
Tissue Regeneration in the Chronically Inflamed Tumor Environment: Implications for Cell Fusion Driven Tumor Progression and Therapy Resistant Tumor Hybrid Cells


Abstract
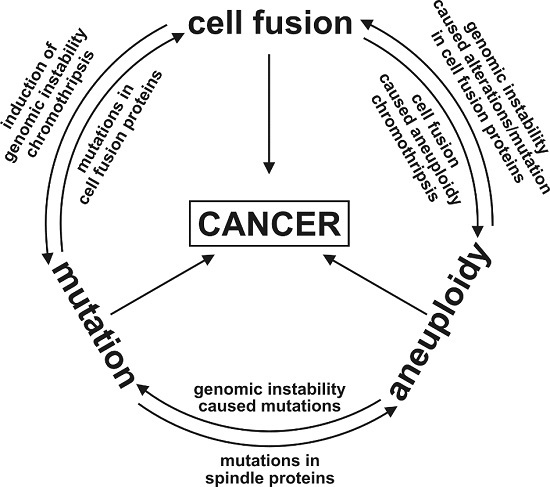
:
1. Introduction

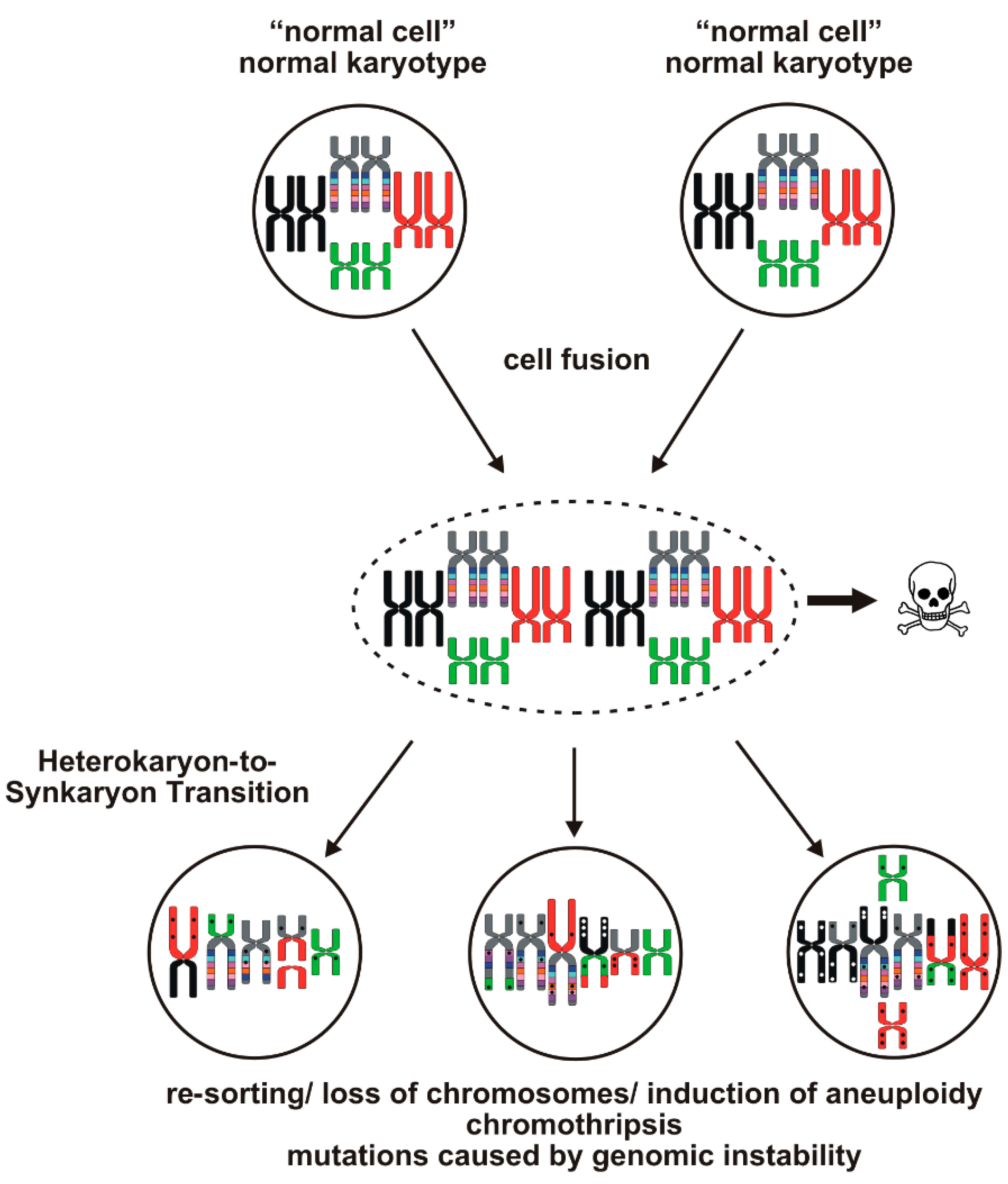
{kind=link}
{kind=link}
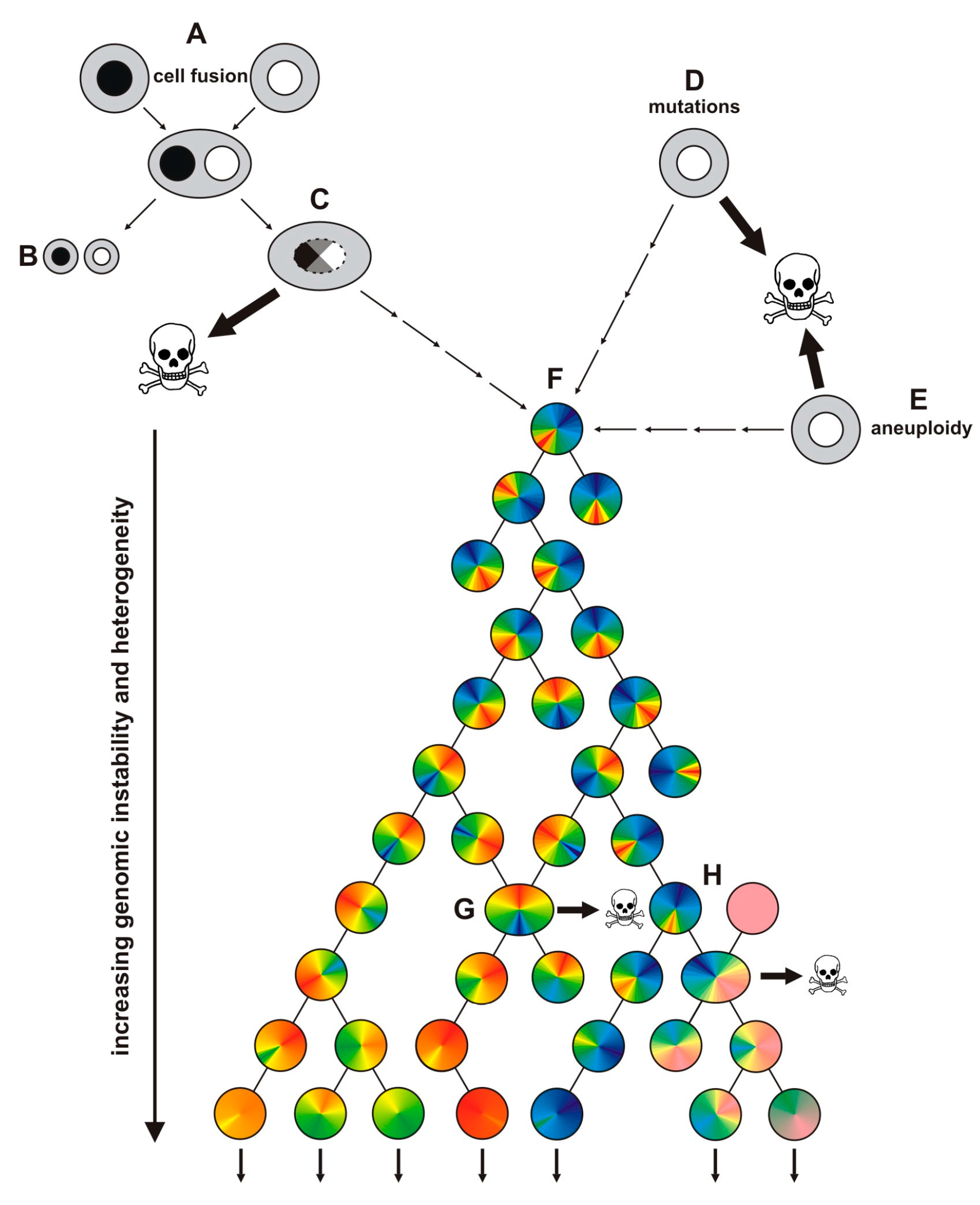{kind=link}
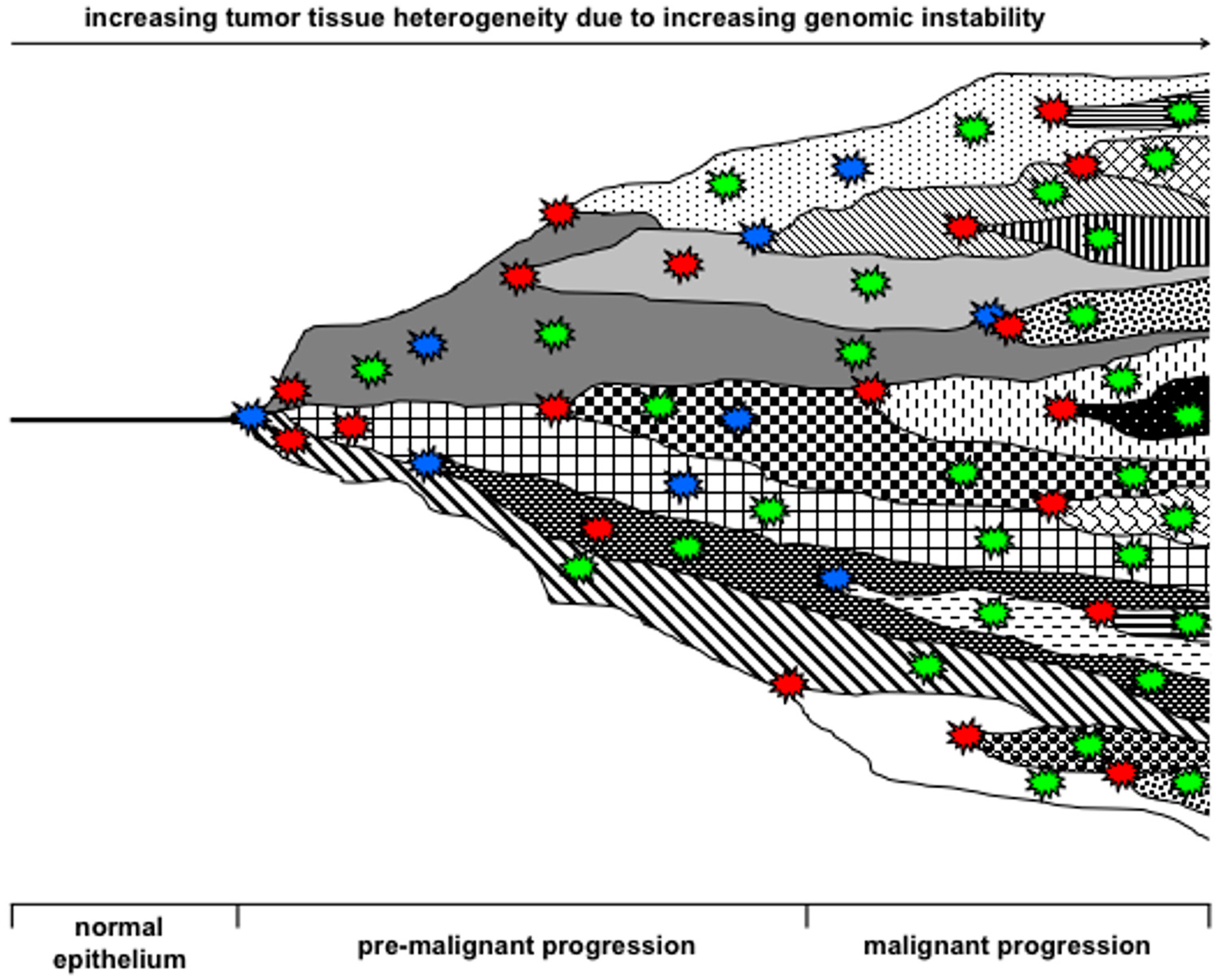{kind=link}
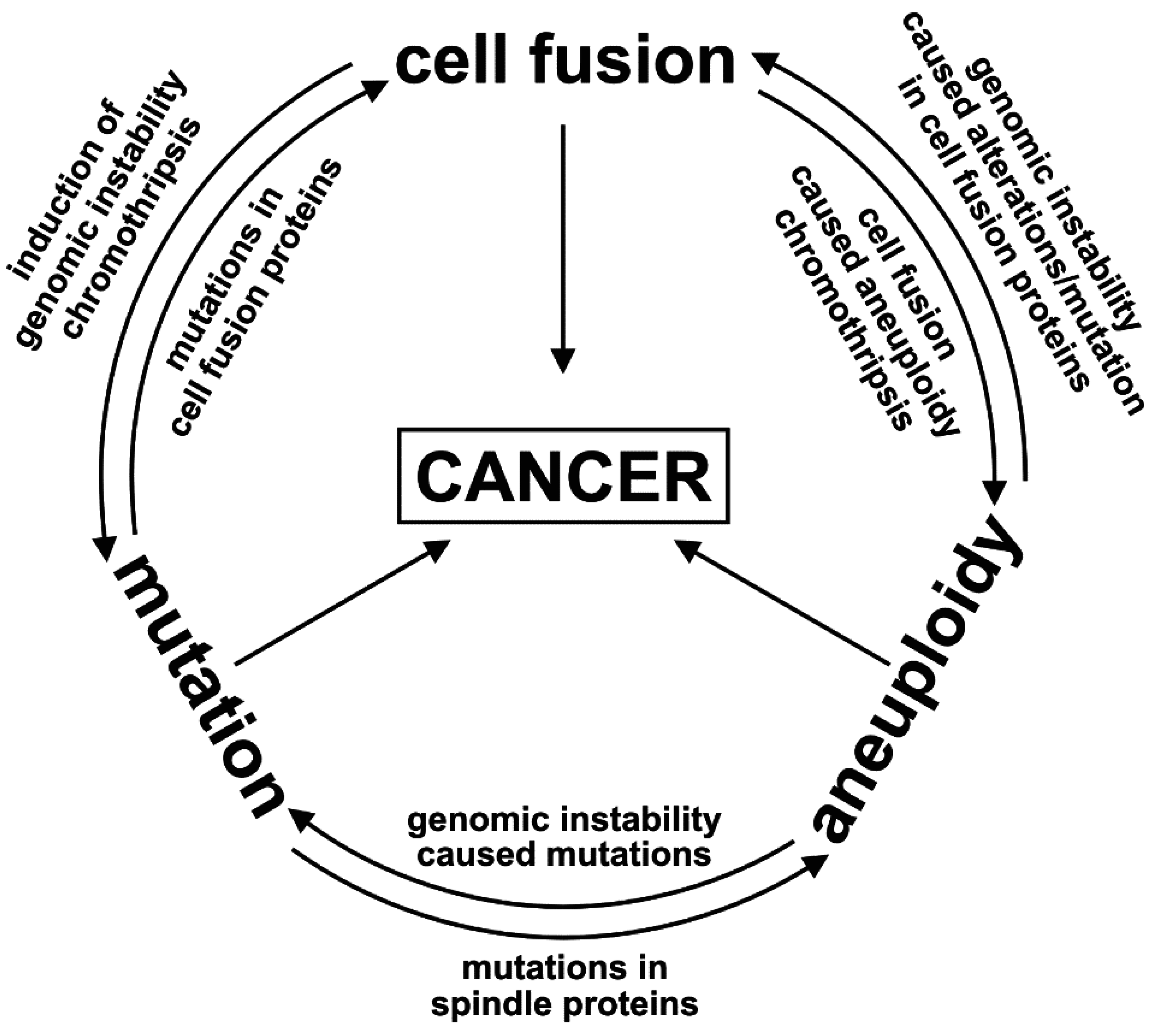{kind=link}
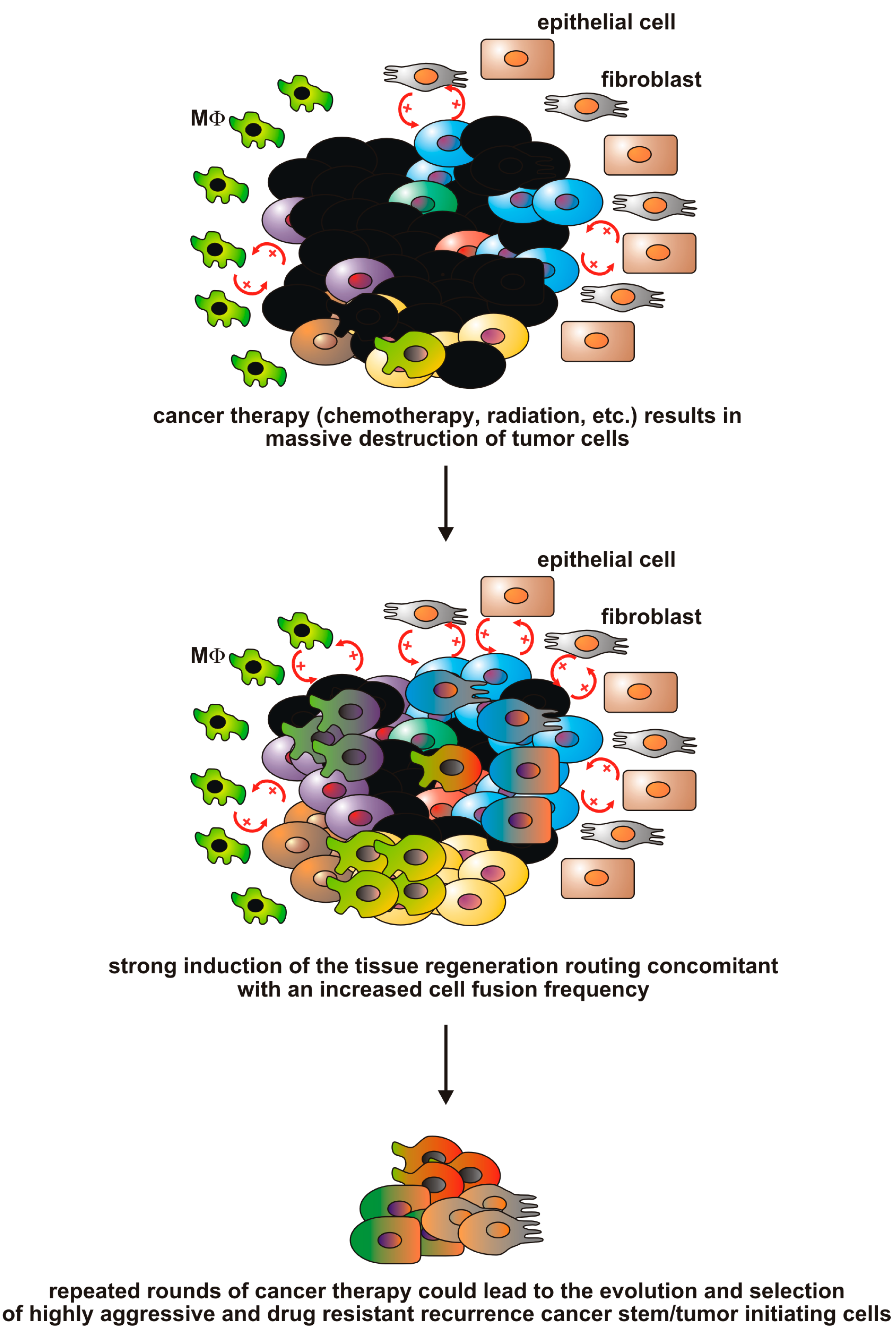{kind=link}
| Authors | Tumor Cells | Normal Cells | Marker | Reference |
|---|---|---|---|---|
| Chakraborty et al., 2004 | Renal cell carcinoma | BMDCs | PCR analysis of blood group alleles * | [23,29] |
| Yilmaz et al., 2004 | Renal cell carcinoma | BMDCs | FISH analysis of Y chromosome * | [24,30] |
| Andersen et al., 2007 | Multiple myeloma | Osteoclasts | FISH analysis | [31,32] |
| Shabo et al., 2008, 2009, 2011, 2013 | Breast cancer, colorectal cancer | Macrophages | CD163, MAC387, DAP12 | [33,34,35,36] |
| Clawson et al., 2012 | Melanoma, colorectal cancer | Macrophages | CD45, CD14, Cytokeratin (KRT) | [24,29] |
| Lazova et al., 2013 | Melanoma cells | BMDCs | STR analysis * | [37] |
| Ramakrishnan et al., 2013 | Epithelial ovarian carcinoma | BMDCs | CD45, CXCR4 | [23] |
| Clawson et al., 2015 | Melanoma | Macrophages | CD14, CD68, CD163, CD204, CD206, CXCR4, CD44, ALCAM, MLANA | [32] |
1.1. Cell Fusion (in Human Cancer): Fact or Fiction?
1.2. The Unpredictable and Random Nature of Cell Fusion; or: Is Cell Fusion an Inducer of the Mutator Phenotype?
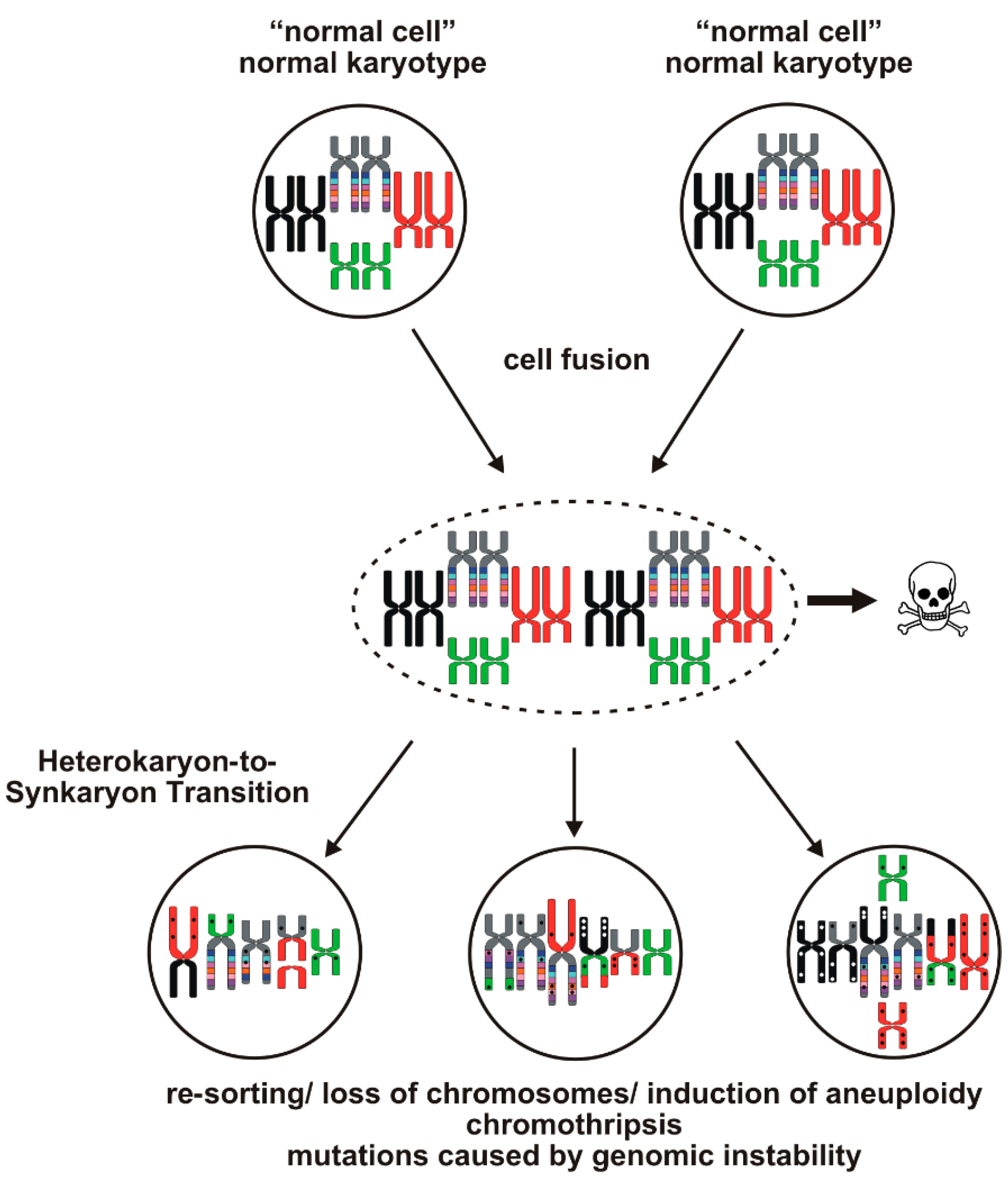
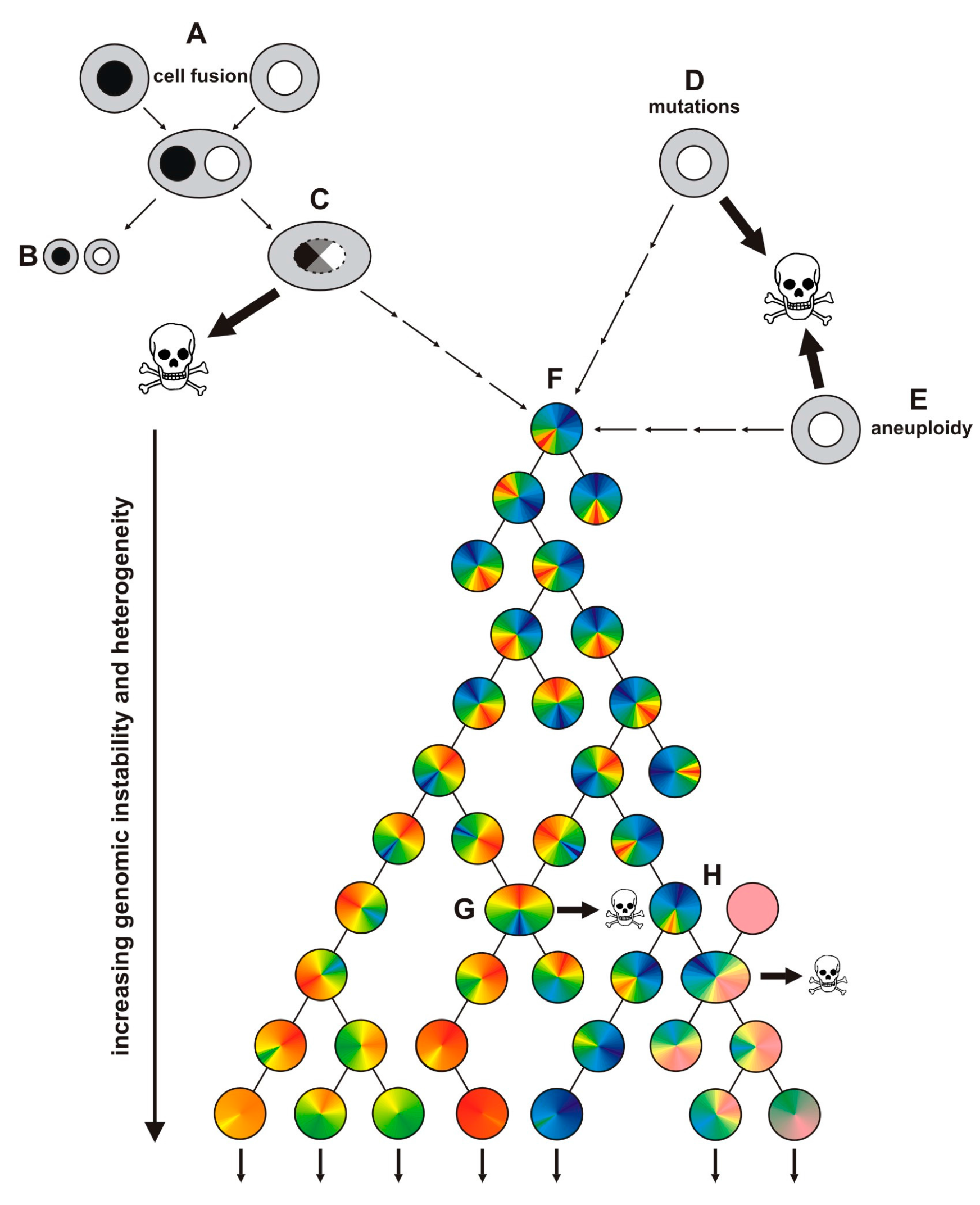
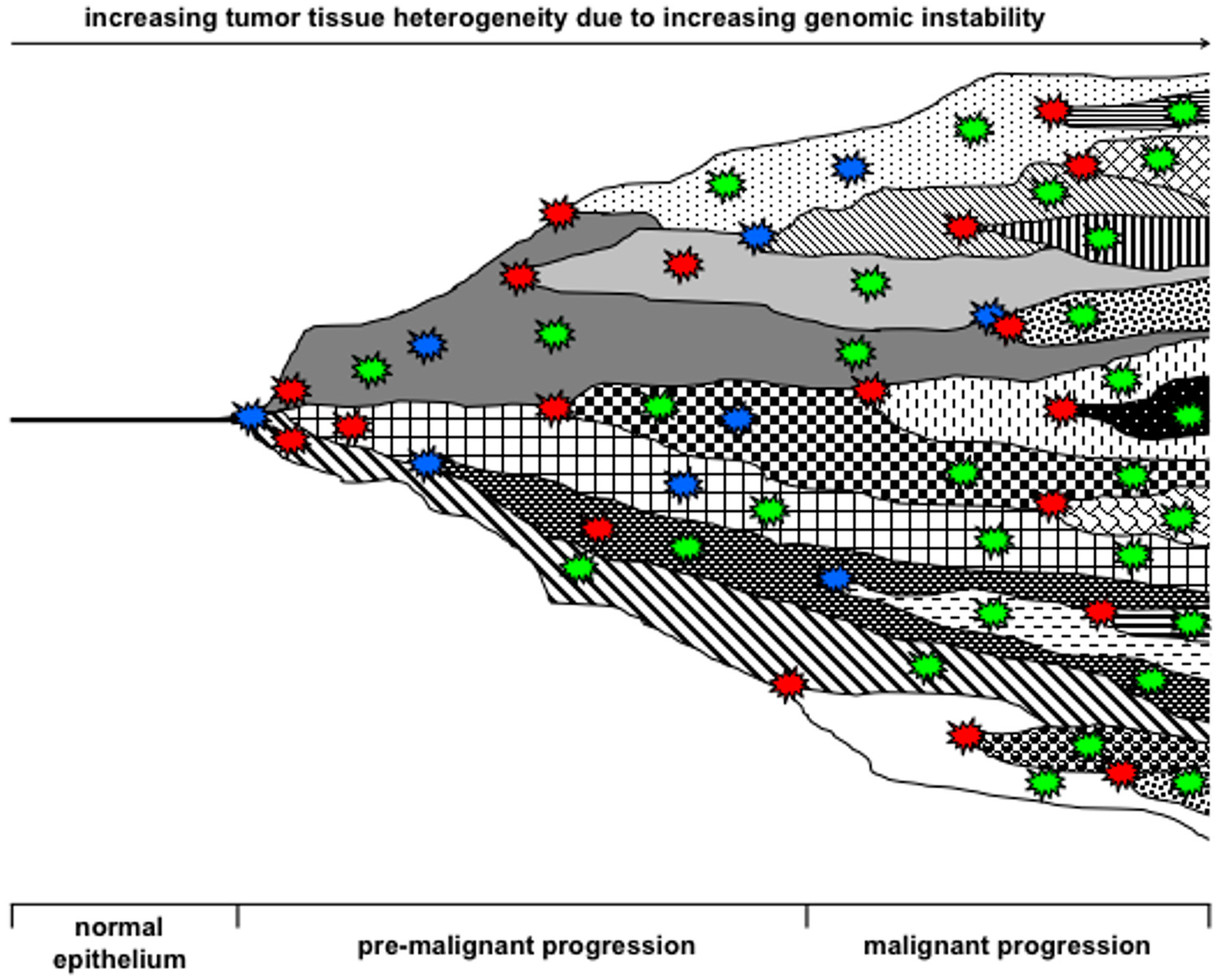
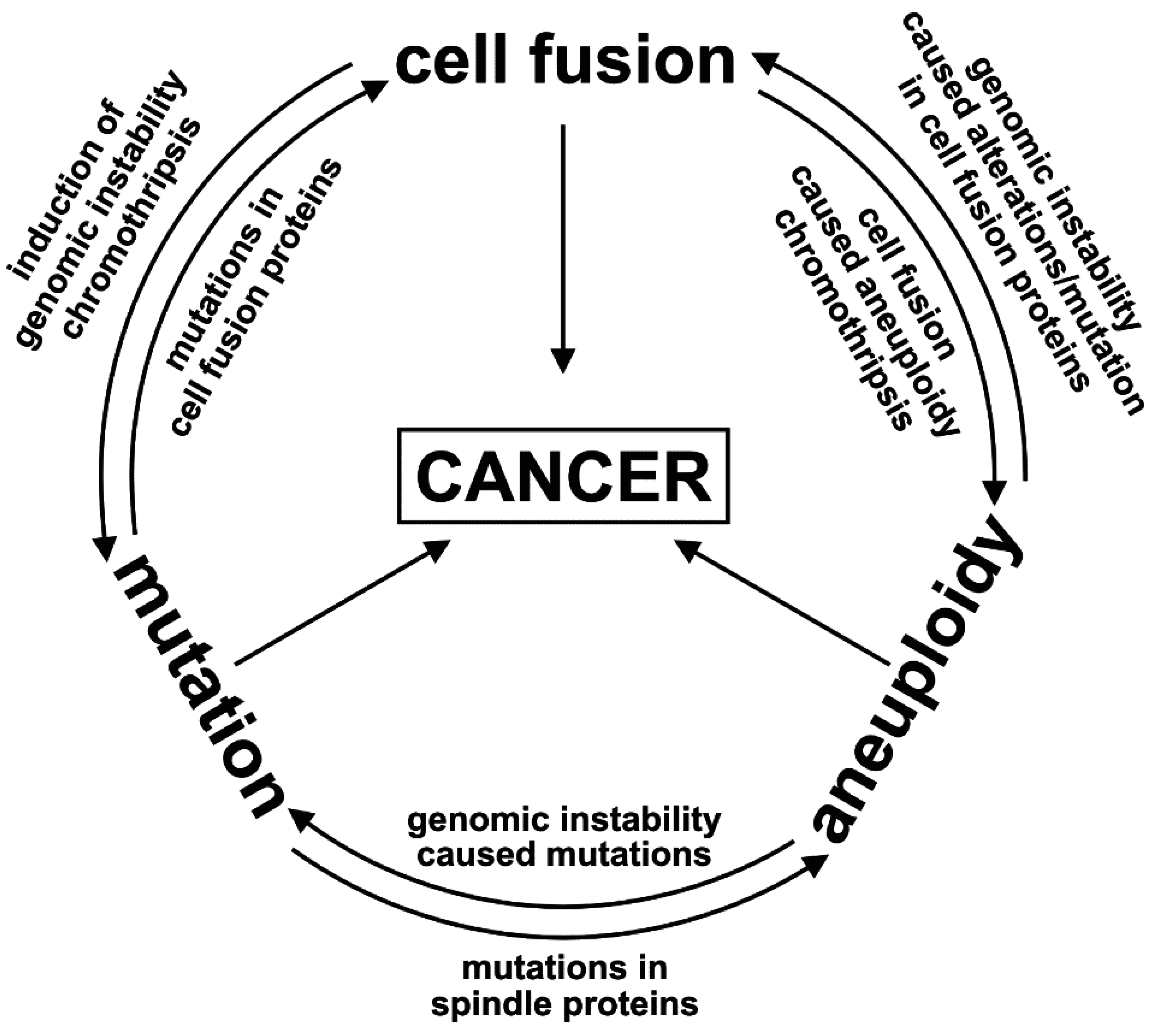
1.3. Evolution of Hybrid Cells Exhibiting Novel Properties by Cell Fusion?
1.4. Are Cancer Therapies Inducers of Cell Fusion and the Origin of Resistant Tumor (Hybrid) Cells?
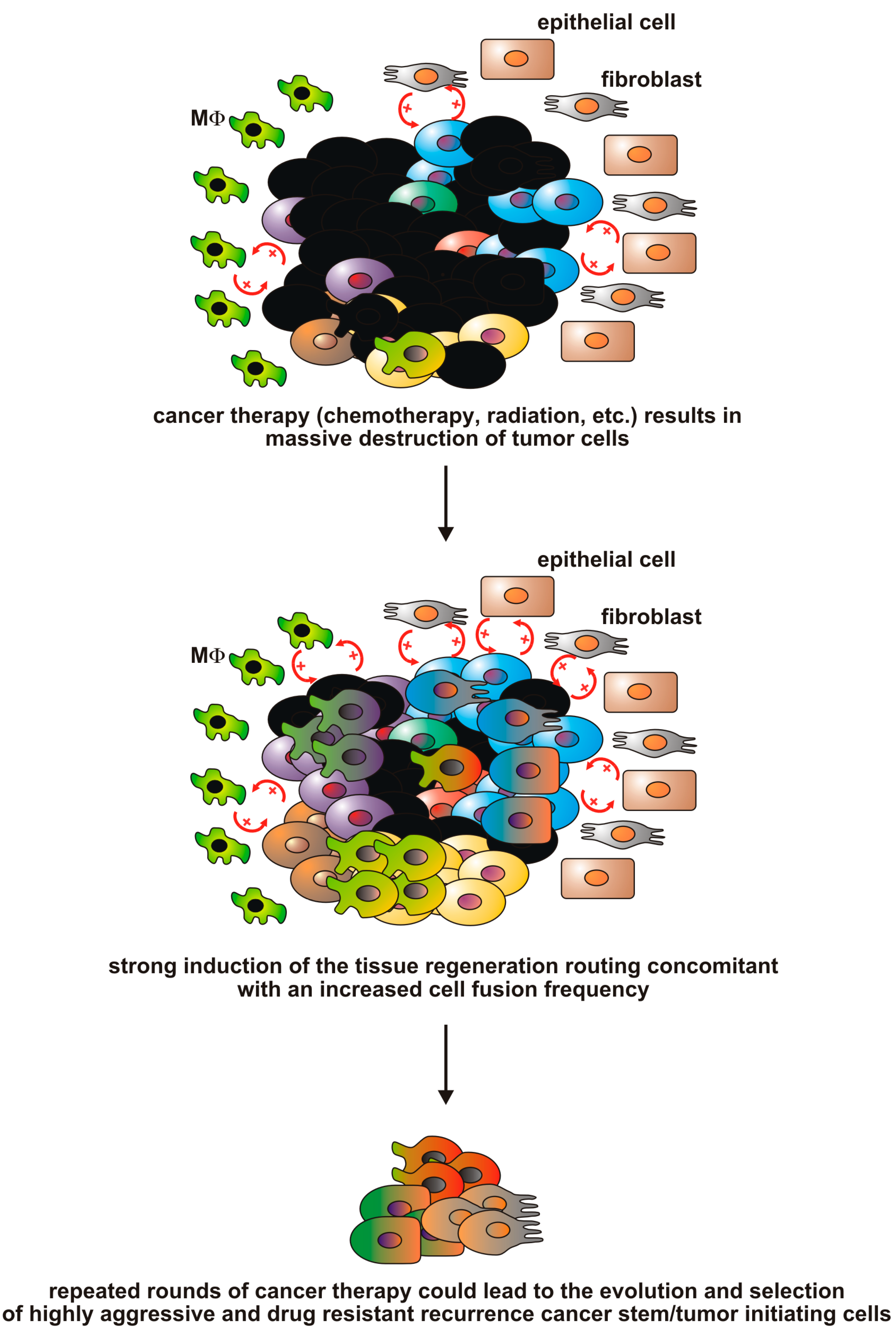
2. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dittmar, T.; Zänker, K.S. Cell fusion in Health and Disease; Springer: Dordrecht, The Netherlands, 2011; Volume 1, pp. 1–198. [Google Scholar]
- Dittmar, T.; Zänker, K.S. Cell fusion in Health and Disease; Springer: Dordrecht, The Netherlands, 2011; Volume 2, pp. 1–202. [Google Scholar]
- Dittmar, T.; Rossbach, M. Human pluripotent stem cells in regenerative and personalized medicine. Curr. Mol. Med. 2013, 13, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef]
- Mohr, M.; Zaenker, K.S.; Dittmar, T. Fusion in cancer: An explanatory model for aneuploidy, metastasis formation, and drug resistance. Methods Mol. Biol. 2015, 1313, 21–40. [Google Scholar] [PubMed]
- Kozorovitskiy, Y.; Gould, E. Stem cell fusion in the brain. Nat. Cell Biol. 2003, 5, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Ogle, B.M.; Cascalho, M.; Platt, J.L. Biological implications of cell fusion. Nat. Rev. Mol. Cell Biol. 2005, 6, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Bustamante, O.; Alvarez-Barrientos, A.; Kofman, A.V.; Fabregat, I.; Bueren, J.A.; Theise, N.D.; Segovia, J.C. Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology 2006, 43, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Ylostalo, J.; Lynch, P.J.; Smith, J.; Perry, A.; Peister, A.; Wang, M.Y.; Prockop, D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc. Natl. Acad. Sci. USA 2003, 100, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Hamazaki, T.; Oka, M.; Hoki, M.; Mastalerz, D.M.; Nakano, Y.; Meyer, E.M.; Morel, L.; Petersen, B.E.; Scott, E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature 2002, 416, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Willenbring, H.; Akkari, Y.; Torimaru, Y.; Foster, M.; Al-Dhalimy, M.; Lagasse, E.; Finegold, M.; Olson, S.; Grompe, M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 2003, 422, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Willenbring, H.; Bailey, A.S.; Foster, M.; Akkari, Y.; Dorrell, C.; Olson, S.; Finegold, M.; Fleming, W.H.; Grompe, M. Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat. Med. 2004, 10, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M.; Pardal, R.; Garcia-Verdugo, J.M.; Fike, J.R.; Lee, H.O.; Pfeffer, K.; Lois, C.; Morrison, S.J.; Alvarez-Buylla, A. Fusion of bone-marrow-derived cells with purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003, 425, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Dey, P. Aneuploidy and malignancy: An unsolved equation. J. Clin. Pathol. 2004, 57, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Sen, S. Aneuploidy and cancer. Curr. Opin. Oncol. 2000, 12, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Berndt, B.; Zanker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, T.; Diaz-Martinez, L.A.; Fair, J.; Elkahloun, A.G.; Harris, B.T.; Toretsky, J.A.; Rosenberg, S.A.; Shukla, N.; Ladanyi, M.; et al. Mutational inactivation of stag2 causes aneuploidy in human cancer. Science 2011, 333, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, R.; Hernando, E.; Diaz-Rodriguez, E.; Teruya-Feldstein, J.; Cordon-Cardo, C.; Lowe, S.W.; Benezra, R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 2007, 11, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Mathur, S.R.; Mukhopadhyay, A. Fusion derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Kimchi, E.; Patrick, S.D.; Xin, P.; Harouaka, R.; Zheng, S.; Berg, A.; Schell, T.; Staveley-O’Carroll, K.F.; Neves, R.I.; et al. Circulating tumor cells in melanoma patients. PLoS ONE 2012, 7, e41052. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Rasnick, D.; Li, R.; Winters, L.; Rausch, C.; Hehlmann, R. How aneuploidy may cause cancer and genetic instability. Anticancer Res. 1999, 19, 4887–4906. [Google Scholar] [PubMed]
- Duesberg, P.; Rausch, C.; Rasnick, D.; Hehlmann, R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc. Natl. Acad. Sci. USA 1998, 95, 13692–13697. [Google Scholar] [CrossRef] [PubMed]
- Sheltzer, J.M.; Blank, H.M.; Pfau, S.J.; Tange, Y.; George, B.M.; Humpton, T.J.; Brito, I.L.; Hiraoka, Y.; Niwa, O.; Amon, A. Aneuploidy drives genomic instability in yeast. Science 2011, 333, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, L.; Flemming, P.; Gebel, M.; Bleck, J.; Terkamp, C.; Wingen, L.; Kreipe, H.; Schlegelberger, B. Induction of aneuploidy by increasing chromosomal instability during dedifferentiation of hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2004, 101, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004, 34, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor y chromosome in renal carcinoma cells of a female bmt recipient: Visualization of putative bmt-tumor hybrids by fish. Bone Marrow Transplant. 2005, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.L.; Boissy, P.; Sondergaard, T.E.; Kupisiewicz, K.; Plesner, T.; Rasmussen, T.; Haaber, J.; Kolvraa, S.; Delaisse, J.M. Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: A new type of cancer-host partnership? J. Pathol. 2007, 211, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS ONE 2015, 10, e0134320. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Stal, O.; Svanvik, J. Breast cancer expression of dap12 is associated with skeletal and liver metastases and poor survival. Clin. Breast Cancer 2013, 13, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Sun, X.F.; Svanvik, J. Expression of the macrophage antigen cd163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 2009, 125, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of cd163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Svanvik, J. Expression of macrophage antigens by tumor cells. Adv. Exp. Med. Biol. 2011, 714, 141–150. [Google Scholar] [PubMed]
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: First evidence for fusion in human cancer. PLoS ONE 2013, 8, e66731. [Google Scholar] [CrossRef] [PubMed]
- Aichel, O. Über zellverschmelzung mit quantitativ abnormer chromosomenverteilung als ursache der geschwulstbildung. In Vorträge und aufsätze über entwicklungsmechanik der organismen; Roux, W., Ed.; Wilhelm Engelmann: Leipzig, Germany, 1911; pp. 1–115. [Google Scholar]
- Clawson, G.A. Cancer. Fusion for moving. Science 2013, 342, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Parris, G.E. Historical perspective of cell-cell fusion in cancer initiation and progression. Crit. Rev. Oncog. 2013, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Camargo, F.D.; Chambers, S.M.; Goodell, M.A. Stem cell plasticity: From transdifferentiation to macrophage fusion. Cell Prolif. 2004, 37, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ferrand, J.; Noel, D.; Lehours, P.; Prochazkova-Carlotti, M.; Chambonnier, L.; Menard, A.; Megraud, F.; Varon, C. Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS ONE 2011, 6, e19569. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, H.; Zhou, B.; Ju, Z.; Yu, L.; Guo, L.; Han, J.; Lu, S. Fusion of human umbilical cord mesenchymal stem cells with esophageal cells. Int. J. Oncol. 2012, 40, 370–377. [Google Scholar] [PubMed]
- Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.M.; Blau, H.M. Extensive fusion of haematopoietic cells with purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Mohr, M.; Tosun, S.; Arnold, W.H.; Edenhofer, F.; Zanker, K.S.; Dittmar, T. Quantification of cell fusion events human breast cancer cells and breast epithelial cells using a cre-loxp-based double fluorescence reporter system. Cell. Mol. Life Sci. 2015, 72, 3769–3782. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Zhu, F.; Zhang, H.Z.; Shang, Z.J. Tumor necrosis factor-alpha enhanced fusions between oral squamous cell carcinoma cells and endothelial cells via vcam-1/vla-4 pathway. Exp. Cell Res. 2012, 318, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Arwert, E.N.; Hoste, E.; Watt, F.M. Epithelial stem cells, wound healing and cancer. Nat. Rev. Cancer 2012, 12, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, S.L.M.; Smits, A.I.P.M.; Driessen-Mol, A.; Baaijens, F.P.T.; Bouten, C.V.C. The immune response in in situ tissue engineering of aortic heart valves. In Calcific Aortic Valve Disease; Aikawa, E., Ed.; InTech: West Palm Beach, FL, USA, 2013; Volume 8, pp. 207–245. [Google Scholar]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The origin and function of tumor-associated macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. New vistas on macrophage differentiation and activation. Eur. J. Immunol. 2007, 37, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Nagler, C.; Niggemann, B.; Zänker, K.S. The dark side of stem cells: Triggering cancer progression by cell fusion. Curr. Mol. Med. 2013, 13, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar] [PubMed]
- Pihan, G.; Doxsey, S.J. Mutations and aneuploidy: Co-conspirators in cancer? Cancer Cell 2003, 4, 89–94. [Google Scholar] [CrossRef]
- Boveri, T. On multipolar mitosis as a means of analysis of the cell nucleus. In Foundations of Experimental Embryology; Willier, B.H., Oppenheimer, J.M., Eds.; Prentice Hall: New York, NY, USA, 1902; Volume 2, pp. 74–97. [Google Scholar]
- Duesberg, P. Chromosomal chaos and cancer. Sci. Am. 2007, 296, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Loeb, K.R.; Loeb, L.A. Significance of multiple mutations in cancer. Carcinogenesis 2000, 21, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.J.; Prindle, M.J.; Loeb, L.A. Do mutator mutations fuel tumorigenesis? Cancer Metastasis Rev. 2013, 32, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A. Mutator phenotype in cancer: Origin and consequences. Semin. Cancer Biol. 2010, 20, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sonik, A.; Stindl, R.; Rasnick, D.; Duesberg, P. Aneuploidy versus gene mutation hypothesis of cancer: Recent study claims mutation, but is found to support aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Li, R.; Fabarius, A.; Hehlmann, R. Aneuploidy and cancer: From correlation to causation. Contrib. Microbiol. 2006, 13, 16–44. [Google Scholar] [PubMed]
- Bhatia, B.; Schneider-Broussard, R.; Calhoun, T.; Tang, S.; Zhou, J.; Schroeder, L.; Shen, J.; Multani, A.S.; Pathak, S.; Chang, S.; et al. Replicative senescence and cell fusion-mediated immortalization and tumorigenic transformation of normal human prostate (nhp) epithelial cells: Implications in prostate tumorigenesis and tumor heterogeneity. Proc. Am. Assoc. Cancer Res. 2005, 46, 2060. [Google Scholar]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell fusion connects oncogenesis with tumor evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Hetzer, M.W. Chromothripsis. Curr. Biol. 2015, 25, R397–R399. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D.M.; Pavia, R.A.; Tsao, M.C. In vivo hybridisation of human tumour and normal hamster cells. Nature 1974, 250, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, B.M.; Harrell, J.C.; Jedlicka, P.; Borges, V.F.; Varella-Garcia, M.; Horwitz, K.B. Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 2006, 66, 8274–8279. [Google Scholar] [CrossRef] [PubMed]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Hernando, E.; Nahle, Z.; Juan, G.; Diaz-Rodriguez, E.; Alaminos, M.; Hemann, M.; Michel, L.; Mittal, V.; Gerald, W.; Benezra, R.; et al. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 2004, 430, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar]
- Schuyler, S.C.; Wu, Y.F.; Kuan, V.J. The mad1-mad2 balancing act—A damaged spindle checkpoint in chromosome instability and cancer. J. Cell Sci. 2012, 125, 4197–4206. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. Bubr1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Yoshinaga, K.; Hisatomi, H.; Sugihara, K.; Hirata, Y. Genetic and epigenetic inactivation of mitotic checkpoint genes hbub1 and hbubr1 and their relationship to survival. Cancer Res. 2002, 62, 13–17. [Google Scholar] [PubMed]
- Babu, J.R.; Jeganathan, K.B.; Baker, D.J.; Wu, X.; Kang-Decker, N.; van Deursen, J.M. Rae1 is an essential mitotic checkpoint regulator that cooperates with bub3 to prevent chromosome missegregation. J. Cell Biol. 2003, 160, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.; Coleman, K.; Reid, S.; Plaja, A.; Firth, H.; Fitzpatrick, D.; Kidd, A.; Mehes, K.; Nash, R.; Robin, N.; et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in bub1b. Nat. Genet. 2004, 36, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [PubMed]
- Fabarius, A.; Hehlmann, R.; Duesberg, P.H. Instability of chromosome structure in cancer cells increases exponentially with degrees of aneuploidy. Cancer Genet. Cytogenet. 2003, 143, 59–72. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64. [Google Scholar] [PubMed]
- Huppertz, B.; Gauster, M. Trophoblast fusion. Adv. Exp. Med. Biol. 2011, 713, 81–95. [Google Scholar] [PubMed]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, T.; Zhao, Z.; Chen, Y.; Zeng, J.; Liu, S.; Zhu, F. Mutations in 3′-long terminal repeat of herv-w family in chromosome 7 upregulate syncytin-1 expression in urothelial cell carcinoma of the bladder through interacting with c-myb. Oncogene 2014, 33, 3947–3958. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 2006, 63, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Sondag, G.R.; Moussa, F.M.; Belcher, J.Y.; Yu, B.; Stinnett, H.; Novak, K.; Mbimba, T.; Khol, M.; Hankenson, K.D.; et al. Mutation in osteoactivin promotes receptor activator of nfkappab ligand (rankl)-mediated osteoclast differentiation and survival but inhibits osteoclast function. J. Biol. Chem. 2015, 290, 20128–20146. [Google Scholar] [CrossRef] [PubMed]
- Silvestris, F.; Ciavarella, S.; Strippoli, S.; Dammacco, F. Cell fusion and hyperactive osteoclastogenesis in multiple myeloma. Adv. Exp. Med. Biol. 2011, 714, 113–128. [Google Scholar] [PubMed]
- Malassine, A.; Pidoux, G.; Gerbaud, P.; Frendo, J.L.; Evain-Brion, D. Human trophoblast in trisomy 21: A model for cell-cell fusion dynamic investigation. Adv. Exp. Med. Biol. 2011, 714, 103–112. [Google Scholar] [PubMed]
- Berndt, B.; Haverkampf, S.; Reith, G.; Keil, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Fusion of ccl21 non-migratory active breast epithelial and breast cancer cells give rise to ccl21 migratory active tumor hybrid cell lines. PLoS ONE 2013, 8, e63711. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, X.; Wang, C.Y.; Hu, P.; Chu, C.Y.; Liu, S.; Zhau, H.E.; Chung, L.W. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhang, Y.; Park, S.C.; Eun, J.R.; Nguyen, N.T.; Tschudy-Seney, B.; Jung, Y.J.; Theise, N.D.; Zern, M.A.; Duan, Y. Cd34 liver cancer stem cells were formed by fusion of hepatobiliary stem/progenitor cells with hematopoietic precursor-derived myeloid intermediates. Stem Cells Dev. 2015, 24, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Mazzocca, A.; Mello, T.; Galli, A.; Capaccioli, S. Cell fusion promotes chemoresistance in metastatic colon carcinoma. Oncogene 2013, 32, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Duelli, D.M.; Padilla-Nash, H.M.; Berman, D.; Murphy, K.M.; Ried, T.; Lazebnik, Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr. Biol. 2007, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Martin-Padura, I.; Marighetti, P.; Gregato, G.; Agliano, A.; Malazzi, O.; Mancuso, P.; Pruneri, G.; Viale, A.; Bertolini, F. Spontaneous cell fusion of acute leukemia cells and macrophages observed in cells with leukemic potential. Neoplasia 2012, 14, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.R.; Mohamed, A.N.; McEachern, D. Production of a more aggressive tumor cell variant by spontaneous fusion of two mouse tumor subpopulations. Cancer Res. 1989, 49, 4316–4321. [Google Scholar] [PubMed]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, W.F.; Greetham, J.; Bennett, D.C. Efficient spontaneous fusion between some co-cultured cells, especially murine melanoma cells. Cell Biol. Int. 1994, 18, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Hsu, Y.; He, X.; Lu, N.; Wei, W.; Zhang, Z.; Zhu, L. Evidence of cell fusion in carcinogen-induced mice gastric carcinoma. Tumour Biol. 2015, 36, 5089–5094. [Google Scholar] [CrossRef] [PubMed]
- Rachkovsky, M.; Sodi, S.; Chakraborty, A.; Avissar, Y.; Bolognia, J.; McNiff, J.M.; Platt, J.; Bermudes, D.; Pawelek, J. Melanoma × macrophage hybrids with enhanced metastatic potential. Clin. Exp. Metastasis 1998, 16, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, M.B.; Dewey, M.J.; Furmanski, P. Cell fusion in tumor development and progression: Occurrence of cell fusion in primary methylcholanthrene-induced tumorigenesis. Int. J. Cancer 1989, 44, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.R.; McInerney, D.; Rogers, C.; Miller, B.E. Spontaneous fusion between metastatic mammary tumor subpopulations. J. Cell. Biochem. 1988, 36, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Stindl, R.; Hehlmann, R. Explaining the high mutation rates of cancer cells to drug and multidrug resistance by chromosome reassortments that are catalyzed by aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 19, 14295–14300. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Wang, J.; Liu, L. Chemotherapy promotes tumour cell hybridization in vivo. Tumour Biol. 2015, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Why therapeutic response may not prolong the life of a cancer patient: Selection for oncogenic resistance. Cell Cycle 2005, 4, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zanker, K.S. Recurrence cancer stem cells--made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Huff, C.A.; Matsui, W.; Smith, B.D.; Jones, R.J. The paradox of response and survival in cancer therapeutics. Blood 2006, 107, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Matsui, W.H.; Smith, B.D. Cancer stem cells: Are we missing the target? J. Natl. Cancer Inst. 2004, 96, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Mullighan, C.G.; Wang, J.C.; Poeppl, A.; Doulatov, S.; Phillips, L.A.; Ma, J.; Minden, M.D.; Downing, J.R.; Dick, J.E. Evolution of human bcr-abl1 lymphoblastic leukaemia-initiating cells. Nature 2011, 469, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct cd44+/cd24- and cd133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R.; Islam, S.; Wright, N.A. Stem cells in cancer: Instigators and propagators? J. Cell Sci. 2010, 123, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Skinner, A.M.; Grompe, M.; Kurre, P. Intra-hematopoietic cell fusion as a source of somatic variation in the hematopoietic system. J. Cell Sci. 2012, 125, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, C.M. Hematopoietic stem cells for transplantation. Nat. Immunol. 2002, 3, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Murch, A.R.; Grounds, M.D.; Marshall, C.A.; Papadimitriou, J.M. Direct evidence that inflammatory multinucleate giant cells form by fusion. J. Pathol. 1982, 137, 177–180. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dittmar, T.; Zänker, K.S. Tissue Regeneration in the Chronically Inflamed Tumor Environment: Implications for Cell Fusion Driven Tumor Progression and Therapy Resistant Tumor Hybrid Cells. Int. J. Mol. Sci. 2015, 16, 30362-30381. https://doi.org/10.3390/ijms161226240
Dittmar T, Zänker KS. Tissue Regeneration in the Chronically Inflamed Tumor Environment: Implications for Cell Fusion Driven Tumor Progression and Therapy Resistant Tumor Hybrid Cells. International Journal of Molecular Sciences. 2015; 16(12):30362-30381. https://doi.org/10.3390/ijms161226240
Chicago/Turabian StyleDittmar, Thomas, and Kurt S. Zänker. 2015. "Tissue Regeneration in the Chronically Inflamed Tumor Environment: Implications for Cell Fusion Driven Tumor Progression and Therapy Resistant Tumor Hybrid Cells" International Journal of Molecular Sciences 16, no. 12: 30362-30381. https://doi.org/10.3390/ijms161226240
APA StyleDittmar, T., & Zänker, K. S. (2015). Tissue Regeneration in the Chronically Inflamed Tumor Environment: Implications for Cell Fusion Driven Tumor Progression and Therapy Resistant Tumor Hybrid Cells. International Journal of Molecular Sciences, 16(12), 30362-30381. https://doi.org/10.3390/ijms161226240