sodium butyrate enhance natriuretic peptide receptor a gene transcription: Role of histone modification. Mol. Pharmacol. 2014, 85, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Mukoyama, M.; Yokoi, H.; Kasahara, M.; Mori, K.; Kato, Y.; Kuwabara, T.; Imamaki, H.; Kawanishi, T.; Koga, K.; et al. Natriuretic peptide receptor guanylyl cyclase-A protects podocytes from aldosterone-induced glomerular injury. J. Am. Soc. Nephrol. 2012, 23, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Inaba-Iemura, C.; Ishimura, K.; Tadokoro, K.; Koshikawa, S.; Ishikawa, K.; Akimoto, K.; Hattori, Y.; Kasai, K.; Minamino, N.; et al. Natriuretic peptide/natriuretic peptide receptor-A (NPR-A) system has inhibitory effects in renal fibrosis in mice. Regul. Pept. 2009, 154, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Makino, H.; Mukoyama, M.; Mori, K.; Suganami, T.; Kasahara, M.; Yahata, K.; Nagae, T.; Yokoi, H.; Sawai, K.; Ogawa, Y.; et al. Transgenic overexpression of brain natriuretic peptide prevents the progression of diabetic nephropathy in mice. Diabetologia 2006, 49, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Mukoyama, M.; Sugawara, A.; Mori, K.; Nagae, T.; Kasahara, M.; Yahata, K.; Makino, H.; Fujinaga, Y.; Ogawa, Y.; et al. Overexpression of brain natriuretic peptide in mice ameliorates immune-mediated renal injury. J. Am. Soc. Nephrol. 2001, 12, 2652–2663. [Google Scholar] [PubMed]
- Kasahara, M.; Mukoyama, M.; Sugawara, A.; Makino, H.; Suganami, T.; Ogawa, Y.; Nakagawa, M.; Yahata, K.; Goto, M.; Ishibashi, R.; et al. Ameliorated glomerular injury in mice overexpressing brain natriuretic peptide with renal ablation. J. Am. Soc. Nephrol. 2000, 11, 1691–1701. [Google Scholar] [PubMed]
- Ameenuddin, S.; Oehler, E.A.; Burnett, J.C., Jr.; Chen, H.H. Treatment with CBA-NP a novel chimeric natriuretic peptide attenuates cardiorenal fibrosis and improves diastolic dysfunction in diabetic rat model. J. Card. Fail. 2011, 17, S22. [Google Scholar] [CrossRef]
- Martin, F.L.; Sangaralingham, S.J.; McKie, P.M.; Huntley, B.K.; Harders, G.E.; Chen, H.H.; Burnett, J.C. Prevention of cardiorenal fibrosis and suppression of proteinuria and aldosterone activation following experimental myocardial infarction with the novel natriuretic peptide CD-NP. J. Card. Fail. 2009, 15, S3. [Google Scholar] [CrossRef]
- Testani, J.M. Renal subanalysis of the acute study of clinical effectiveness of nesiritide in decompensated heart failure (ASCEND-HF): The end of nesiritide as a cardiorenal therapeutic? Circulation 2014, 130, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Judge, P.; Haynes, R.; Landray, M.J.; Baigent, C. Neprilysin inhibition in chronic kidney disease. Nephrol. Dial. Transplant. 2014. [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Mora, I.; Cortes, M.A.; Calleros, L.; Garcia-Jerez, A.; Ortiz, A.; Rodriguez-Puyol, M.; Rodriguez-Puyol, D.; Olmos, G. Relevant role of PKG in the progression of fibrosis induced by TNF-like weak inducer of apoptosis. Am. J. Physiol. Renal Physiol. 2014, 307, F75–F85. [Google Scholar] [CrossRef] [PubMed]
- Faria-Costa, G.; Leite-Moreira, A.; Henriques-Coelho, T. Cardiovascular effects of the angiotensin type 2 receptor. Rev. Port. Cardiol. 2014, 33, 439–449. [Google Scholar] [PubMed]
- Persson, F.; Lewis, J.B.; Lewis, E.J.; Rossing, P.; Hollenberg, N.K.; Hans-Henrik, P. Impact of aliskiren treatment on urinary aldosterone levels in patients with type 2 diabetes and nephropathy: An AVOID substudy. J. Renin Angiotensin Aldosterone Syst. 2012, 13, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Nadler, J.L.; Hsueh, W.; Horton, R. Therapeutic effect of calcium channel blockade in primary aldosteronism. J. Clin. Endocrinol. Metab. 1985, 60, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Bledsoe, G.; Kato, K.; Chao, L.; Chao, J. Tissue kallikrein attenuates salt-induced renal fibrosis by inhibition of oxidative stress. Kidney Int. 2004, 66, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Yayama, K.; Chao, L.; Chao, J. Human kallikrein gene delivery protects against gentamycin-induced nephrotoxicity in rats. Kidney Int. 1998, 53, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.-F.; Bledsoe, G.; Chao, L.; Chao, J. Kallikrein gene transfer reduces renal fibrosis, hypertrophy, and proliferation in DOCA-salt hypertensive rats. Am. J. Physiol. Renal Physiol. 2005, 289, F622–F631. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Periyasamy, R.; Das, S.; Pandey, K. Retinoic acid and sodium butyrate attenuate renal fibrosis and inflammation in guanylyl cyclase-A/natriuretic peptide receptor-A gene-targeted mice (796.13). FASEB J. 2014, 28 Supplement 796-13. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schinner, E.; Wetzl, V.; Schlossmann, J. Cyclic Nucleotide Signalling in Kidney Fibrosis. Int. J. Mol. Sci. 2015, 16, 2320-2351. https://doi.org/10.3390/ijms16022320
Schinner E, Wetzl V, Schlossmann J. Cyclic Nucleotide Signalling in Kidney Fibrosis. International Journal of Molecular Sciences. 2015; 16(2):2320-2351. https://doi.org/10.3390/ijms16022320
Chicago/Turabian StyleSchinner, Elisabeth, Veronika Wetzl, and Jens Schlossmann. 2015. "Cyclic Nucleotide Signalling in Kidney Fibrosis" International Journal of Molecular Sciences 16, no. 2: 2320-2351. https://doi.org/10.3390/ijms16022320
APA StyleSchinner, E., Wetzl, V., & Schlossmann, J. (2015). Cyclic Nucleotide Signalling in Kidney Fibrosis. International Journal of Molecular Sciences, 16(2), 2320-2351. https://doi.org/10.3390/ijms16022320