
De Novo Transcriptome Sequencing of Oryza officinalis Wall ex Watt to Identify Disease-Resistance Genes

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
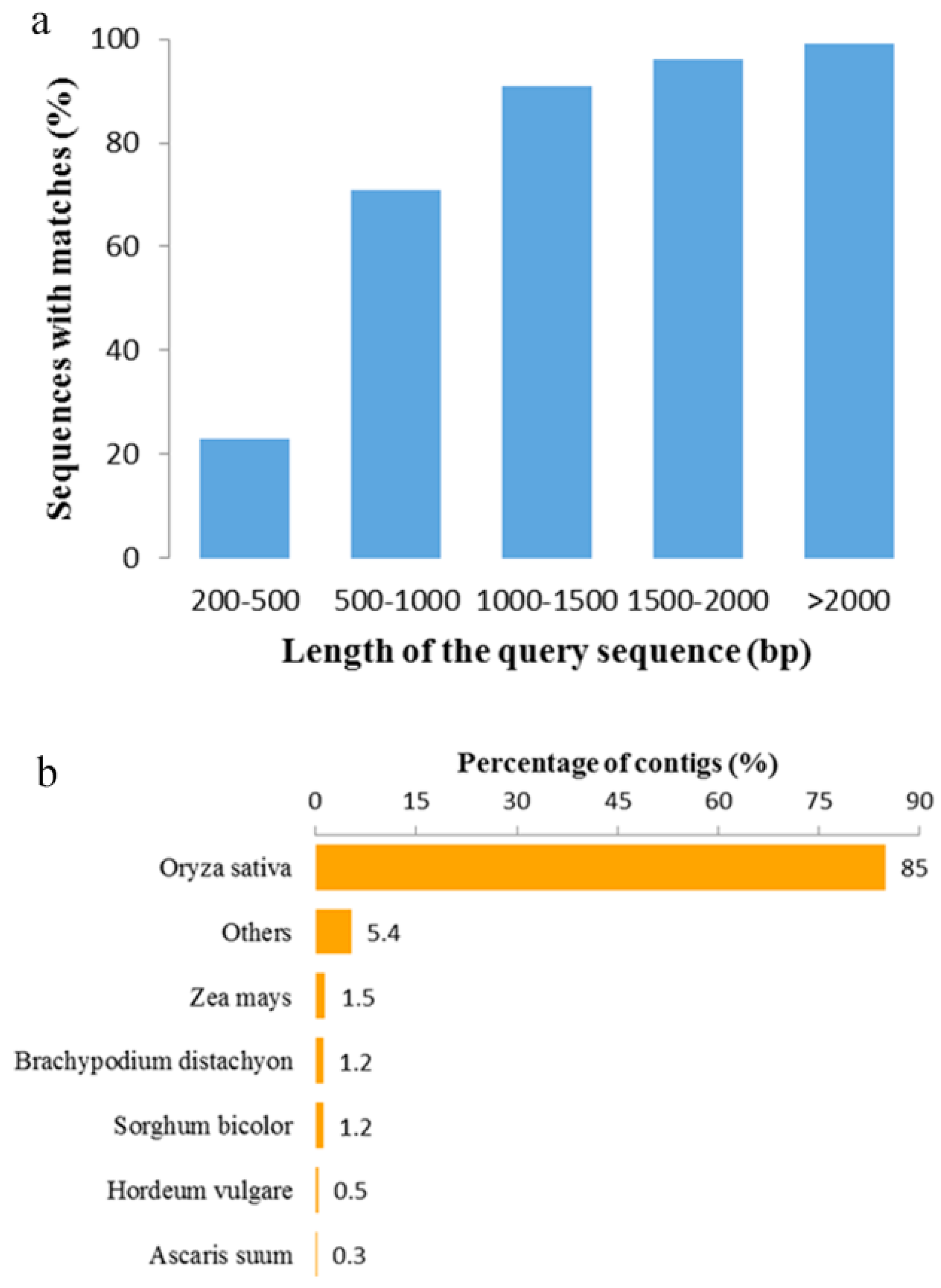
2.1. De Novo Assembly and Quality Assessment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | Number of Sequences |
|---|---|
| Assembly | |
| Total number of contigs | 137,299 |
| Mean length (bp) | 1459 |
| Maximum contig length (bp) | 19,214 |
| N50 (bp) | 2331 |
| Number of contigs (≥1 kb) | 72,521 |
| Annotation | |
| Number of predicted ORFs | 82,983 |
| Number of predicted ORFs (≥900 bp) | 53,239 |
| Transcript BLASTx against NR | 88,249 |
| Transcript BLASTx against Pfam | 79,310 |
| Transcripts annotated with GO terms | 75,589 |
| Transcript annotations against KEGG | 21,191 |
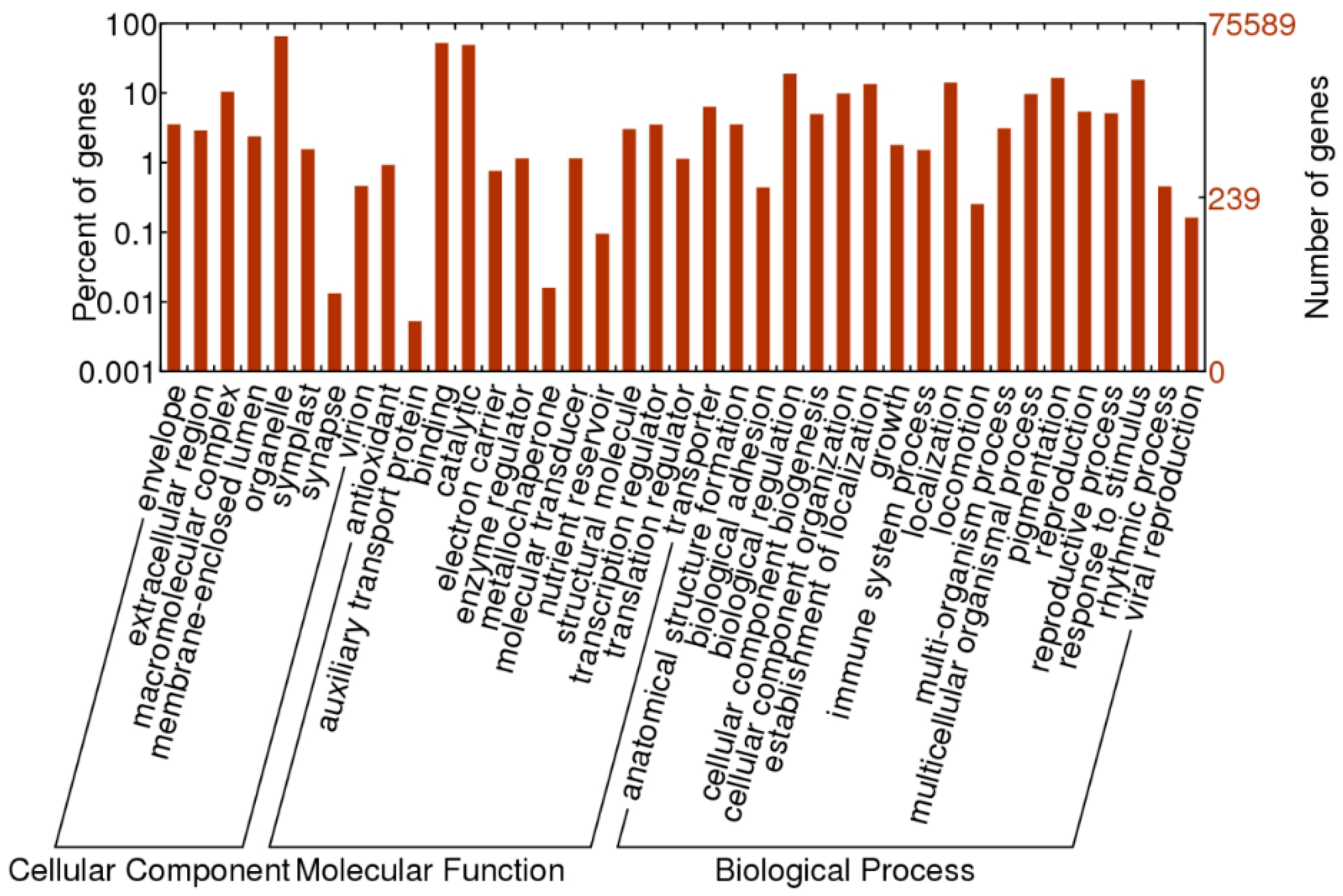
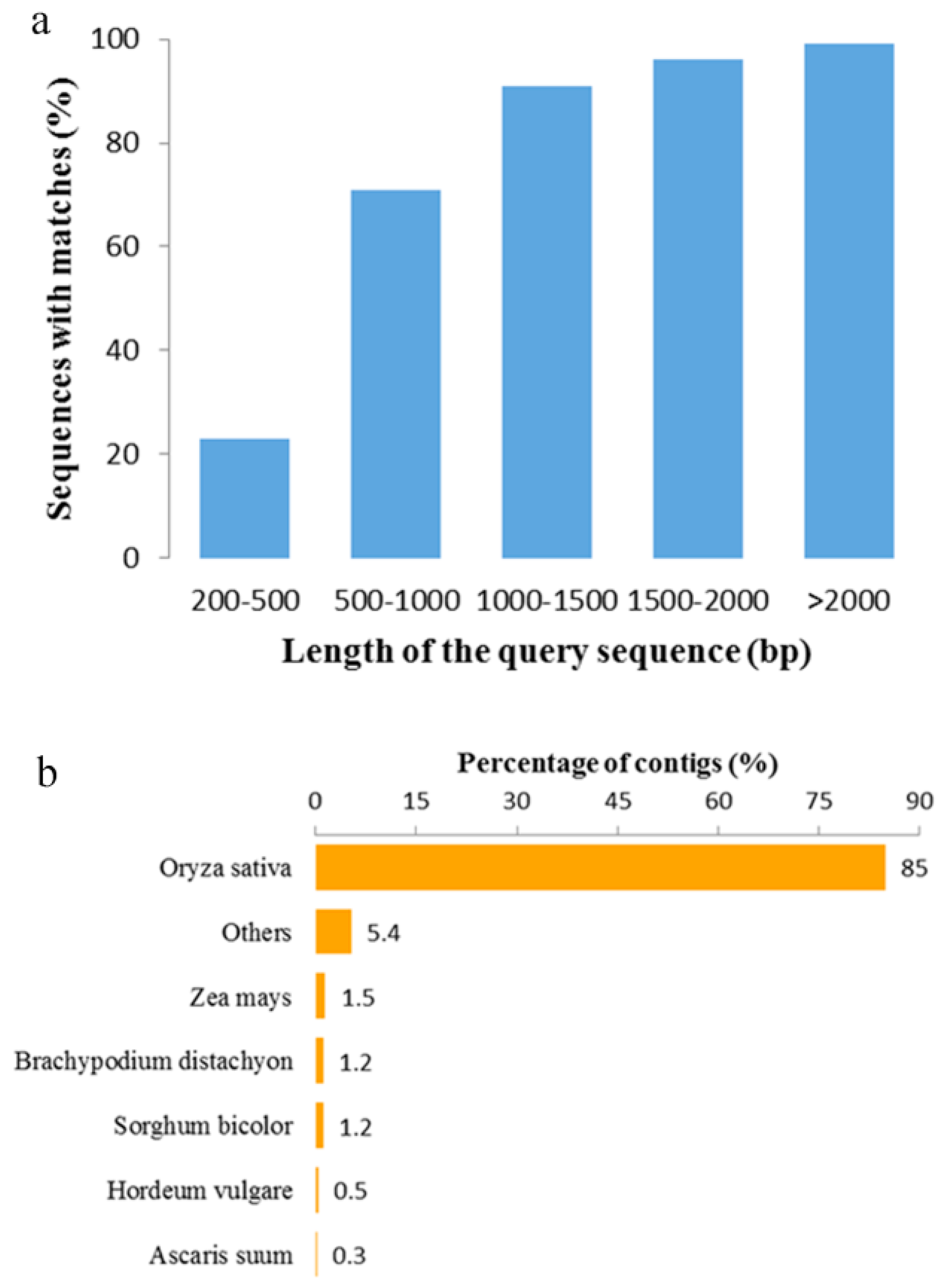
2.2. Functional Annotation of O. officinalis Transcriptome

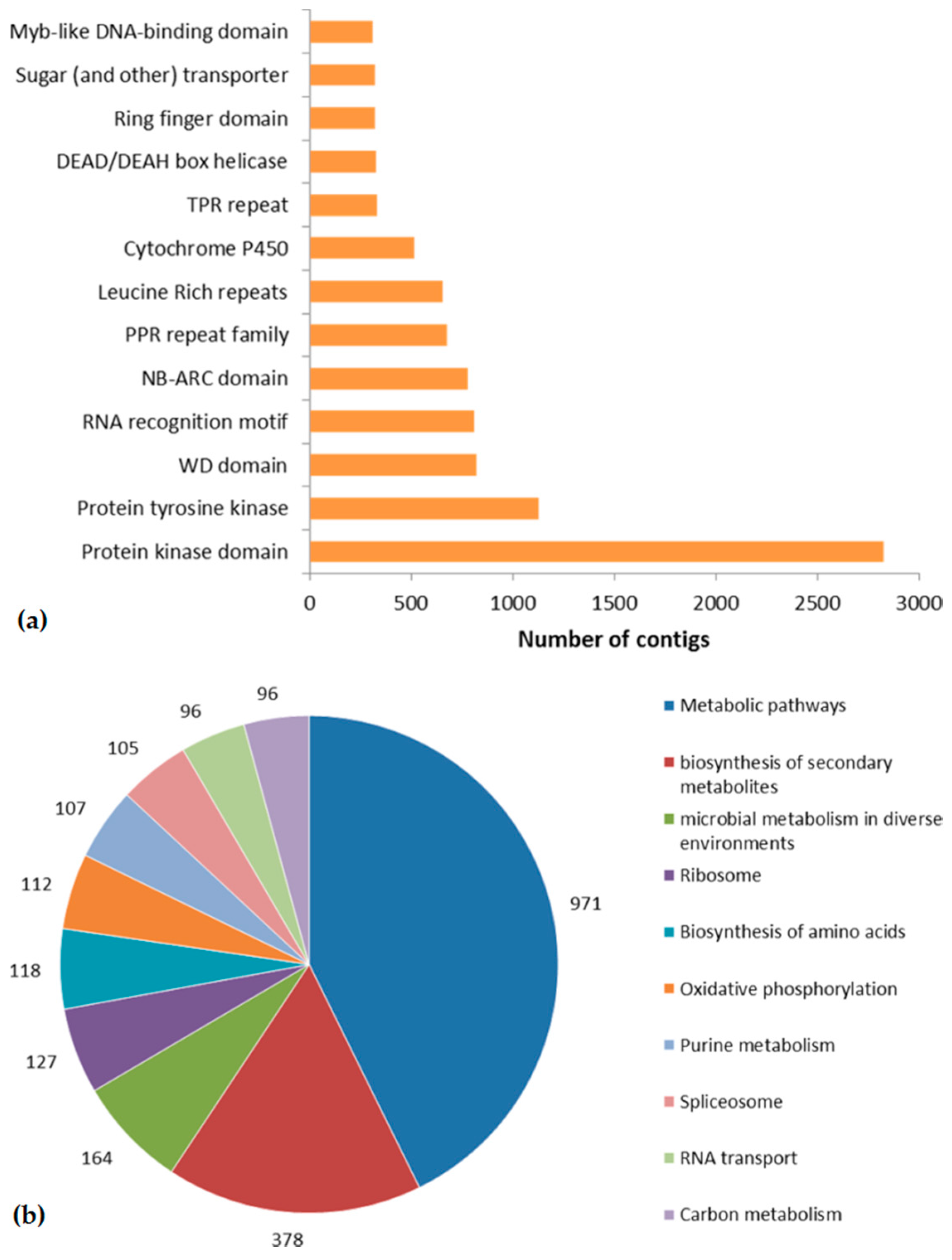
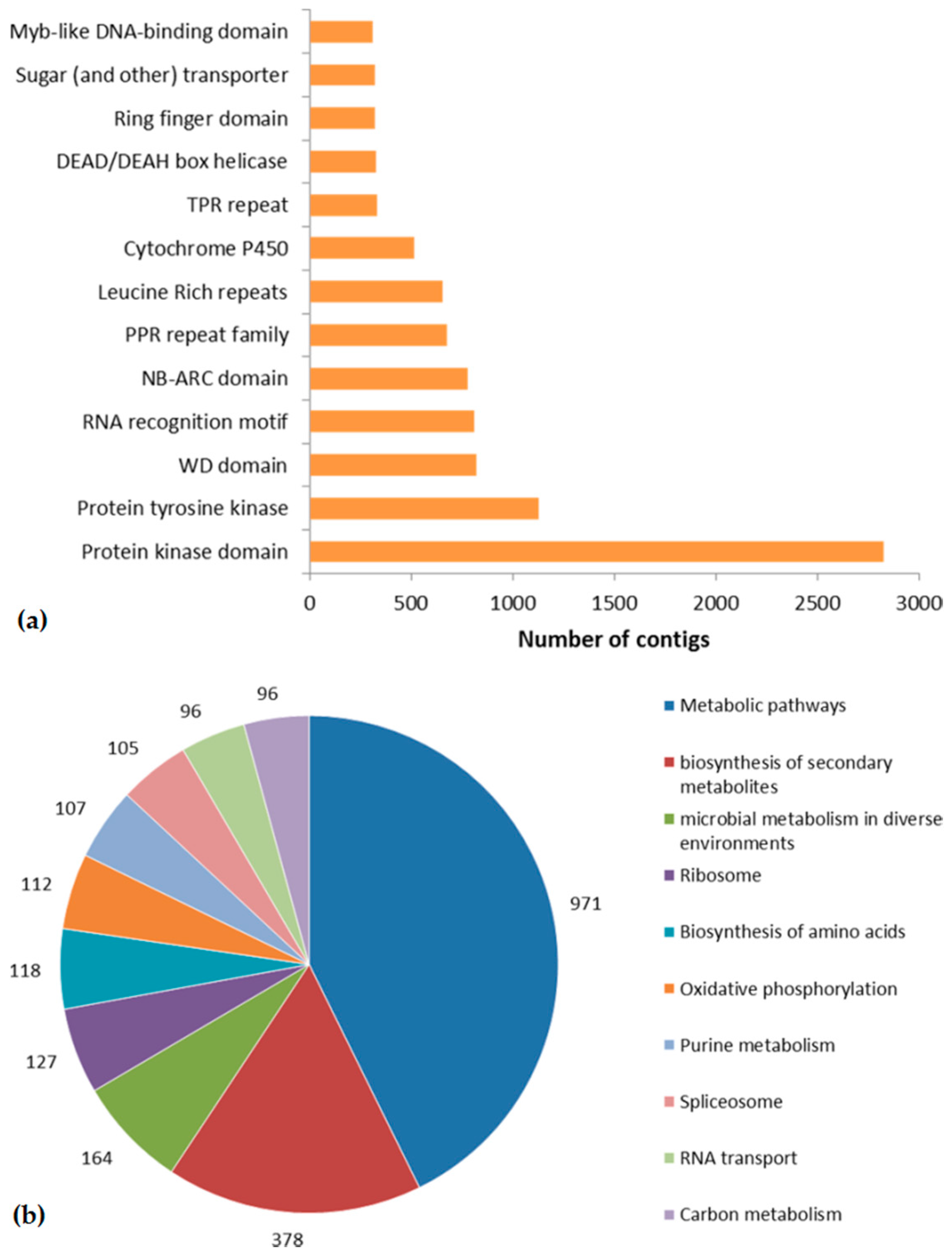
2.3. Domain Prediction and KEGG Pathway Mapping
2.4. Genes Related to Disease Resistance
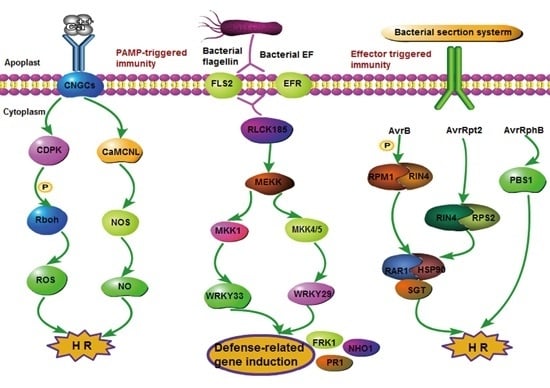
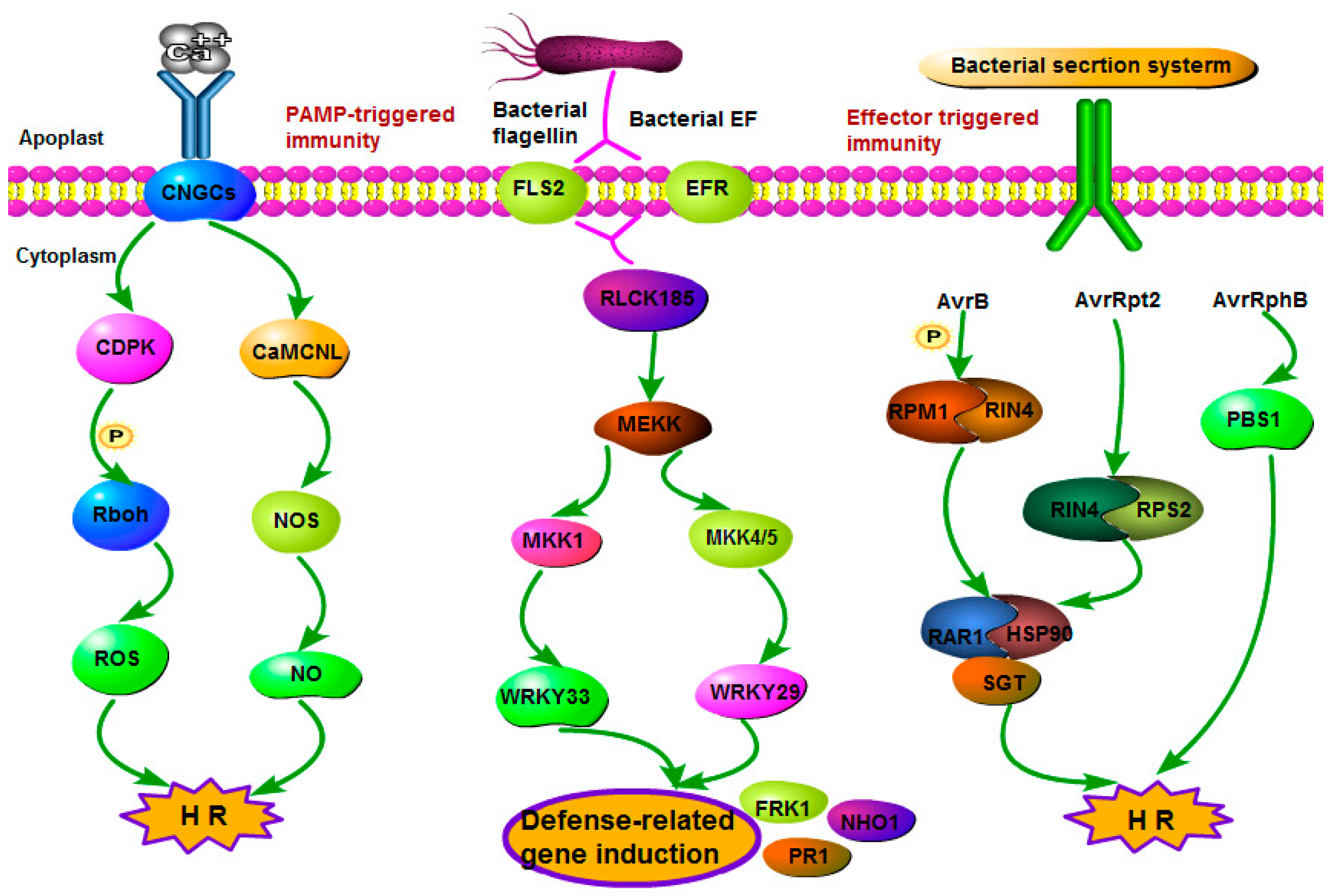
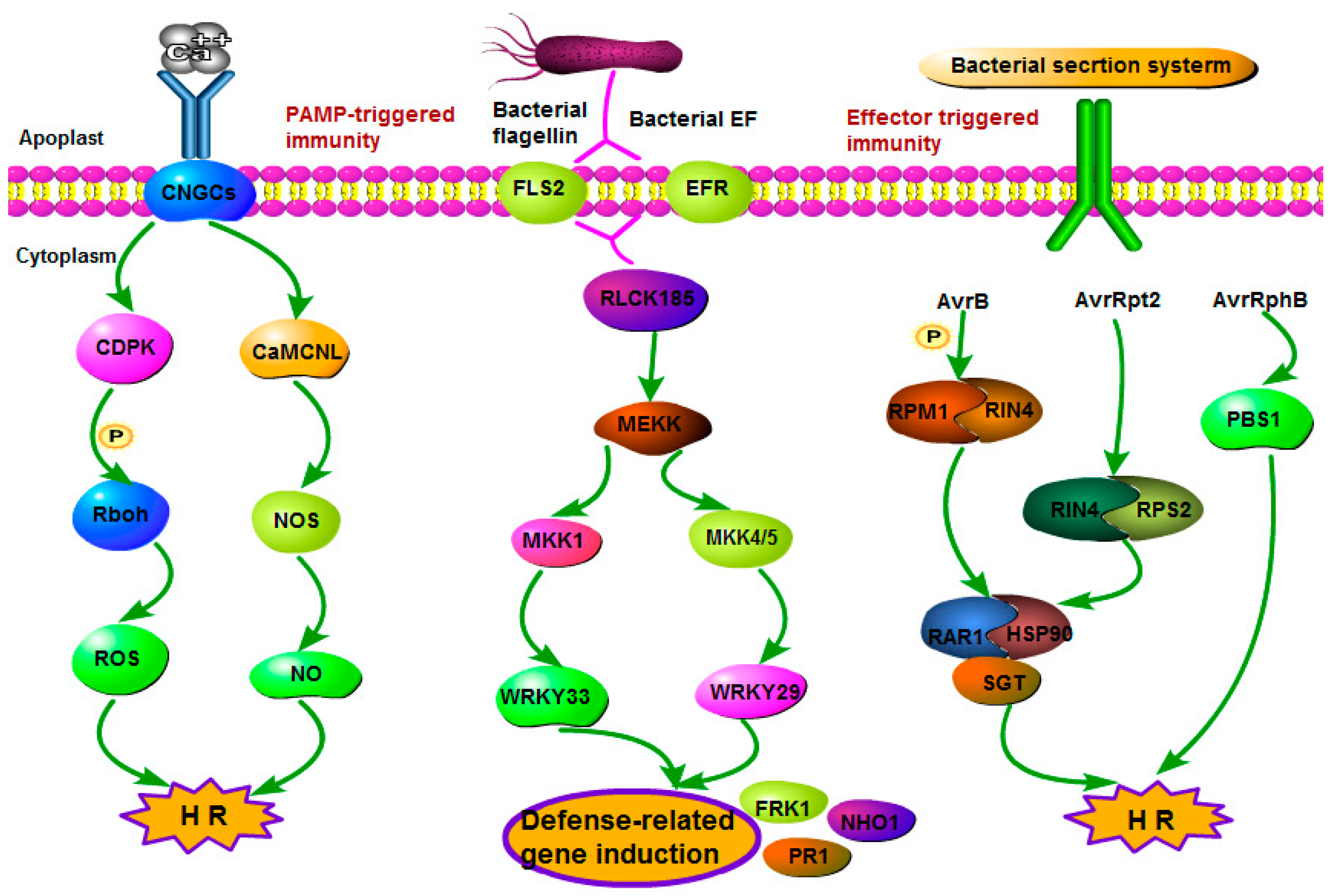
2.4.1. Signaling Events in the Plant–Pathogen/Insect Interaction
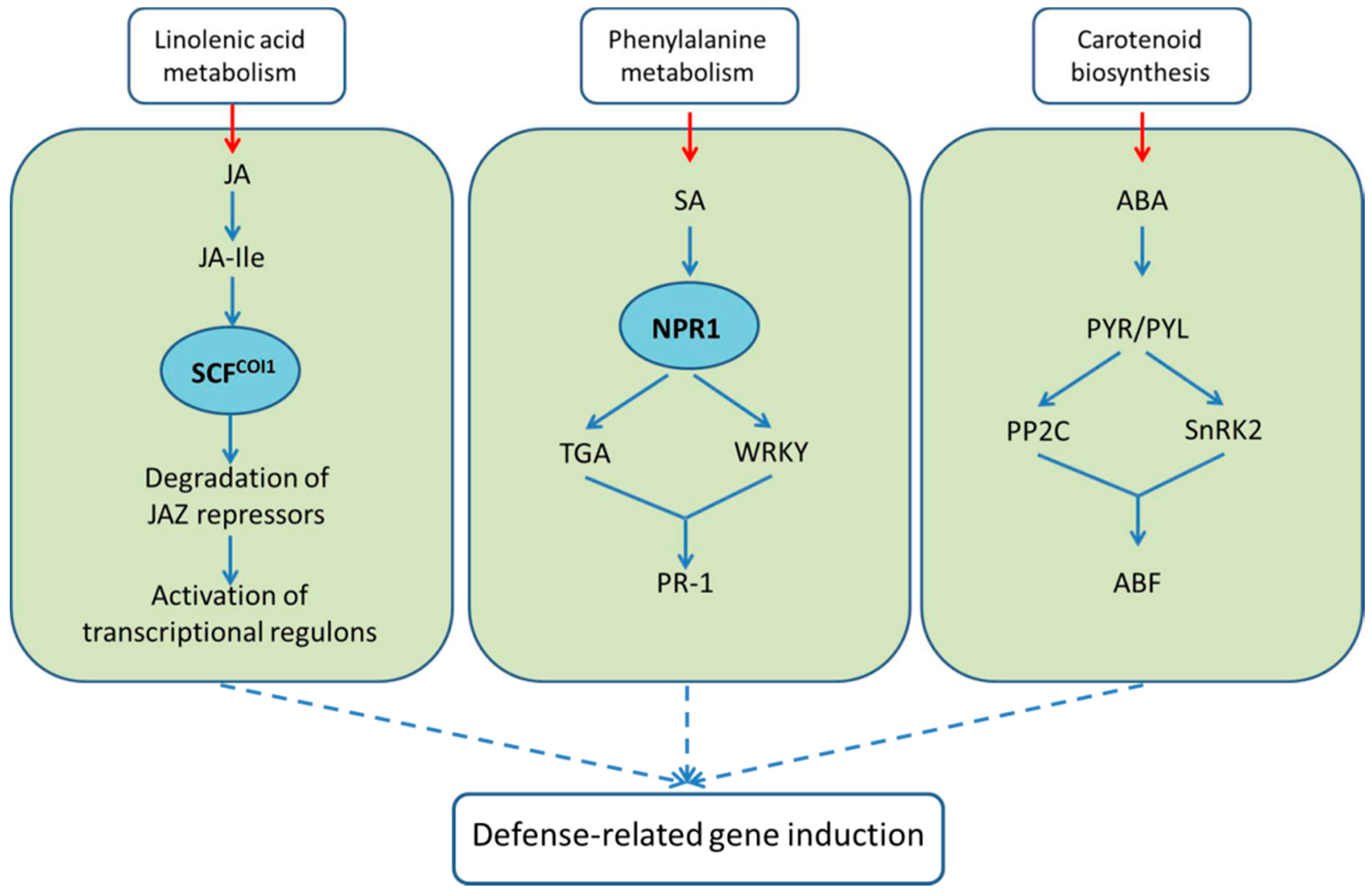
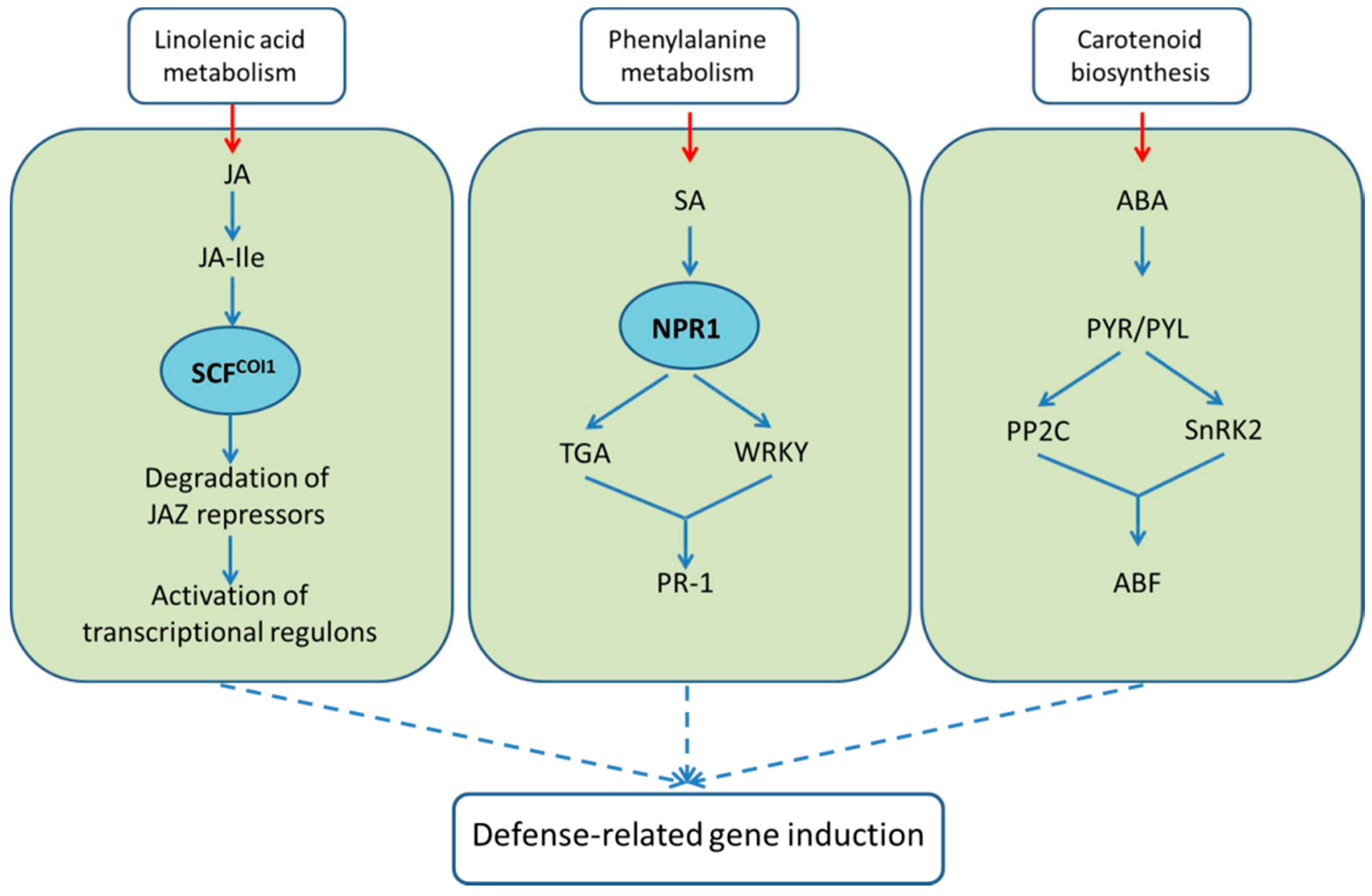
2.4.2. Regulation of Defense Responses by Plant Hormones
2.4.3. Genes Related to Bacterial Blight Resistance
3. Materials and Methods
3.1. Plant Materials and RNA Extraction
3.2. cDNA Library Construction and Sequencing
3.3. Data-Preprocessing, De Novo Assembly and Assembly Assessment
3.4. Gene Annotation and Differential Gene Expression Analysis
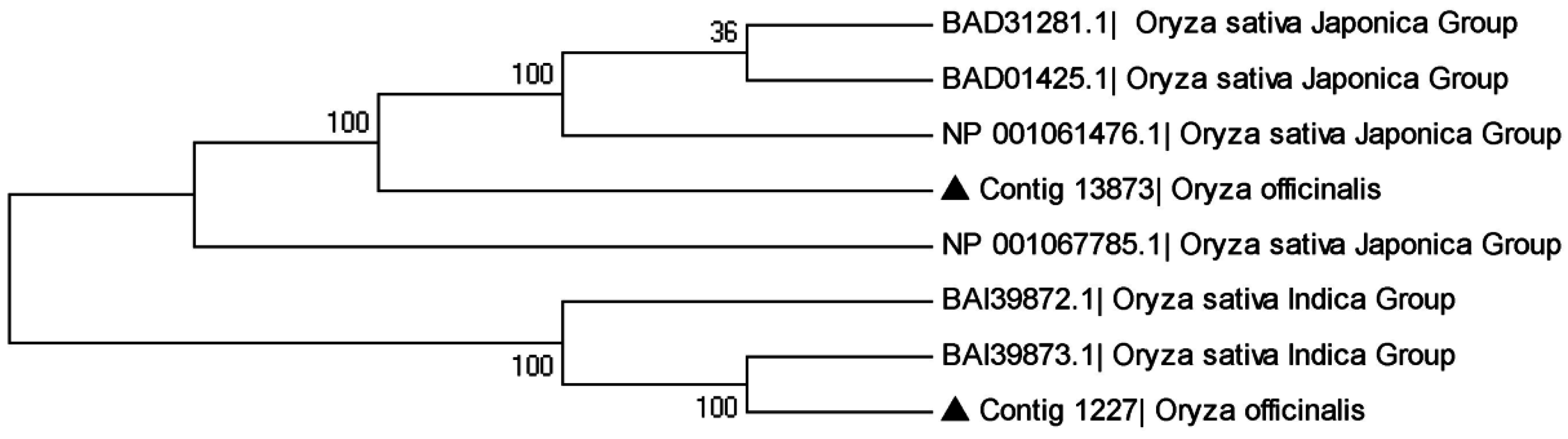
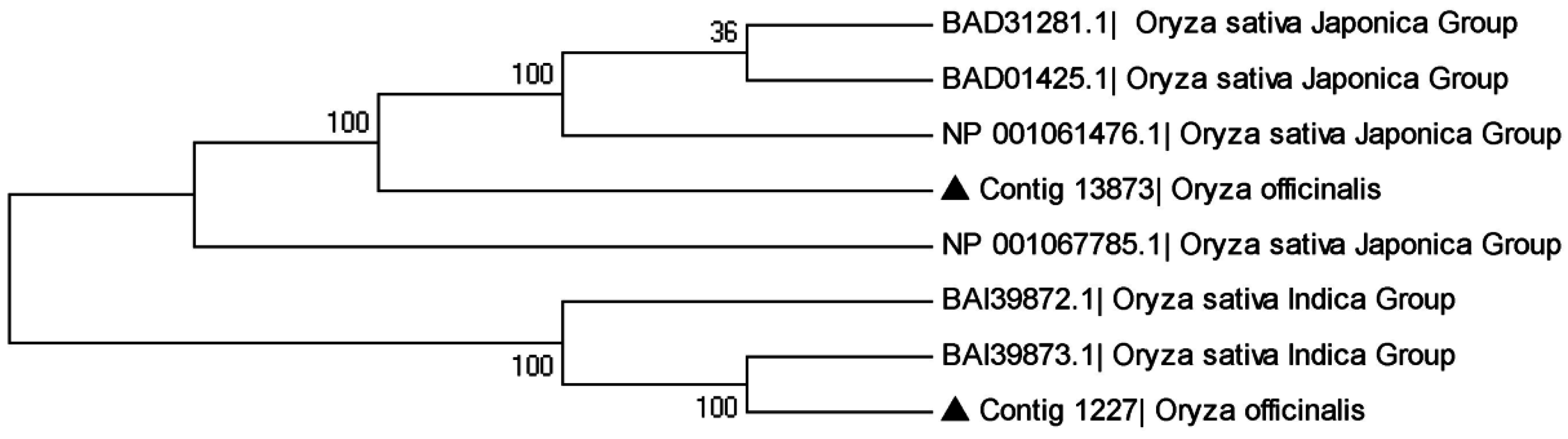
3.5. The Phylogenetic Relationship of Sequence
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Availability of Supporting Data
References
- Fu, X.L.; Lu, Y.G.; Liu, X.D.; Li, J.Q.; Zhao, X.J. Cytological behavior of hybridization barriers between Oryza sativa and Oryza officinalis. Agric. Sci. China 2011, 10, 1489–1500. [Google Scholar]
- Zhang, W.; Dong, Y.; Yang, L.; Ma, B.; Ma, R.; Huang, F.; Wang, C.; Hu, H.; Li, C.; Yan, C.; et al. Small brown planthopper resistance loci in wild rice (Oryza officinalis). Mol. Genet. Genom. 2014, 289, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Devanna, N.B.; Vijayan, J.; Sharma, T.R. The blast resistance gene Pi54of cloned from Oryza officinalis interacts with Avr-Pi54 through its novel non-LRR domains. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, D.R.; Wang, L.; Liu, G.J.; Zhuang, J.Y. Identification of quantitative trait loci for resistance to whitebacked planthopper, Sogatella furcifera, from an interspecific cross Oryza sativa × O. rufipogon. Breed. Sci. 2010, 60, 153–159. [Google Scholar] [CrossRef]
- Tan, G.X.; Ren, X.; Weng, Q.-M.; Shi, Z.-Y.; Zhu, L.-L.; He, G.-C. Mapping of a new resistance gene to bacterial blight in rice line introgressed from Oryza officinalis. Acta Genet. Sin. 2004, 31, 724–729. [Google Scholar] [PubMed]
- Jacquemin, J.; Bhatia, D.; Singh, K.; Wing, R.A. The international Oryza map alignent project: Development of a genus-wide comparative genomics platform to help solve the 9 billion-people question. Curr. Opin. Plant Biol. 2013, 16, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Xu, S.; Jing, X.; Meng, L.; Qin, Z. De novo assembly and characterization of Oryza officinalis leaf transcriptome by using RNA-seq. BioMed Res. Int. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jeung, J.; Kim, B.; Cho, Y.; Han, S.; Moon, H.; Lee, Y.; Jena, K. A novel gene, Pi40 (t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice. Theor. Appl. Genet. 2007, 115, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.L.; Wing, R.A.; Brar, D.S. The wild relative of rice: genomes and genomics. In Genetics and Genomics of Rice; Springer: New York, NY, USA, 2013; pp. 9–25. [Google Scholar]
- Huang, Z.; He, G.; Shu, L.; Li, X.; Zhang, Q. Identification and mapping of two brown planthopper resistance genes in rice. Theor. Appl. Genet. 2001, 102, 929–934. [Google Scholar] [CrossRef]
- Boller, T.; He, S.Y. Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 2009, 324, 742. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, W.; Han, H.; Song, L.; Bai, L.; Gao, Z.; Zhang, Y.; Yang, X.; Li, X.; Gao, A.; et al. De novo transcriptome sequencing of Agropyron cristatum to identify available gene resources for the enhancement of wheat. Genomics 2015, 106, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A.; Ricke, D.; Lan, T.-H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Project IRGS. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar]
- Cho, S.; Ok, S.; Jeung, J.; Shim, K.; Jung, K.; You, M.; Kang, K.; Chung, Y.; Choi, H.; Moon, H.; et al. Comparative analysis of 5,211 leaf ESTs of wild rice (Oryza minuta). Plant Cell Rep. 2004, 22, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yu, S.; Fan, D.; Mu, J.; Shangguan, Y.; Wang, Z.; Minobe, Y.; Lin, Z.; Han, B. Collection and comparative analysis of 1888 full-length cDNAs from wild rice Oryza rufipogon Griff. W1943. DNA Res. 2008, 15, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hu, L.; Hurek, T.; Reinhold-Hurek, B. Global characterization of the root transcriptome of a wild species of rice, Oryza longistaminata, by deep sequencing. BMC Genom. 2010, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhao, S.; Chen, Y.; Cao, Q.; Wei, C.; Cheng, X.; Zhang, Y. Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms. BMC Genom. 2015, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-B.; Xia, E.-H.; Huang, H.; Jiang, J.-J.; Liu, B.-Y.; Gao, L.-Z. De novo transcriptome assembly of the wild relative of tea tree (Camellia taliensis) and comparative analysis with tea transcriptome identified putative genes associated with tea quality and stress response. BMC Genom. 2015, 16, 298. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Hwang, B.K. Molecular and cellular control of cell death and defense signaling in pepper. Planta 2015, 241, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, C.; Robatzek, S.; Navarro, L.; Oakeley, E.J.; Jones, J.D.; Felix, G.; Boller, T. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 2004, 428, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Yamaguchi, K.; Sakamoto, K.; Yoshimura, S.; Inoue, K.; Tsuge, S.; Kojima, C.; Kawasaki, T. Bacterial effector modulation of host E3 ligase activity suppresses PAMP-triggered immunity in rice. Nat. Commun. 2014, 5, 5430. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yamada, K.; Ishikawa, K.; Yoshimura, S.; Hayashi, N.; Uchihashi, K.; Ishihama, N.; Kishi-Kaboshi, M.; Takahashi, A.; Tsuge, S.; et al. A receptor-like cytoplasmic kinase targeted by a plant pathogen effector is directly phosphorylated by the chitin receptor and mediates rice immunity. Cell Host Microbe 2013, 13, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.-F.; Ao, Y.; Qu, J.; Li, Z.; Su, J.; Zhang, Y.; Liu, J.; Feng, D.; Qi, K.; et al. Lysin motif–containing proteins LYP4 and LYP6 play dual roles in peptidoglycan and chitin perception in rice innate immunity. Plant Cell 2012, 24, 3406–3419. [Google Scholar] [CrossRef] [PubMed]
- Katsir, L.; Chung, H.S.; Koo, A.J.; Howe, G.A. Jasmonate signaling: a conserved mechanism of hormone sensing. Curr. Opin. Plant Biol. 2008, 11, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lugtenberg, B.J.; Chin-A-Woeng, T.F.; Bloemberg, G.V. Microbe–plant interactions: Principles and mechanisms. Antoni. Leeuwe. 2002, 81, 373–383. [Google Scholar] [CrossRef]
- Katou, S.; Asakura, N.; Kojima, T.; Mitsuhara, I.; Seo, S. Transcriptome analysis of WIPK/SIPK-suppressed plants reveals induction by wounding of disease resistance-related genes prior to the accumulation of salicylic acid. Plant Cell Physiol. 2013, 54, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Loake, G.; Grant, M. Salicylic acid in plant defence—The players and protagonists. Curr. Opin. Plant Biol. 2007, 10, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, F.; Wang, Z.; Huang, Z.; Xiong, A.; Hou, X. Characterization and co-expression analysis of WRKY orthologs involved in responses to multiple abiotic stresses in Pak-choi (Brassica campestris ssp. chinensis). BMC Plant Biol. 2013, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Liu, H.; Qiu, D.; Zhou, Y.; Li, X.; Xu, C.; Wang, S. A pair of allelic WRKY genes play opposite roles in rice-bacteria interactions. Plant Physiol. 2009, 151, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Mauch-Mani, B.; Mauch, F. The role of abscisic acid in plant–pathogen interactions. Curr. Opin. Plant Biol. 2005, 8, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, T.; Sugiyama, N.; Mizoguchi, M.; Hayashi, S.; Myouga, F.; Yamaguchi-Shinozaki, K.; Ishihama, Y.; Hirayama, T.; Shinozaki, K. Type 2C protein phosphatases directly regulate abscisic acid-activated protein kinases in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 17588–17593. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, R.M.; Chatterjee, S.; Oliva, R.; Laha, G.S.; Cruz, C.V.; Leach, J.E.; Sonti, R.V. Update on Bacterial Blight of Rice: Fourth. Rice 2014, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Gu, Y.-H.; Wang, H.-Y.; Zheng, W.; Li, X.; Zhao, C.W.; Zhang, Y.Z. Digital gene expression analysis based on integrated de novo transcriptome assembly of sweet potato [Ipomoea batatas (L.) Lam.]. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.P.; van Dongen, S.; Abreu-Goodger, C.; Bartonicek, N.; Enright, A.J. Kraken: A set of tools for quality control and analysis of high-throughput sequence data. Methods 2013, 63, 41–49. [Google Scholar] [CrossRef] [PubMed]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 November 2015).
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Method 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ruhlman, T.A.; Mower, J.P.; Jansen, R.K. Comparative analyses of two Geraniaceae transcriptomes using next-generation sequencing. BMC Plant Biol. 2013, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- WEGO. Available online: http://wego.genomics.org.cn/cgi-bin/wego/index.pl (accessed on 1 November 2015).
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- KAAS. Available online: http://www.genome.jp/kegg/kaas/ (accessed on 1 November 2015).
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.; et al. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.-M. The significance of digital gene expression profiles. Genom. Res. 1997, 7, 986–995. [Google Scholar]
- NCBI. Available online: http://www.ncbi.nlm.nih.gov (accessed on 1 November 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, B.; Gu, Y.; Tao, X.; Cheng, X.; Wei, C.; Fu, J.; Cheng, Z.; Zhang, Y. De Novo Transcriptome Sequencing of Oryza officinalis Wall ex Watt to Identify Disease-Resistance Genes. Int. J. Mol. Sci. 2015, 16, 29482-29495. https://doi.org/10.3390/ijms161226178
He B, Gu Y, Tao X, Cheng X, Wei C, Fu J, Cheng Z, Zhang Y. De Novo Transcriptome Sequencing of Oryza officinalis Wall ex Watt to Identify Disease-Resistance Genes. International Journal of Molecular Sciences. 2015; 16(12):29482-29495. https://doi.org/10.3390/ijms161226178
Chicago/Turabian StyleHe, Bin, Yinghong Gu, Xiang Tao, Xiaojie Cheng, Changhe Wei, Jian Fu, Zaiquan Cheng, and Yizheng Zhang. 2015. "De Novo Transcriptome Sequencing of Oryza officinalis Wall ex Watt to Identify Disease-Resistance Genes" International Journal of Molecular Sciences 16, no. 12: 29482-29495. https://doi.org/10.3390/ijms161226178
APA StyleHe, B., Gu, Y., Tao, X., Cheng, X., Wei, C., Fu, J., Cheng, Z., & Zhang, Y. (2015). De Novo Transcriptome Sequencing of Oryza officinalis Wall ex Watt to Identify Disease-Resistance Genes. International Journal of Molecular Sciences, 16(12), 29482-29495. https://doi.org/10.3390/ijms161226178