
Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants

Abstract
:
1. Introduction
2. Heavy Metals
2.1. Mercury (Hg)
2.1.1. Mercury Induced Nephrotoxicity

{kind=link}
{kind=link}
| Metal | Form(s) | Sources | Route of Entry | Symptoms | Health Effects | References | |
|---|---|---|---|---|---|---|---|
| Acute | Chronic | ||||||
| Mercury, At. No: 80, At. Mass: 200.6 | Hg, Hg2+, Hg+, Hg-organic Oxidation state: +1, +2 | Fossil fuel combustion, mining, smelting, solid waste combustion, fertilizers industrial wastewater, use in electrical switches, fluorescent bulbs Mercury arc lamps, incineration of municipal wastes, emissions from mercury products: batteries, thermometers, Mercury amalgams | Inhalation, ingestion and absorption through skin | GI pain, vomiting, diuresis, anemia, hypovolemic shock, renal toxicity, tension, irritability, intention tremors, insomnia, fatigue | Gingivitis, tachycardia, goiter, high urine Hg | Disruption of the nervous system, damage to brain functions, DNA damage and chromosomal damage, allergic reactions, tiredness and headaches, negative reproductive effects, such as sperm damage, birth defects and miscarriages | [22] |
| Arsenic, At. No: 33, At. Mass: 74.92 | AsIII, AsV, Oxidation state: +3, +5 | Pesticides, mining, smelting of gold, Lead, Copper and Nickel, Production of iron and steel, combustion of coal, tobacco smoke | Inhalation and ingestion | Mucosal damage, hypovolemic shock, fever, sloughing, gastro-intestinal pain, anorexia | Weakness, hepatomegaly, melanosis, arrhythmias, peripheral neuropathy, peripheral vascular disease, carcinogenicity, liver angiosarcoma, skin and lung cancer | Birth defects, Carcinogen: lung, skin, liver, bladder, Kidneys, Gastrointestinal damage, Severe vomiting, diarrhea, death | [25] |
| Lead, At. No: 82, At. Mass: 207.19 | Pb2+, Oxidation state: +2, +4 | Application of lead in gasoline, fuel combustion, industrial processes, solid waste combustion, used in paints, used in ceramics and dishware, Lead is used in some types of PVC mini-blinds | Inhalation and ingestion | Nausea, vomiting, thirst, diarrhea/constipation, abdominal pain, hemoglobinuria, oligura leading to hypovolemic shock | Lead colic, lead palsy and lead encephalopathy | Aanemia (less Hb), hypertension, kidney damage, miscarriages, disruption of nervous systems, brain damage, infertility, intellectual disorders | [26,27] |
2.1.2. Mercury Induced Neurotoxicity
2.2. Arsenic (As)
2.3. Lead (Pb)
3. Cytotoxic Mechanisms of Heavy Metals
3.1. ROS/RNS and Protein Destruction
3.2. ROS/RNS and Lipid Peroxidation
3.3. ROS/RNS and Nucleic Acid Destabilization
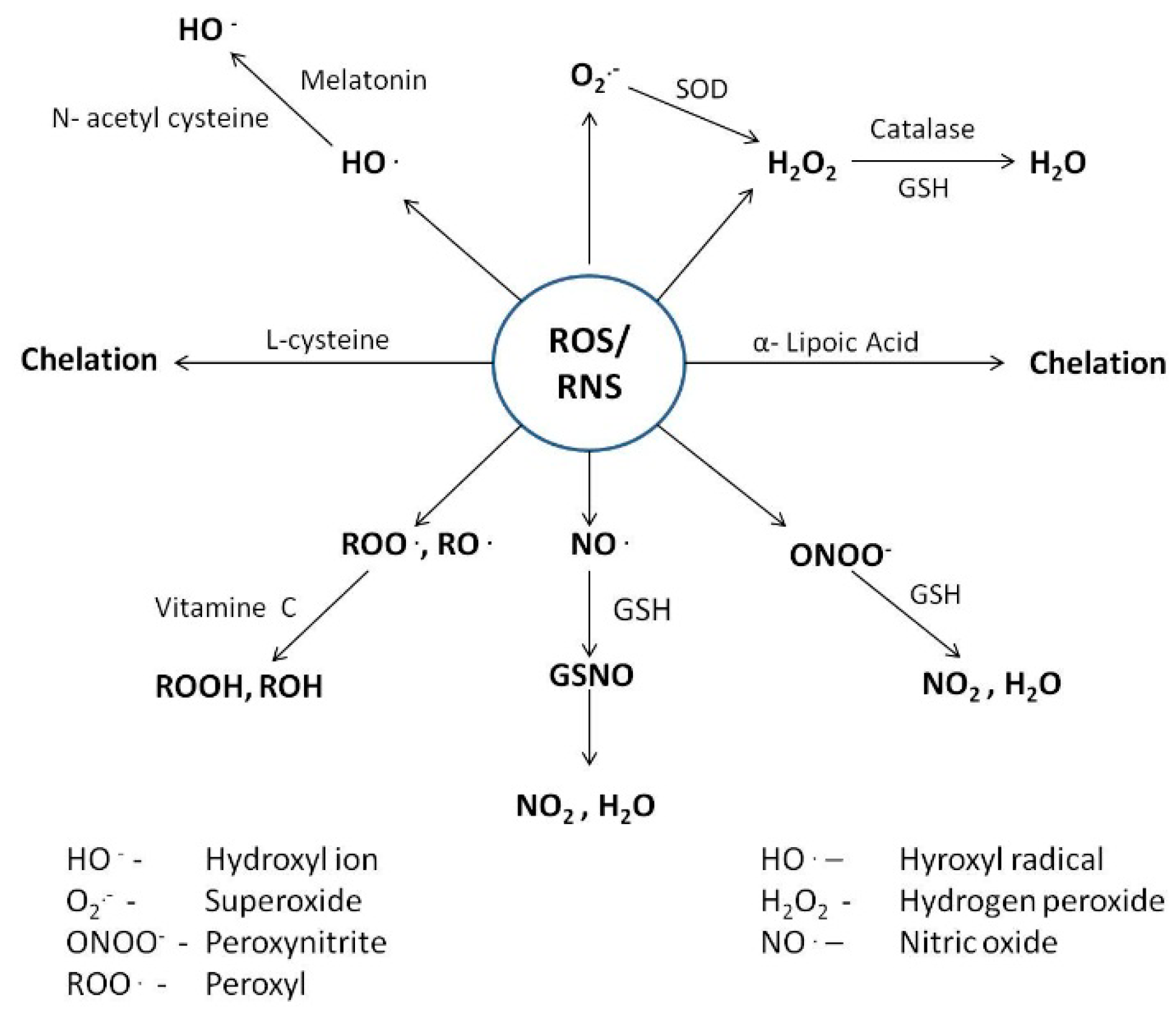
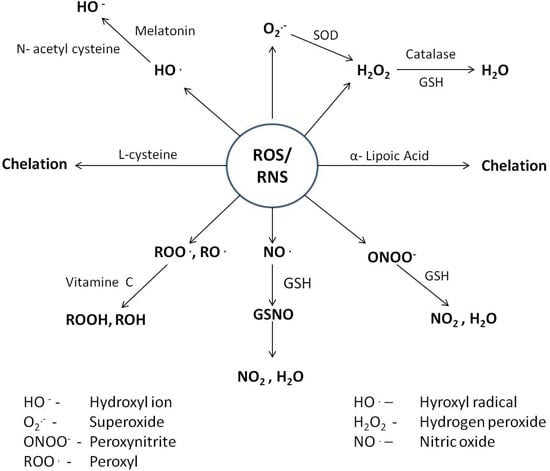
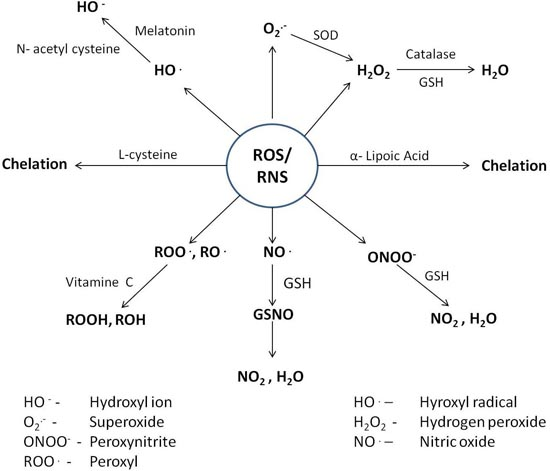
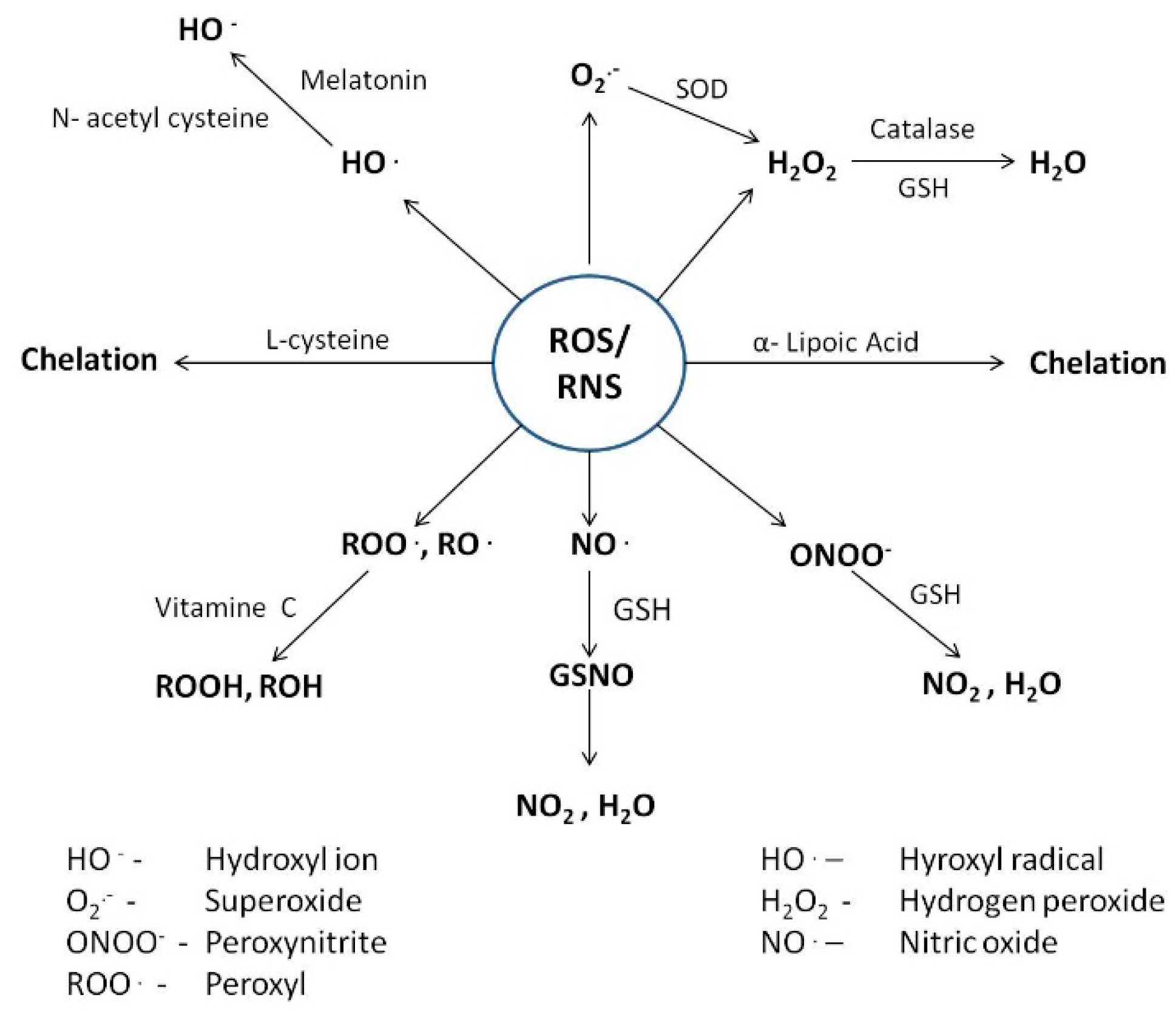
4. Counteractive Antioxidant Defence
4.1. Cellular reductants
4.1.1. Glutathione
| Metal | Mechanism of toxicity | Biomarkers of Toxicity | Antioxidants | Mechanism of action | Health effects | Ref. |
|---|---|---|---|---|---|---|
| Mercury Arsenic Lead | Oxidative and nitrative stress, alteration of thiol dependent pathways, depletion of intracellular antioxidants, binding to specific location and dislocation of essential ion, damage to macromolecules, inhibition of repair machinery, chromosomal abnormalities and altered gene expression, binding to –SH group and inhibition of enzymatic activity, membrane damage, inhibition of oxidative phosphorylation, inhibition of heme biosynthesis, disruption of protein structure, hypertension | Malondialdehyde (MDA), 8-OH-2-OxoG, Hg-GSH, albumin, transferrin, α1-microglobulin (α1-MG), β2-microglobulin (β2-MG), retinol binding protein (RBP), enhanced deposition in hair, bones and soft tissues, lipid peroxides, methylated products of arsenic (MMAV, DMAV), increased B-Pb level, increased disposal of δ-ALA and ZPP | Endogenous thiols (GSH, l-Cys, NAC, Taurine, Melatonin) | Scavenging of free radicals, interrupt radical chain reactions, formation of stable complexes with metals | Reduces metal availability, decreases damage to cell organs and biological macromolecules, Promotes detoxification | [28,29,30,31,59,60,61,86,96,114,115,116,138,139,140,141,147,148,149,150,162,163,164,171,172,173,174,175,176,177,178,179] |
| Minerals (Se, Fe, Cu, Zn) | Competes with intestinal absorption, decreases replacement of essential ions, formation of insoluble metal-mineral complexes, induces production of metal binding proteins (MTs) | decreases GI absorption and as such its distribution, prevents redistribution and accumulation in tissues, reduces metal availability thereby decreases toxicity, Stabilizes cell membranes, decreases damage to biological macromolecules, decreases teratogenic toxicity | ||||
| Enzymatic (SOD, GPx CAT) | Neutralize free radicals and as such attenuates oxidative damage | Protects cell organs and biological macromolecules, Stabilizes cell membranes | ||||
| Vitamins (α-LA, Vit C, Carotenoids) | Scavenging of free radicals, decrease in cellular oxidative stress | Reduces plasma to lipid peroxidation, decreases risk of having stroke, reduces incidences of chronic and degenerative diseases, reduces sperm ROS generation and prevents loss of motility and oocyte penetration |
4.1.2. l-cysteine and N-acetyl Cysteine
4.1.3. Taurine
4.1.4. Melatonin
4.2. Essential Mineral Ions
4.2.1. Selenium
4.2.2. Iron
| Group | Age | Estimated Average Requirement | Tolerable Upper Intake Level | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Selenium | Iron | Copper | Zinc | Selenium | Iron | Copper | Zinc | ||
| Children | 1–3 yr | 17 μg/d | 3 mg/d | 260 μg/d | 2.5 mg/d | 90 μg/d | 40 mg/d | 1000 μg/d | 7 mg/d |
| 4–8 yr | 23 μg/d | 4.1 mg/d | 340 μg/d | 4 mg/d | 150 μg/d | 40 mg/d | 3000 μg/d | 12 mg/d | |
| Males | 9–18 yr | 35–45 μg/d | 5.9–7.7 mg/d | 540–685 μg/d | 7–8.5 mg/d | 280–400 μg/d | 40–45 mg/d | 5000–8000 μg/d | 23–34 mg/d |
| 19–70 yr | 45 μg/d | 6 mg/d | 700 μg/d | 9.4 mg/d | 400 μg/d | 45 mg/d | 10000 μg/d | 40 mg/d | |
| Females | 9–18 yr | 35–45 μg/d | 5.7–7.9 mg/d | 540–685 μg/d | 7–7.3 mg/d | 280–400 μg/d | 40–45 mg/d | 5000–8000 μg/d | 23–34 mg/d |
| 19–70 yr | 45 μg/d | 5–8.1 mg/d | 700 μg/d | 6.8 mg/d | 400 μg/d | 45 mg/d | 10000 μg/d | 40 mg/d | |
| Pregnancy (19–30 yr) | 49 μg/d | 22 mg/d | 800 μg/d | 9.5 mg/d | 400 μg/d | 45 mg/d | 10000 μg/d | 40 mg/d | |
| Lactation (19–30yr) | 59 μg/d | 6.5 mg/d | 1000 μg/d | 10.4 mg/d | 400 μg/d | 45 mg/d | 10000 μg/d | 40 mg/d | |
4.2.3. Copper
4.2.4. Zinc
4.3. Enzymatic Antioxidants
4.4. Dietary Antioxidants
4.4.1. α-Lipoic Acid
4.4.2. Vitamin C
4.4.3. Carotenoids
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bennett, L.E.; Burkhead, J.L.; Hale, K.L.; Tery, N.; Pilon, M.; Pilon-Smits, E.A.H. Analysis of transgenic Indian mustard plants for Phytoremediation of metal contaminated mine tailings. J. Environ. Qual. 2003, 32, 432–440. [Google Scholar] [CrossRef]
- Ikhuoria, E.U.; Okieimen, F.E. Scavenging cadmium, copper, lead, nickel and zinc ions from aqueous solution by modified cellulosic sorbent. Int. J. Environ. Stud. 2000, 57, 401–409. [Google Scholar] [CrossRef]
- Sahu, R.K.; Arora, N.K. Bioassay as a tool for assessing susceptibility and resistant plant species for field contaminated with industrial effluents. World J. Microbiol. Biotechnol. 2008, 24, 143–148. [Google Scholar] [CrossRef]
- Jaishankar, M.; Mathew, B.B.; Shah, M.S.; Murthy, K.T.P.; Gowda, S.K.R. Biosorption of few heavy metal ions using agricultural wastes. J. Environ. Pollut. Hum. Health 2014, 2, 1–6. [Google Scholar]
- Nagajyoti, P.C.; Lee, K.D.; Sreekanth, T.V.M. Heavy metals, occurrence and toxicity for plants: A review. Environ. Chem. Lett. 2010, 8, 199–216. [Google Scholar] [CrossRef]
- Meharg, A.A. Arsenic in rice–understanding a new disaster for south-east Asia. Trends Plant. Sci. 2004, 9, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.P. Heavy metal pollution in China: Origion, pattern and control. Environ. Sci. Pollut. Res. Int. 2003, 10, 192–198. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, C.; Peretiatko, R. The nexus between industrialization and environment: A case study of Indian enterprises. Environ. Manag. Health 2002, 13, 80–97. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Mittal, M.; Mehta, A. Heavy metal induced oxidative stress and its reversal by chelation therapy. Ind. J. Med. Res. 2008, 128, 501–523. [Google Scholar]
- Jan, A.T.; Ali, A.; Haq, Q.M.R. Glutathione as an antioxidant in inorganic mercury induced nephrotoxicity. J. Postgrad. Med. 2011, 57, 72–77. [Google Scholar] [PubMed]
- Chen, C.W.; Chen, C.F.; Dong, C.D. Distribution and Accumulation of Mercury in Sediments of Kaohsiung River Mouth, Taiwan. APCBEE Procedia 2012, 1, 153–158. [Google Scholar] [CrossRef]
- Jan, A.T.; Murtaza, I.; Ali, A.; Haq, Q.M.R. Mercury pollution: An emerging problem and potential bacterial remediation strategies. World J. Microbiol. Biotechnol. 2009, 25, 1529–1537. [Google Scholar] [CrossRef]
- Chang, L.W. Neurotoxic effects of mercury: A review. Environ. Res. 1977, 14, 329–373. [Google Scholar] [CrossRef]
- Asano, S.; Eto, K.; Kurisaki, E.; Gunji, H.; Hiraiwa, K.; Sato, M.; Sato, H.; Hasuike, M.; Hagiwara, N.; Wakasa, H. Acute inorganic mercury vapour inhalation poisoning. Pathol. Int. 2000, 50, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Bates, N. Metallic and inorganic mercury poisoning. Emerg. Nurse 2003, 11, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, T.W. Mercury: Major issues in environmental health. Environ. Health Perspect. 1993, 100, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Aschner, M.; Rocha, J.B.T. Oxidative stress in methylmercury induced neurotoxicity. Toxicol. Appl. Pharm. 2011, 256, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; King, P.G.; Hooker, B.S.; Dorea, J.G.; Kern, J.K.; Sykes, L.K.; Geier, M.R. Thimerosal: Clinical, epidemiologic and biochemical studies. Clin. Chim. Acta 2015, 444, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Hintelmann, H. Organomercurials: Their formation and pathways in the environment. Met Ions Life Sci. 2010, 7, 365–401. [Google Scholar] [PubMed]
- Patrick, L. Mercury toxicity and antioxidants: Part 1: role of glutathione and α-lipoic acid in the treatment of mercury toxicity. Altern. Med. Rev. 2002, 7, 456–471. [Google Scholar] [PubMed]
- Carneiro, M.F.H.; Oliveira Souza, J.M.; Grotto, D.; Batista, B.L.; de Oliveira Souza, V.C.; Barbosa, F., Jr. A systemic study of the deposition and metabolism of mercury species in mce after exposure to low levels of Thimerosal (ethylmercury). Environ. Res. 2014, 134, 218–227. [Google Scholar] [CrossRef] [PubMed]
- ASTDR. Toxicological profile for mercury. Available online: http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=115&tid=24 (accessed on 22 September 2015).
- WHO. Guidelines for drinking-water quality. Sixty-first meeting, Rome, 10–19 June 2003. Joint FAO/WHO Expert Committee on Food Additives, 2004. Available online: http://ftp.fao.org/es/esn/jecfa/jecfa61sc.pdf (accessed on 2 October 2015).
- Zalups, R.K.; Lash, L.H. Advances in understanding the renal transport and toxicity of mercury. J. Toxicol. Environ. Health 1994, 42, 1–44. [Google Scholar] [CrossRef] [PubMed]
- ATSDR. Toxicological profile for arsenic. 2007. Available online: http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=22&tid=3 (accessed on 22 September 2015). [Google Scholar]
- EPA. An overview of Airborne Metal Regulations, Exposure Limits, Health Effects and Contemporary Research (Appendix C). 2010. Available online: http://www3.epa.gov/ttnemc01/prelim/otm31appC.pdf (accessed on 2 October 2015). [Google Scholar]
- ATSDR. Toxicological profile for lead. 2007. Available online: http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=96&tid=22 (accessed on 22 September 2015). [Google Scholar]
- Langworth, S.; Elinder, C.G.; Sundquist, K.G.; Vesterberg, O. Renal and immunological effects of occupational exposure to inorganic mercury. Br. J. Ind. Med. 1992, 49, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Roels, H.; Bernard, A.M.; Barbon, R.; Lauwerys, R.R.; Rosello, J.; Hotter, G.; Mutti, A.; Franchini, I.; Fels, L.M.; et al. Markers of early renal changes induced by industrial pollutants. I. Application to workers exposed to mercury vapor. Br. J. Ind. Med. 1993, 50, 17–27. [Google Scholar] [PubMed]
- Franko, A.; Budihna, M.V.; Dodic-Fikfak, M. Long-term effects of elemental mercury on renal function in miners of the Idrija Mercury mine. Ann. Occup. Hyg. 2005, 49, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, I.; Al-Sedairi, A.; Elkhatib, R. Effect of mercury (Hg) dental amalgam fillings on renal and oxidative stress biomarkers in children. Sci. Total Environ. 2012, 431, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, B.; Yang, L.; Li, H. Blood mercury concentration among residents of a historic mercury mine and possible effects on renal function: a cross sectional study in south western China. Environ. Monit. Assess. 2013, 185, 3049–3055. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Molecular intereactions with mercury in the kidney. Pharmacol. Rev. 2000, 52, 113–143. [Google Scholar] [PubMed]
- Zalups, R.K.; Barfuss, D.W. Accumulation of inorganic mercury along the renal proximal tubule of the rabbit. Toxicol. Appl. Pharmacol. 1990, 106, 245–253. [Google Scholar] [CrossRef]
- Zalups, R.K. Method for studying the in vivo accumulation of inorganic mercury in segments of the nephron in the kidneys of rats treated with mercuric chloride. J. Pharmacol. Methods 1991, 26, 89–104. [Google Scholar] [CrossRef]
- Tanaka-Kagawa, T.; Naganuma, A.; Imura, N. Tubular secretion and reabsorption of mercury compounds in mouse kidney. J. Pharmacol. Exp. Ther. 1993, 264, 776–782. [Google Scholar] [PubMed]
- DeCeaurriz, J.; Payan, J.P.; Morel, G.; Brondeau, M.T. Role of extracellular glutathione and γ-glutamyltranspeptidase in the disposition and kidney toxicity of inorganic mercury in rats. J. Appl. Toxicol. 1994, 14, 201–206. [Google Scholar] [CrossRef]
- Zalups, R.K. Organic anion transport and action of g-glutamyltranspeptidase in kidney linked mechanistically to renal tubular uptake of inorganic mercury. Toxicol. Appl. Pharmacol. 1995, 132, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Barfuss, D.W. Participation of mercuric conjugates of cysteine, homocysteine, and N-acetylcysteine in mechanisms involved in the renal tubular uptake of inorganic mercury. J. Am. Soc. Nephrol. 1998, 9, 551–561. [Google Scholar] [PubMed]
- Cannon, V.T.; Zalups, R.K.; Barfuss, D.W. Molecular homology and the luminal transport of Hg2+ in the renal proximal tubule. J. Am. Soc. Nephrol. 2000, 11, 394–402. [Google Scholar] [PubMed]
- Zalups, R.K. Basolateral uptake of inorganic mercury in the kidney. Toxicol. Appl. Pharmacol. 1998, 150, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005, 204, 274–308. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.M.; Dnyanmote, A.V.; Bush, K.T.; Wu, W.; Nigam, S.K. Deletion of multispecific organic anion transporter (Oat1/Slc22a6) protects from mercury-induced kidney injury. J. Biol. Chem. 2011, 286, 26391–26395. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Transport of inorganic mercury and methylmercury in target tissues and organs. J. Toxicol. Environ. Health Crit. Rev. 2010, 13, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Joshee, L.; Zalups, R.K. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J. Pharmacol. Exp. Ther. 2008, 324, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Compeau, G.C.; Bartha, R. Sulfate-reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Appl. Environ. Microbiol. 1985, 50, 498–502. [Google Scholar] [PubMed]
- Clarkson, T.W.; Magos, L.; Myers, G.J. Toxicology of mercury—current exposures and clinical manifestations. N. Engl. J. Med. 2003, 349, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- National Research Council. Toxicological Effect of Methylmercury; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- European Food Safety Authority (EFSA). Panel on contaminants in food chain: Scientific opinion on the risk for public health related to presence of mercury and methylmercury in food. EFSA J. 2012, 10, 2985. [Google Scholar]
- Zareba, G.; Cernichiari, E.; Hojo, R.; Nitt, S.M.; Weiss, B.; Mumtaz, M.M.; Jones, D.E.; Clarkson, T.W. Thimerosal distribution and metabolism in neonatal mice: Comparison with methyl mercury. J. Appl. Toxicol. 2007, 27, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Jiang, H.; Syversen, T.; Rocha, J.B.; Farina, M.; Aschner, M. The methylmercury-l-cysteine conjugate is a substrate for the l-type large neutral amino acid transporter. J. Neurochem. 2008, 107, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Castoldi, A.F.; Onishchenko, N.; Manzo, L.; Vahter, M.; Ceccatelli, S. Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox. Res. 2007, 11, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Atchison, W.D. Mercurial-induced alterations in neuronal divalent cation homeostasis. Neurotoxicology 1996, 17, 47–61. [Google Scholar] [PubMed]
- Gasso, S.; Cristofol, R.M.; Selema, G.; Rosa, R.; Rodriguez-Farre, E.; Sanfeliu, C. Antioxidant compounds and Ca2+ pathway blockers differentially protect against methylmercury and mercuric chloride neurotoxicity. J. Neurosci. Res. 2001, 66, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Limke, T.L.; Heidemann, S.R.; Atchison, W.D. Disruption of intraneuronal divalent cation regulation by methylmercury: Are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? Neurotoxicology 2004, 25, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Sudati, J.H.; Nogueira, C.W.; Rocha, J.B. In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. Biometals 2010, 23, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Canario, J.; Holmgren, A.; Carvalho, C. Inhibition of the thioredoxin system in the brain and liver of zebra sea breams exposed to waterborne methylmercury. Toxicol. Appl. Pharmacol. 2011, 251, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Campos, F.; Vendrell, I.; Berenguer, J.; Barzi, M.; Pons, S.; Sunol, C. Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicol. Sci. 2009, 112, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Vargas, A.P.; Roos, D.H.; Morel, A.F.; Farina, M.; Nogueira, C.W.; Aschner, M.; Rocha, J.B. Comparative study of quercetin and its two glycoside derivatives quercitrin and rutin against methylmercury (MeHg)-induced ROS production in rat brain slices. Arch. Toxicol. 2010, 84, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Lu, J.; Zhang, X.; Arner, E.S.; Holmgren, A. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: Implications for treatment of mercury poisoning. FASEB J. 2010, 25, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Aschner, M.; Syversen, T. Glutathione modulation influences methylmercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology 2006, 27, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.L.; Braga, H.C.; Stringari, J.; Missau, F.C.; Posser, T.; Mendes, B.G.; Leal, R.B.; Santos, A.R.; Dafre, A.L.; Pizzolatti, M.G.; et al. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem. Res. Toxicol. 2007, 20, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Amonpatumrat, S.; Sakurai, H.; Wiriyasermkul, P.; Khunweeraphong, N.; Nagamori, S.; Tanaka, H.; Piyachaturawat, P.; Kanai, Y. l-glutamate enhances methylmercury toxicity by synergistically increasing oxidative stress. J. Pharmacol. Sci. 2008, 108, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.H.; Puntel, R.L.; Farina, M.; Aschner, M.; Bohrer, D.; Rocha, J.B.; De Vargas Barbosa, N.B. Modulation of methylmercury uptake by methionine: prevention of mitochondrial dysfunction in rat liver slices by a mimicry mechanism. Toxicol. Appl. Pharmacol. 2011, 252, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Fujimura, M. Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. J. Biol. Chem. 2009, 286, 6641–6649. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Mutkus, L.A.; Aschner, M. Methylmercury-mediated inhibition of 3 HD-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001, 902, 92–100. [Google Scholar] [CrossRef]
- Mori, N.; Yasutake, A.; Hirayama, K. Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch. Toxicol. 2007, 81, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Dorea, J.G. Making sense of epidemiological studies of young children exposed to thimerosal in vaccines. Clin. Chim. Acta 2010, 411, 1580–1586. [Google Scholar] [CrossRef] [PubMed]
- ASTDR. Priority list of hazardous substances. 2013. Available online: http://www.atsdr.cdc.gov/spl/ (accessed on 22 September 2015). [Google Scholar]
- Hughes, M.F.; Beck, B.D.; Chen, Y.; Lewis, A.S.; Thomas, D.J. Arsenic exposure and toxicology: A historical perspective. Toxicol. Sci. 2011, 123, 305–332. [Google Scholar] [CrossRef] [PubMed]
- Mandal, B.K.; Suzuki, K.T. Arsenic round the world: A Review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, D.; Sahu, A. Arsenic in the environment: Effects on human health and possible prevention. J. Environ. Biol. 2007, 28, 359–365. [Google Scholar] [PubMed]
- Kaur, S.; Kamli, M.R.; Ali, A. Role of arsenic and its resistance in nature. Can. J. Microbiol. 2011, 57, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E. The toxic side of rice. Nature 2014, 514, 62–63. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Jarouliya, U.; Prasad, G.B.K.S.; Flora, S.J.S.; Bisen, P.S. Arsenic-induced abnormalities in glucose metabolism: Biochemical basis and potential therapeutic and nutritional interventions. World J. Transl. Med. 2014, 3, 96–111. [Google Scholar] [CrossRef]
- Naujokas, M.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Chavaz, L.A.; Rendon-Lopez, C.R.R.; Zepeda, A.; Silva-Adaya, D.; Razo, L.M.D.; Gonsebatt, M.E. Neurological effects of inorganic arsenic exposure: Altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell. Neurosci. 2015, 9, 1–12. [Google Scholar]
- Villa-Bellosta, R.; Sorribas, V. Role of rat sodium/phosphate cotransporters in the cell membrane transport of arsenate. Toxicol. Appl. Pharmacol. 2008, 232, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Calatayud, M.; Barrios, J.A.; Velez, D.; Devesa, V. In vitro study of transporters involved in intestinal absorption of inorganic arsenic. Chem. Res. Toxicol. 2012, 25, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Maciaszczyk-Dziubinska, E.; Wawrzycka, D.; Wysocki, R. Arsenic and antimony transporters in eukaryotes. Int. J. Mol. Sci. 2012, 13, 3527–3548. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S. Arsenic-induced oxidative stress and its reversibility. Free Radic. Biol. Med. 2011, 51, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Styblo, M.; del Razo, L.M.; Vega, L.; Germolec, D.R.; LeCluyse, E.L.; Hamilton, G.A.; Reed, W.; Wang, C.; Cullen, W.R.; Thomas, D.J. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 2000, 74, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Singh, S.; Siddiqi, N.J. Biomedical implications of heavy metal induced imbalances in redox systems. BioMed Res. Int. 2014, 2014, 640754. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.H.; Lingas, E.O.; Rahman, M. Contamination of drinking-water by arsenic in Bangladesh: A public health emergency. Bull. World Health Organ. 2000, 78, 1093–1103. [Google Scholar] [PubMed]
- Shi, H.; Shi, X.; Liu, K.J. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol. Cell. Biochem. 2004, 255, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamauchi, H.; Fan Sun, G. Chronic health effects in people exposed to arsenic via the drinking water: Dose-response relationships in review. Toxicol. Appl. Pharmacol. 2004, 198, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Hopenhayn, C. Arsenic in drinking water: impact on human health. Elements 2006, 2, 103–107. [Google Scholar] [CrossRef]
- Gordon, J.J.; Quastel, G.H. Effect of organic arsenicals on enzyme system. Biochem. J. 1948, 42, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, G. Chronic arsenic toxicity and human health. Indian J. Med. Res. 2008, 128, 436–447. [Google Scholar]
- Lin, S.; Shi, Q.; Nix, F.B.; Styblo, M.; Beck, M.A.; Herbin-Davis, K.M.; Hall, L.L.; Simeonsson, J.B.; Thomas, D.J. A novel S-adenosyl-l-methionine: ArsenicIII methyltransferase from rat liver cytosol. J. Biol. Chem. 2002, 277, 10795–10803. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.B.; Devesa, V.; DelRazo, L.M.; Styblo, M.; Thomas, D.J. Endogenous reductants support the catalytic function of recombinant rat cyt19, an arsenic methyltransferase. Chem. Res. Toxicol. 2004, 17, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.J. Unraveling arsenic-glutathione connections. Toxicol. Sci. 2009, 107, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.S.; Jagadish, B.; Mash, E.A.; Aposhian, H.V. Monomethylarsonous acid (MMAIII) and arsenite: Ld(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem. Res. Toxicol. 2001, 14, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Gamble, M.V. Nutritional manipulation of one-carbon metabolism: Effects on arsenic methylation and toxicity. J. Toxicol. 2012, 2012, 595307. [Google Scholar] [CrossRef] [PubMed]
- Karrari, P.; Mehrpour, O.; Abdollahi, M. A systemic review on status of lead pollution and toxicity in Iran; Guidance for preventive measures. DARU J. Pharm. Sci. 2012, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Malekirad, A.A.; Oryan, S.; Fani, A.; Babapor, V.; Hashemi, M.; Baeeri, M.; Bayrami, Z.; Abdollahi, M. Study on clinical and biochemical toxicity biomarkers in a zinc-lead mine workers. Toxicol. Ind. Health 2010, 26, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Hernberg, S. Lead poisoning in a historical perspective. Am. J. Ind. Med. 2000, 38, 244–254. [Google Scholar] [CrossRef]
- Jalali, M.; Khanlari, Z.V. Enviromental contamination of Zn, Cd, Ni, Cu and Pb from industrial areas in Hamadan Province, western Iran. Environ. Geol. 2008, 55, 1537–1543. [Google Scholar] [CrossRef]
- Parizanganeh, A.; Hajisoltani, P.; Zamani, A. Assessment of heavy metal pollution in surficial soils surrounding Zinc Industrial Complex in Zanjan-Iran. Procedia Environ. Sci. 2010, 2, 162–166. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Amin, M.M.; Hashemi, H.; Foladifard, R.; Vahiddastjerdi, M. A survey of groundwater chemical quality in Sajad Zarinshahr. Health Syst. Res. 2011, 6, 918–926. [Google Scholar]
- Advisory Committee on Childhood Lead Poisoning Prevention. Recommendations for blood lead screening of young children enrolled in Medicaid: Targeting a group at high risk. MMWR Morb. Mortal. Wkly. Rep. 2000, 49, 1–13. [Google Scholar]
- Abdollahi, M.; Ebrahimi-Mehr, M.; Nikfar, S.; Jalali, N. Monitoring of lead poisoning in simple workers of a copying center by flame atomic absorption spectroscopy. MJIRI 1996, 10, 69–72. [Google Scholar]
- Nelson, L.; Lewin, N.; Howland, M.A.; Hoffman, R.; Goldfrank, L.; Flomenbaum, N. Goldfrank’s Toxicologic Emergencies, 9th ed.; Mc Graw Hill: New York, NY, USA, 2011; pp. 1266–1280. [Google Scholar]
- Pourmand, A.; Al-tiae, T.K.; Mazer-Amirshahi, M. Perspective of lead toxicity, a comparison between the United States and Iran. DARU J. Pharm. Sci. 2012, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Rogan, W.J.; Dietrich, K.N.; Ware, J.H.; Dockery, D.W.; Salganik, M.; Radcliffe, J.; Jones, R.L.; Ragan, N.B.; Chisolm, J.J., Jr.; Rhoads, G.G. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N. Eng. J. Med. 2001, 344, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Neal, A.P.; Guilarte, T.R. Mechanism of lead and manganese neurotoxicity. Toxic Res. 2013, 2, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, N.C.; Hatzidaki, E.G.; Belivanis, S.; Tzanakakis, G.N.; Tsatsakis, A.M. Lead toxicity update. A brief review. Med. Sci. Monitor 2005, 11, 329. [Google Scholar]
- Azizi, M.H.; Azizi, F. Lead Poisoning in the world and Iran. Int. J. Occup. Environ. Med. 2010, 1, 81–87. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention. Preventing Lead Poisoning in Young Children; Centers for Disease Control: Atlanta, GA, USA, 2005.
- Ziegler, E.E.; Edwards, B.B.; Jensen, R.L.; Mahaffey, K.R.; Fomon, S.J. Absorption and retention of lead by infants. Pediatr. Res. 1978, 12, 29–34. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM. European food safety authority panel on contaminants in the food chain (CONTAM); scientific opinion on lead in food. EFSA J. 2010, 8, 1570. [Google Scholar]
- Gulson, B.L.; Mizon, K.J.; Korsch, M.J.; Palmer, J.M.; Donnelly, J.B. Mobilization of lead from human bone tissue during pregnancy and lactation—A summary of long-term research. Sci. Total Environ. 2003, 303, 79–104. [Google Scholar] [CrossRef]
- Skoczynska, A. Genetic aspects of hypertensive effect of lead. Med. Pr. 2008, 59, 325–332. [Google Scholar] [PubMed]
- Ahamed, M.; Siddiqui, M.K.J. Low level lead exposure and oxidative stress: Current opinions. Clin. Chem. Acta 2007, 383, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, W.E.; Eisinger, J.; Lamola, A.A.; Zuckerman, D.M. Principles and applications of hematofluorometry. J. Clin. Lab. Autom. 1984, 4, 29–42. [Google Scholar]
- Douki, T.; Onuki, J.; Medeiros, M.H.; Bechara, E.J.H.; Cadet, J.; DiMascio, P. DNA alkylation by 4,5-dioxovaleric acid, the final oxidation product of 5-aminolevulinic acid. Chem. Res. Toxicol. 1998, 11, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Onuki, J.; Medeiros, M.H.; Bechara, E.J.H.; Cadet, J.; Di Mascio, P. Hydroxy radicals are involved in the oxidation of isolated and cellular DNA bases by 5-aminolevulinic acid. FEBS Lett. 1998, 428, 93–96. [Google Scholar] [CrossRef]
- Donaldson, W.E.; Knowles, S.O. Is lead toxicosis a reflection of altered fatty acid composition ofmembrane? Comp. Biochem. Physiol. Part C Comp. Pharm. 1993, 104, 377–379. [Google Scholar] [CrossRef]
- Navas-Acien, A.; Guallar, E.; Silbergeld, E.K.; Rothenberg, S.J. Pb exposure and cardiovascular disease—A systematic review. Environ. Health Perspect. 2007, 115, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Poreba, R.; Gac, P.; Poreba, M.; Andrzejak, R. Environmental and occupational exposure to lead as a potential risk factor for cardiovascular diseases. Environ. Toxicol. Pharm. 2011, 31, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Poreba, R.; Poreba, M.; Gac, P.; Andrzejak, R. Ambulatory blood pressure monitoring and structural changes in carotid arteries in normotensive workers occupationally exposed to lead. Hum. Exp. Toxicol. 2011, 30, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Poreba, R.; Gac, P.; Poreba, M.; Derkacz, A.; Pilecki, W.; Antonowicz-juchniewicz, J.; Andrzejak, R. Relationship between chronic exposure to lead, cadmium and manganese, blood pressure values and incidence of arterial hypertension. Med. Pr. 2010, 61, 5–14. [Google Scholar] [PubMed]
- Khalil-Manesh, F.; Gonick, H.C.; Weiler, E.J.; Prins, B.; Weber, M.A.; Purdy, R. Lead induced hypertension: Possible role of endothelial factors. Am. J. Hypertens. 1993, 6, 723–729. [Google Scholar] [PubMed]
- Gonick, H.C.; Ding, Y.; Bondy, S.C.; Ni, Z.; Vaziri, N.D. Lead-induced hypertension I: Interplay of nitric oxide and reactive oxygen species. Hypertension 1997, 30, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Zawadzki, M.; Poreba, R.; Gac, P. Mechanisms and toxic effects of lead on the cardiovascular system. Med. Pr. 2006, 57, 543–549. [Google Scholar] [PubMed]
- Vargas, H.; Castillo, C.; Posadas, F.; Escalante, B. Acute lead exposure induces renal heme oxygenase-1 and decreases urinary Na+ excretion. Hum. Exp. Toxicol. 2003, 22, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.B.; Tiwari, A.; Jatawa, S.K. Free radicals and antioxidants: A review. J. Pharm. Res. 2011, 4, 4340–4343. [Google Scholar]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of lead: A review with recent updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Brochin, R.; Leone, S.; Phillips, D.; Shepard, N.; Zisa, D.; Angerio, A. The cellular effect of lead poisoning and its clinical picture. GUJHS 2008, 5, 1–8. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Babior, B.M. NADPH oxidase: An update. Blood 1999, 93, 1464–1476. [Google Scholar] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Cadenas, E. Biochemistry of oxygen toxicity. Annu. Rev. Biochem. 1989, 58, 79–110. [Google Scholar] [CrossRef] [PubMed]
- Pastor, N.; Weinstein, H.; Jamison, E.; Brenowitz, M. A detailed interpretation of OH radical footprints in a TBP–DNA complex reveals the role of dynamics in the mechanism of sequence specific binding. J. Mol. Biol. 2000, 304, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ischiropoulus, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Dicov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signalling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Non-Native Proteins as Newly Identified Targets of Heavy Metals and Metalloids. In Cellular Effects of Heavy Metals; Bánfalvi, G., Ed.; Springer: Heidelberg, Germany, 2011; pp. 263–274. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M.T.D. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Role of oxidant species in aging. Curr. Med. Chem. 2004, 11, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Oxidoreduction of protein thiols in redox regulation. Biochem. Soc. Trans. 2005, 33, 1378–1381. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Goloubinoff, P.; Christen, P. Heavy metal ions are potent inhibitors of protein folding. Biochem. Biophys. Res. Commun. 2008, 372, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.; Navarrete, C.; Sharma, S.K.; Sideri, T.C.; Ibstedt, S.; Priya, S.; Grant, C.M.; Christen, P.; Goloubinoff, P.; Tamas, M.J. Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci. 2012, 125, 5073–5083. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.L.; Molina-Jijon, E.; Rodriguez-Munoz, R.; Bautista-Gracia, P.; Debray-Gracia, Y.; Namorada, M.C. Tight junction proteins and oxidative stress in heavy metals induced nephrotoxicity. BioMed Res. Int. 2013, 2013, 730789. [Google Scholar]
- Tamas, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic. Biol. Med. 2009, 47, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, J.S.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Uchida, K. 4-hydroxy-2-nonenal: A product and modulator of oxidative stress. Prog. Lipid Res. 2003, 42, 318–343. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Leonarduzzi, G. 4-hydroxynonenal—protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol. Asp. Med. 2008, 29, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Lucesoli, F.; Fraga, C.G. Oxidative damage to lipids and DNA concurrent with decrease of antioxidants in rat testes after acute iron intoxication. Arch. Biochem. Biophys. 1995, 316, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Sole, J.; Huguet, J.; Arola, L.; Romeu, A. In vivo effects of nickel and cadmium in rats on lipid peroxidation and ceruloplasmin activity. Bull. Environ. Contam. Toxicol. 1990, 44, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Kasprzak, K.S. Possible role of oxidative damage in metalinduced carcinogenesis. Cancer Investig. 1995, 13, 411–430. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.T.; Azam, M.; Ali, A.; Haq, Q.M.R. Prospects for exploiting bacteria for bioremediation of metal pollution. Crit. Rev. Environ. Sci. Technol. 2014, 44, 1–42. [Google Scholar] [CrossRef]
- O’Neill, P.; Chapman, P.W. Potential repair of free radical adducts of dGMP and dG by a series of reductants. A pulse radiolytic study. Int. J. Radiat. Biol. Relat. Study Phys. Chem. Med. 1985, 47, 71–80. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidative damage to DNA in mammalian chromatin. Mutat. Res. 1992, 275, 331–342. [Google Scholar] [CrossRef]
- Breen, A.P.; Murphy, J.A. Reactions of oxyl radicals with DNA. Free Radic. Biol. Med. 1995, 18, 1033–1077. [Google Scholar] [CrossRef]
- Burcham, P.C. Internal hazards: Baseline DNA damage by endogenous products of normal metabolism. Mutat. Res. 1999, 443, 11–36. [Google Scholar] [CrossRef]
- Laval, F.; Wink, D.A.; Laval, J. A discussion of mechanisms of NO genotoxicity: Implication of inhibition of DNA repair proteins. Rev. Physiol. Biochem. Pharmacol. 1997, 131, 175–191. [Google Scholar] [PubMed]
- Jaiswal, M.; LaRusso, N.F.; Nishioka, N.; Nakabeppu, Y.; Gores, G.J. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001, 61, 6388–6393. [Google Scholar] [PubMed]
- Haghdoost, S.; Czene, S.; Naslund, I.; Skog, S.; Harms-Ringdahl, M. Extracellular 8-oxo-dG as a sensitive parameter for oxidative stress in vivo and in vitro. Free Radic. Res. 2005, 39, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Jaruga, P.; Dizdaroglu, M. Repair of products of oxidative DNA base damage in human cells. Nucleic Acids Res. 1996, 24, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- DeBont, R.; Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Masella, R.; DiBenedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Deleve, L.D.; Kaplowitz, N. Glutathione metabolism and its role in hapatotoxicity. Pharmacol. Ther. 1991, 52, 287–305. [Google Scholar] [CrossRef]
- Rabenstein, D.L. Metal complexes of glutathione and their biological significance. In Glutathione: Chemical, Biochemical and Medical Aspects; Dolphin, D., Avramomic, O., Poulson, R., Eds.; John Wiley and Sons: New York, NY, USA, 1998; pp. 147–186. [Google Scholar]
- Lund, B.O.; Miller, D.M.; Woods, J.S. Studies on mercury (II) induced H2O2 formation and oxidative stress in vivo and in vitro in rat kidney mitochondria. Biochem. Pharmacol. 1993, 45, 2017–2024. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.; Monco, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Niki, E. Assesment of antioxidant capacity in vitro and in vivo. Free Radic. Biol. Med. 2010, 49, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.; Chan, H.M. The influence of nutrition on methylmercury intoxication. Environ. Health Perspect. 2000, 108, 29–56. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Rooney, J.P.K. The role of thiols, dithiols, nutritional factors and interacting ligands in the toxicology of mercury. Toxicology 2007, 234, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Peraza, M.A.; Ayala Fierro, F.; Barber, D.S.; Casarez, E.; Rael, L.T. Effects of micronutrients on metal toxicity. Environ. Health Perspect. 1998, 106, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.C.; Guo, Y.L. Antioxidant nutrients and lead toxicity. Toxicology 2002, 180, 33–44. [Google Scholar] [CrossRef]
- Oroian, M.; Escriche, I. Antioxidants: Characterization, natural sources, extraction and analysis. Food Res. Int. 2015, 74, 10–36. [Google Scholar] [CrossRef]
- Wojcik, M.; Burzynska-pedziwiatr, I.; Woznaik, L.A. A review of natural and synthetic antioxidants important for health and longevity. Curr. Med. Chem. 2010, 17, 3262–3288. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.G.; Incerpi, S.; Ahmed, F.; Gaber, A. The developmental and physiological interaction between free radicals and antioxidant defense system: Effect of environmental pollutants. J. Natl. Sci. Res. 2013, 3, 74–110. [Google Scholar]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, L.; Gazzaniga, A. Toxicological, pharmacokinetic and metabolic studies on acetylcysteine. Eur. J. Res. Dis. 1980, 61, 45–51. [Google Scholar]
- DeVries, N.; DeFlora, S. N-acetylcysteine. J. Biol. Chem. 1993, 17, 270–277. [Google Scholar]
- DeFlora, S.; Rossi, G.A.; DeFlora, A. Metabolic, dismutagenic and anticarcinogenic effects of N-acetylcysteine. Respiration 1986, 50, 43–49. [Google Scholar] [CrossRef]
- Huxtable, R. Physiological action of taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar] [PubMed]
- Huxtable, R. Expanding the circle 1975–1999: Sulfur biochemistry and insights on the biological functions of taurine. Adv. Exp. Med. Biol. 2000, 483, 1–25. [Google Scholar] [PubMed]
- Zelikovic, I.; Chesney, R.W.; Friedman, A.L.; Ahlfors, C.E. Taurine depletion in very low birth weight infants receiving prolonged total parenteral nutrition: role of renal immaturity. J. Pediatr. 1990, 116, 301–306. [Google Scholar] [CrossRef]
- Howard, D.; Thompson, D.F. Taurine: An essential amino acid to prevent cholestasis in neonates. Ann. Pharmacother. 1992, 26, 1390–1392. [Google Scholar] [PubMed]
- Okamoto, E.; Rassin, D.K.; Zucker, C.L.; Salen, G.S.; Heird, W.C. Role of taurine in feeding the low-birth-weight infant. J. Pediatr. 1984, 104, 936–940. [Google Scholar] [CrossRef]
- Vinton, N.E.; Laidlaw, S.A.; Ament, M.E.; Kopple, J.D. Taurine concentrations in plasma, blood cells and urine of children undergoing long-term parenteral nutrition. Pediatr. Res. 1987, 21, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.R.; Dawson, R. Decreased plasma taurine in aged rats. Gerontology 1990, 36, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.D.; Wang, J.H.; Fennessy, F.; Redmond, H.P.; Bouchier-Hayes, H.D. Taurine prevents high-glucose-induced human vascular endothelial cell apoptosis. Am. J. Physiol. 1999, 277, 1229–1238. [Google Scholar]
- Son, M.; Ko, J.I.; Kim, W.B.; Kang, H.K.; Kim, B.K. Taurine can ameliorate inflammatory bowel disease in rats. Adv. Exp. Med. Biol. 1998, 442, 291–298. [Google Scholar] [PubMed]
- Kendler, B.S. Recent nutritional approaches to the prevention and therapy of cardiovascular disease. Prog. Cardiovasc. Nurs. 1997, 12, 3–23. [Google Scholar] [PubMed]
- Hwang, D.F.; Wang, L.C.; Cheng, H.M. Effect of taurine on toxicity of copper in rats. Food Chem. Toxicol. 1998, 36, 239–244. [Google Scholar] [CrossRef]
- Neal, R.; Cooper, K.; Kellogg, G.; Gwer, H.; Ercal, N. Effect of some sulfur containing antioxidants on lead exposed lenses. Free Radic. Biol. Med. 1999, 26, 239–243. [Google Scholar] [CrossRef]
- McGowan, C.; Donaldson, W.E. Effect of lead toxicity on the organ concentration of glutathione and glutathione-related free amino acids in the chick. Toxicol. Lett. 1987, 38, 265–270. [Google Scholar] [CrossRef]
- Herrero, M.C.; Alvarez, C.; Cartana, J.; Blade, C.; Arola, L. Nickel effects on hepatic amino acids. Res. Commun. Chem. Pathol. Pharmacol. 1993, 79, 243–248. [Google Scholar] [PubMed]
- Menendez-Pelaez, A.; Poeggeler, B.; Reiter, R.J.; Barlow-Walden, L.; Pablos, M.I.; Tan, D.X. Nuclear localization of melatonin in different mammalian tissues: Immunocytochemical and radioimmunoassay evidence. J. Cell. Biochem. 1993, 53, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Okatani, Y.; Okamoto, K.; Hayashi, K.; Wakatsuki, A.; Tamura, S.; Sagara, Y. Maternal-fetal transfer of melatonin in pregnant women near term. J. Pineal Res. 1998, 25, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, Y.; Ferrebuz, A.; Romero, F.; Vaziri, N.D.; Rodriguez-Iturbe, B. Melatonin ameliorates oxidative stress, inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction. Am J. Physiol. Ren. Physiol. 2008, 294, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Kotler, M.; Rodriguez, C.; Sainz, R.M.; Antolin, I.; Menendez-Pelaez, A. Melatonin increases gene expression for antioxidant enzymes in rat brain cortex. J. Pineal Res. 1998, 24, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Rodriquez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Pieri, C.; Marra, M.; Moroni, F.; Recchioni, R.; Marcheselli, F. Melatonin—A peroxide radical scavenger more effective than vitamin E. Life Sci. 1994, 55, 271–276. [Google Scholar] [CrossRef]
- Tan, D.X.; Chen, L.D.; Poeggler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: A potent endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Melchiorri, R.J.; Reiter, A.M.; Atlia, M.; Hara, A.; Burgos-Nistico, G. Potent protective effect of melatonin on in vivo paraquat induced oxidative damage in rats. Life Sci. 1995, 56, 83–89. [Google Scholar] [CrossRef]
- Othman, A.; Sharawy, S.A.; Missiry, M.A.E. Role of melatonin in ameliorating lead induced haematotoxicity. Pharmacol. Res. 2004, 50, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Cano, P.; Poliandri, A.H.B.; Jimenez, V.; Cardinali, D.P.; Esquifino, A.I. Cadmium induced changes in Per 1 and Per 2 gene expression in rat hypothalamus and anterior pitutory: Effect of melatonin. Toxicol. Lett. 2007, 172, 131–136. [Google Scholar] [CrossRef] [PubMed]
- National Academy of Sciences. Dietary Reference Intake: Application in dietary assessment. 2000. Available online: http://www.nap.edu/ (accessed on 28 September 2015).
- Letavayova, L.; Vlckova, V.; Brozmanova, J. Selenium: From cancer prevention to DNA damage. Toxicology 2006, 227, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.B.; Aswath, K.; Chanock, S.J.; McKay, H.F.; Peters, U. Polymorphism analysis of six selenoprotein genes: support for a selective sweep at the glutathione peroxidase 1 locus (3p21) in Asian populations. BMC Genet. 2006, 7, 56. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Trueba, G.P.; Sanchez, G.M.; Giuliani, A. Oxygen free radical and antioxidant defense mechanism in cancer. Front. Biosci. 2004, 9, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; El-Missiry, M.A. Role of selenium against lead toxicity in male rats. J. Biochem. Mol. Toxicol. 1998, 12, 345–349. [Google Scholar] [CrossRef]
- Abdulla, M.; Chmielnicka, J. New aspects on the distribution and metabolism of essential trace elements after dietary exposure to toxic metals. Biol. Trace Elem. 1990, 23, 25–53. [Google Scholar] [CrossRef]
- Whagner, P.D. Selenium in the treatment of heavy metal poisoning and chemical carcinogenesis. J. Trace Elem. Electro. Health Dis. 1992, 6, 209–221. [Google Scholar]
- Suzuki, K.T. Equimolar Hg-Se complex binds to selenoprotein-P. Biochem. Biophy. Res. Commun. 1997, 231, 7–11. [Google Scholar]
- Kalia, K.; Flora, S.J.S. Safe and effective therapeutic measures for chronic arsenic and lead poisoning. J. Occup. Health 2005, 47, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Combs, G.F., Jr.; Midthune, D.N.; Patterson, K.Y.; CanWeld, W.K.; Hill, A.D.; Levander, O.A.; Taylor, P.R.; Moler, J.E.; Patterson, B.H. Effects of selenomethionine supplementation on selenium status and thyroid hormone concentrations in healthy adults. Am. J. Clin. Nutr. 2009, 89, 1808–1814. [Google Scholar] [CrossRef] [PubMed]
- Bronzetti, G.; Cini, M.; Andreoli, E.; Caltavuturo, L.; Panunzio, M.; Croce, C.D. Protective effects of vitamins and selenium compounds in yeast. Mutat. Res. 2001, 496, 105–115. [Google Scholar] [CrossRef]
- Fang, Y.; Yang, S.; Wu, G. Free radicals, antioxidants and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar] [CrossRef]
- Stoltzfus, R.J. Defining iron-deficiency anemia in public health terms: Time for reflection. J. Nutr. 2001, 131, 565–567. [Google Scholar]
- WHO. WHO World Wide Prevalence of Anaemia 1993–2005: WHO Global Database on Anaemia; De Benoist, B., McLean, E., Egli, I., Cogswell, M., Eds.; Centers for Disease Control: Atlanta, GA, USA, 2008. [Google Scholar]
- Gutteridge, J. Ferrous ion-EDTA-stimulated phospholipids peroxidation. A reaction changing from alkoxyl-radical to hydroxyl-radical dependent initiation. Biochem. J. 1984, 224, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Tadolini, B.; Hakim, G. The mechanism of iron (III) stimulation of lipid peroxidation. Free Radic. Res. 1996, 25, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.; Mackenzie, G.; Verstraeten, S. Metals in neurodegeneration: involvement of oxidants and oxidant-sensitive transcription factors. Mol. Asp. Med. 2004, 25, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Labbe, R. Lead poisoning mechanisms. Clin. Chem. 1990, 36, 1870. [Google Scholar] [PubMed]
- Briner, W. The alchemists approach to metal poisoning: Transforming the metal burden. Toxics 2014, 2, 364–376. [Google Scholar] [CrossRef]
- DeFeo, C.J.; Aller, S.G.; Unger, V.M. A structural perspective on copper uptake in eukaryotes. Biometals 2007, 20, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Behari, J.R.; Tandon, S.K. Protective role of trace metals in lead intoxication. Toxicol. Lett. 1982, 13, 51–56. [Google Scholar] [CrossRef]
- Millier, G.D.; Massaro, T.F.; Massaro, E.J. Interaction between lead and essential elements—A review. Neurotoxicology 1990, 11, 99–120. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar] [PubMed]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed]
- O’Halloran, T.V.; Culotta, V.C. Metallochaperones: an intracellular shuttle service for metal ions. J. Biol. Chem. 2000, 275, 25057–25060. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Muzzioli, M. Zinc, metallothioneins, immune response, survival and ageing. Biogerontology 2000, 1, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Muzzioli, M.; Giacconi, R. Zinc and immunoresistance to infections in ageing: New biological tools. Trends Pharmacol. Sci. 2000, 21, 205–208. [Google Scholar] [CrossRef]
- Vasak, M.; Hasler, D.W. Metallothioneins: New functional and structural insights. Curr. Opin. Chem. Biol. 2000, 4, 177–183. [Google Scholar] [CrossRef]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, J.A.; Taylor, C.G.; Weiler, H.A. Marginal zinc deficiency exacerbates bone lead accumulation and high dietary zinc attenuates lead accumulation at the expense of bone density in growing rats. Toxicol. Sci. 2006, 92, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Singh, S.; Tandon, S.K. Thiamine and zinc in prevention of lead intoxication. J. Int. Med. Res. 1989, 17, 68–75. [Google Scholar] [PubMed]
- Flora, S.J.S.; Kumar, D.; Gupta, D. Intereaction of zinc, methionine or their combination with lead at gastrointestinal or post-absorptive levels in rats. Pharmacol. Toxicol. 1999, 68, 3–7. [Google Scholar] [CrossRef]
- Franciscato, C.; Silva, L.M.; Duarte, F.A.; Oliveira, C.S.; Ineu, R.P.; Flores, E.M.M.; Dressler, V.L.; Piexoto, N.C.; Pereira, M.E. Delayed biochemical changes induced by mercury intoxication are prevented by zinc exposure. Ecotoxicol. Environ. Saf. 2011, 74, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, G.; LaPorta, C.A.M. Molecular mechanisms triggered by mercury. Toxicology 2008, 244, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Menvielle-Bourg, F.J. Superoxide Dismutase (SOD), a Powerful Antioxidant, is now available orally. Phytotherapie 2005, 3, 1–4. [Google Scholar]
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Perez-Gomez, C.; DeCastro, I.N. Antioxidant enzymes and human diseases. Clin. Biochem. 1999, 32, 595–603. [Google Scholar] [CrossRef]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Lodge, J.; Packer, L. Natural source of lipoic acid in plant and animal tissues. In Antioxidant Food Supplements in Human Health; Packer, L., Hiramatsu, M., Yoshikawa, T., Eds.; Academic Press: San Diego, CA, USA, 1999; p. 121. [Google Scholar]
- Wollin, S.D.; Jones, P.L. α-lipoic acid and cardiovascular disease. J. Nutr. 2003, 133, 3327–3330. [Google Scholar] [PubMed]
- Marquet, A.; TseSum, B.B.; Florentin, D. Biosynthesis of biotin and lipoic acid. Vitam. Horm. 2001, 61, 51–94. [Google Scholar] [PubMed]
- Ou, P.; Tritscher, H.J.; Wolff, S.P. Thioctic (Lipoic) acid: A therapeutic metal chelating antioxidant. Biochem. Pharmacol. 1995, 50, 123–126. [Google Scholar] [CrossRef]
- Bustamante, J.; Lodge, J.K.; Marcocci, L.; Tritschler, H.J.; Packer, L.; Rihn, B.H. α-lipoic acid in liver metabolism and disease. Free Radic. Biol. Med. 1998, 24, 1023–1039. [Google Scholar] [CrossRef]
- Arivazhagan, P.; Ramanathan, K.; Panneerselvam, C. Effect of d,l-α-lipoic acid on mitochondrial enzymes in aged rats. Chem. Biol. Interact. 2001, 138, 189–198. [Google Scholar] [CrossRef]
- Reed, L.J. Multienzyme complex. Acc. Chem. Res. 1974, 7, 40–46. [Google Scholar] [CrossRef]
- Packer, L.; Kraemer, K.; Rimbach, G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 2001, 17, 888–895. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. α-lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Biewenga, G.P.; Haenen, G.R.M.M.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Cremer, D.R.; Rabeler, R.; Roberts, A.; Lynch, B. Long-term safety of α-lipoic acid (ALA) consumption: A 2-year study. Long Regul. Toxicol. Pharmacol. 2006, 46, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Tiechert, J.; Kern, J.; Tritschler, H.J.; Ulrich, H.; Preib, R. Investigations on the pharmacokinetics of α-lipoic acid in healthy volunteers. Int. J. Clin. Pharm. Ther. 1998, 36, 625–628. [Google Scholar]
- Ali, Y.F.; Desouky, O.S.; Selim, N.S.; Ereiba, M. Assessment of the role of α-lipoic acid against the oxidative stress of induced iron overload. J. Radiat. Res. Appl. Sci. 2015, 8, 26–35. [Google Scholar] [CrossRef]
- Podda, M.; Tritschler, H.J.; Ulrich, H.; Packer, L. α-lipoic acid supplementation prevents symptoms of vitamin E deficiency. Biochem. Biophys. Res. Commun. 1994, 204, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Tritscher, H.J.; Packer, L. α-lipoic acid increases intracellular glutathione in a human T-lymphocyte jurkat cell line. Biochem. Biophys. Res. Commun. 1995, 207, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent l-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [PubMed]
- Expert Group on Vitamins and Minerals (EVM). Safe Upper Levels for Vitamins and Minerals; Food Standard Agency: London, UK, 2003.
- European Food Safety Authority (EFSA). Opinion of the scientific panel on dietetic products, nutrition and allergies (NDA) on the upper tolerable intake of Vitamin C (l-ascorbic acid, its calcium, potassium and sodium salts and l-ascorbyl-6-palmitate). EFSA J. 2004, 59, 1–21. [Google Scholar]
- Pokorski, M.; Marczak, M.; Dymecka, A.; Suchocki, P. Ascorbyl palmitate as a carrier of ascorbate into neural tissues. J. Biomed. Sci. 2003, 10, 193198. [Google Scholar] [CrossRef]
- Cuzzorcrea, S.; Thiemermann, C.; Salvemini, D. Potential therapeutic effect of antioxidant therapy in shock and inflammation. Curr. Med. Chem. 2004, 11, 1147–1162. [Google Scholar] [CrossRef]
- Kasparova, S.; Brezova, V.; Valko, M.; Horecky, J.; Mlynarik, V.; Liptaj, T.; Vancova, O.; Ulicna, O.; Dobrota, D. Study of the oxidative stress in a rat model of chronic brain hypoperfusion. Neurochem. Int. 2005, 46, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. New aspects of glutathione biochemistry and transport-selective alteration of glutathione metabolism. Nutr. Rev. 1984, 42, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione-ascorbic acid antioxidant system in animals. J. Biol. Chem. 1994, 269, 9397–9400. [Google Scholar] [PubMed]
- Winkler, B.S.; Orselli, S.M.; Rex, T.S. The redox couple between glutathione and ascorbic acid: A chemical and physiological perspective. Free Radic. Biol. Med. 1994, 17, 333–349. [Google Scholar] [CrossRef]
- Henson, D.E.; Block, G.; Levine, M. Ascorbic acid: Biological functions and relation to cancer. J. Natl. Cancer Inst. 1991, 83, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals and antioxidants: A personal view. Nutr. Rev. 1994, 52, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Kagan, V.E.; Aust, S.D.; Reed, D.J.; Omaye, S.T. Impact of nutrients on cellular lipid peroxidation and antioxidant defense system. Toxicol. Sci. 1995, 26, 1–7. [Google Scholar] [CrossRef]
- Houston, D.K.N.; Johnson, M.A. Does vitamin C intake protects against lead toxicity? Nutr. Rev. 2000, 58, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Knekt, P.; Jarvinen, R.; Seppanen, R.; Rissanen, A.; Aromaa, A.; Heinonen, O.P.; Albanes, D.; Heinonen, M.; Pukkala, E.; Teppo, L. Dietary antioxidants and the risk of lung-cancer. Am. J. Epidemiol. 1991, 134, 471–479. [Google Scholar] [PubMed]
- Carr, A.; Frei, B. Does Vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar] [PubMed]
- Kojo, S. Vitamin C: Basic metabolism and its function as an index of oxidative stress. Curr. Med. Chem. 2000, 11, 1041–1064. [Google Scholar] [CrossRef]
- Ramanathan, K.; Anusuyadevi, M.; Shila, S.; Panneerselvam, C. Ascorbic acid and tocopherol as potent modulators of apoptosis on arsenic induced toxicity in rats. Toxicol. Lett. 2005, 156, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Flora, S.J.S.; Tandon, S.K. Prevention and therapeutic effects of thiamine, ascorbic acid and their combination in lead intoxication. Acta. Pharmacol. Toxicol. 1986, 58, 374–378. [Google Scholar] [CrossRef]
- Dhawan, M.; Kachru, D.N.; Tandon, S.K. Influence of thiamine and ascorbic acid supplementation on antidotal afficacy of thiol chelators in lead intoxication. Arch. Toxicol. 1988, 62, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, G. Carotenoid biosynthesis in microorganisms and plants. Eur. J. Biochem. 1994, 223, 7–24. [Google Scholar] [CrossRef] [PubMed]
- McNulty, H.P.; Jacob, R.F.; Mason, R.P. Biologic activity of carotenoids related to distinct membrane physicochemical interactions. Am. J. Cardiol. 2008, 101, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Braun, C.L.; Ernst, H. The chemistry of novel xanthophyll carotenoids. Am. J. Cardiol. 2008, 101, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Schweigert, F. Metabolism of Carotenoids in Mammals; Birkhauser Verlag: Basel, Switzerland, 1998. [Google Scholar]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applicationsfor human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications: A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Fassett, R.G.; Coombes, J.S. Astaxanthin: A potential therapeutic agent in cardiovascular disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Naquib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Olson, J.A. Carotenoids: Absorption, transport and metabolism in humans. Pure Appl. Chem. 2004, 66, 1011–1016. [Google Scholar] [CrossRef]
- Miki, W. Biological function and activities of animal carotenoids. Pure Appl. Chem. 1991, 63, 141–146. [Google Scholar] [CrossRef]
- Ranga Rao, A.; Raghunath Reddy, R.L.; Baskaran, V.; Sarada, R.; Ravishankar, G.A. Characterization of microalgal carotenoids by mass spectrometry and their bioavailability and antioxidant properties elucidated in rat models. J. Agric. Food Chem. 2010, 58, 8553–8559. [Google Scholar] [CrossRef] [PubMed]
- Sharoni, Y.; Danilenko, M.; Dubi, N.; Ben-Dor, A.; Levy, J. Carotenoids and transcription. Arch. Biochem. Biophys. 2004, 430, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, A.; Skibsted, L.H.; Truscott, T.G. The interaction of dietary carotenoids with radical species. Arch. Biochem. Biophys. 2001, 385, 13–19. [Google Scholar] [CrossRef] [PubMed]
- El-Agamey, A.; Lowe, G.M.; McGarvey, D.J.; Mortensen, A.; Phillip, D.M.; Truscott, T.G. Carotenoid radical chemistry and antioxidant/pro-oxidant properties. Arch. Biochem. Biophys. 2004, 430, 37–48. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jan, A.T.; Azam, M.; Siddiqui, K.; Ali, A.; Choi, I.; Haq, Q.M.R. Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants. Int. J. Mol. Sci. 2015, 16, 29592-29630. https://doi.org/10.3390/ijms161226183
Jan AT, Azam M, Siddiqui K, Ali A, Choi I, Haq QMR. Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants. International Journal of Molecular Sciences. 2015; 16(12):29592-29630. https://doi.org/10.3390/ijms161226183
Chicago/Turabian StyleJan, Arif Tasleem, Mudsser Azam, Kehkashan Siddiqui, Arif Ali, Inho Choi, and Qazi Mohd. Rizwanul Haq. 2015. "Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants" International Journal of Molecular Sciences 16, no. 12: 29592-29630. https://doi.org/10.3390/ijms161226183
APA StyleJan, A. T., Azam, M., Siddiqui, K., Ali, A., Choi, I., & Haq, Q. M. R. (2015). Heavy Metals and Human Health: Mechanistic Insight into Toxicity and Counter Defense System of Antioxidants. International Journal of Molecular Sciences, 16(12), 29592-29630. https://doi.org/10.3390/ijms161226183