
Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges

 ,
,
Abstract
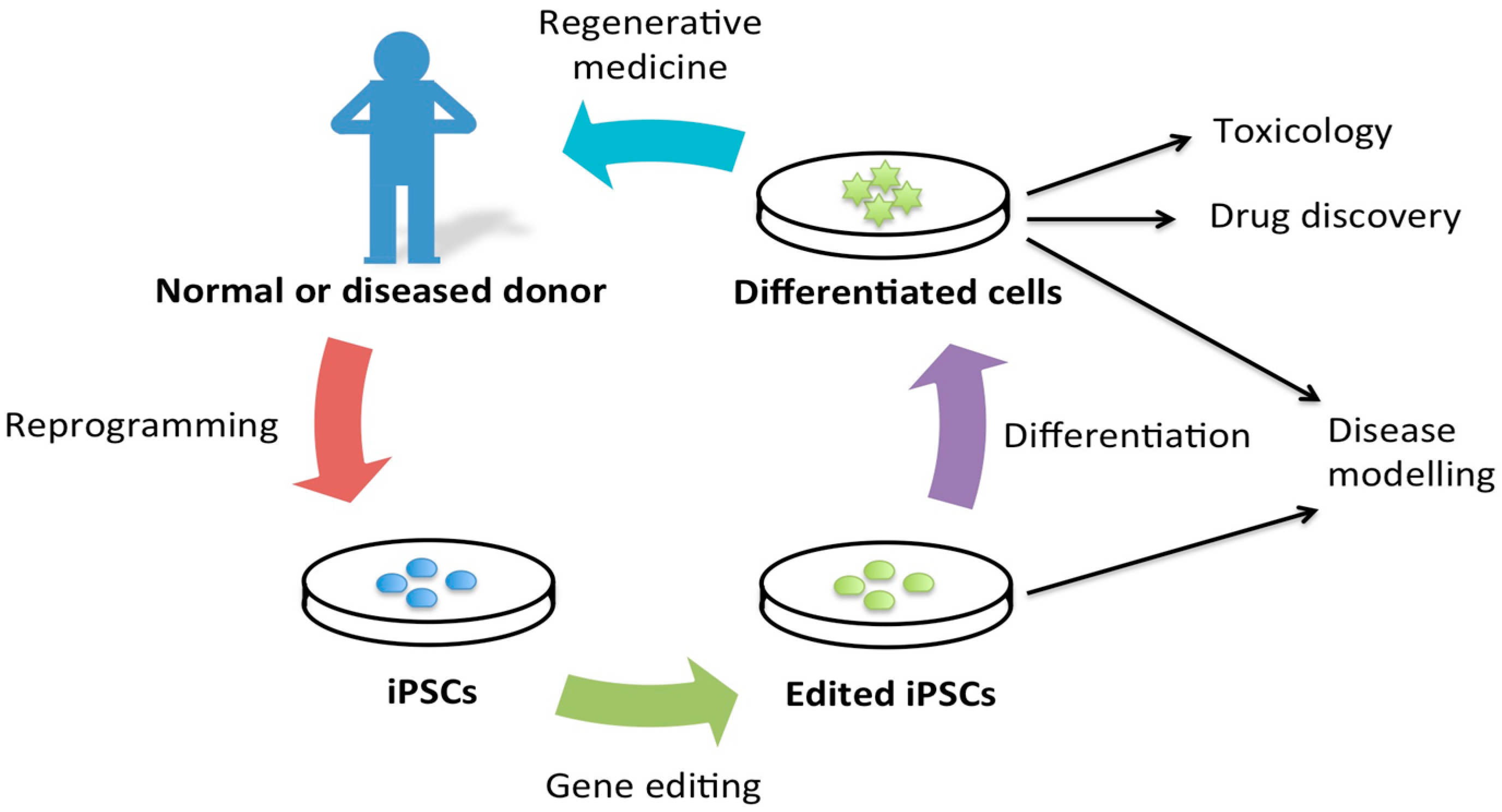
:1. Introduction
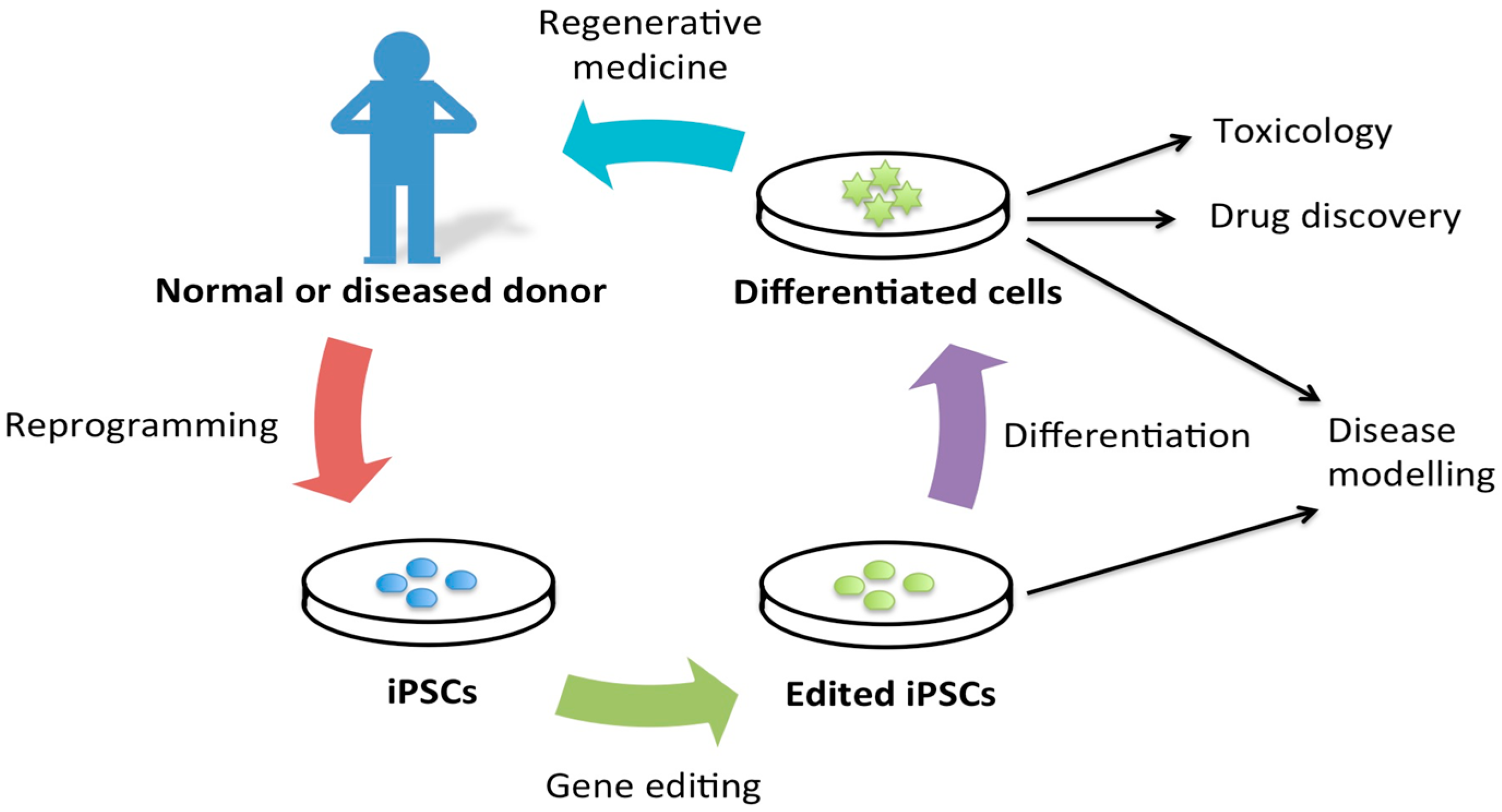
2. Disease Modeling
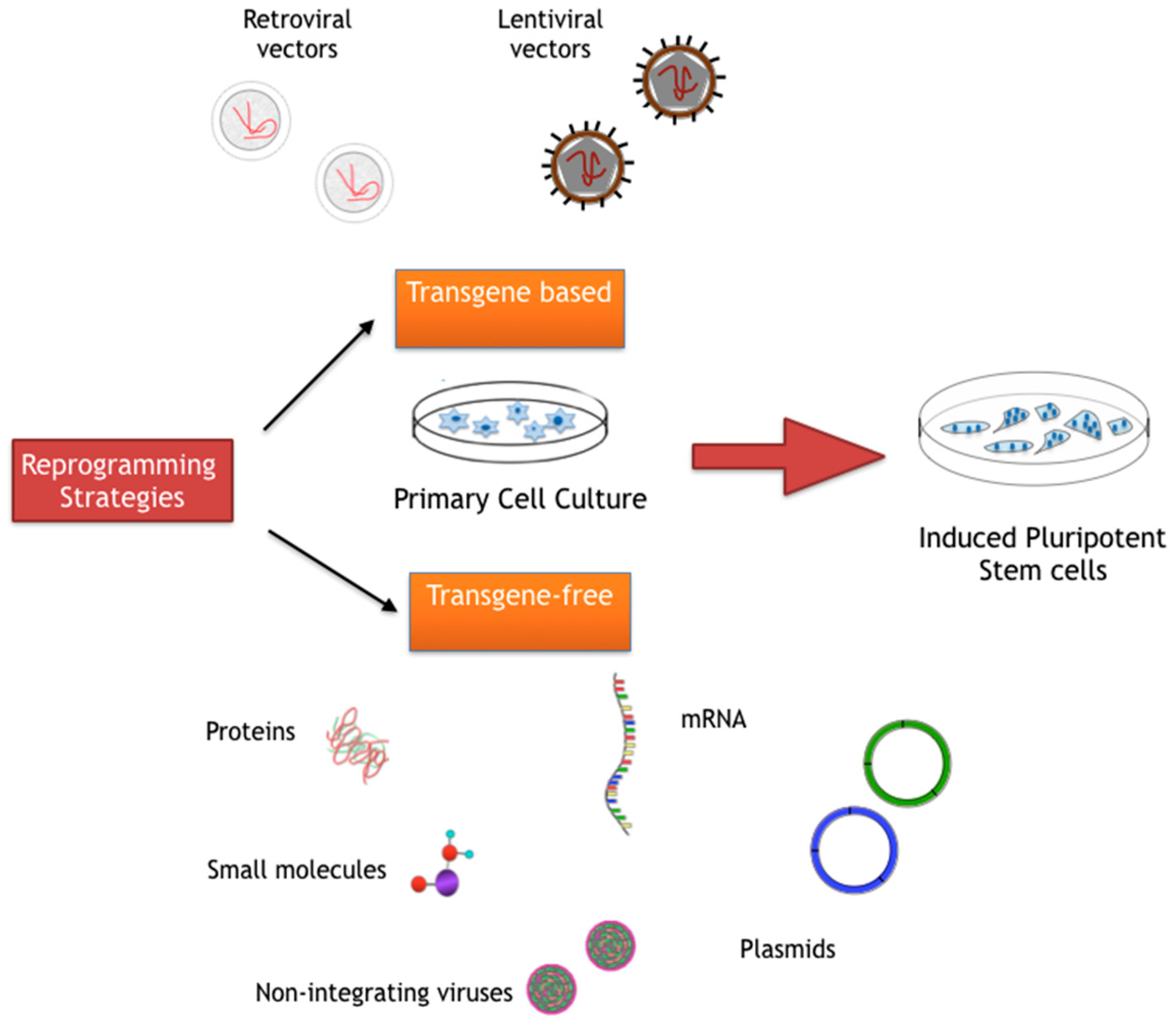
3. Cellular Reprogramming
3.1. Transgene-Based Cellular Reprogramming Methods
3.2. Transgene-Free Cellular Reprogramming Methods


{kind=link}
{kind=link}
{kind=link}
| Method Type | Method | Factors/Other Agents | Cell Type | References | Efficiency |
|---|---|---|---|---|---|
| Transgene-based | Retroviral | OSKM | Mouse fibroblasts | [6] | 0.02% |
| Retroviral + small molecules | OSKM + SB431542 + PD0325901 | Human fibroblasts | [23] | ~1% | |
| Lentiviral | OSNL | Mouse fibroblasts, human fibroblasts | [18] | 0.0095% | |
| Inducible lentiviral | OSKMN | Human fibroblasts, keratinocytes | [19] | 1%–3% | |
| Integrating, non-viral inducible plasmid vectors | OSKM | Rat fibroblasts | [20] | 0.0027%–0.0078% | |
| Transgene-free | Small molecules | C6FZ or VC6TFZ | Mouse fibroblasts | [29] | 0.2% |
| Episomal plasmids | OSKM | Mouse fibroblasts | [17] | ~0.1% | |
| Sendai viruses | OSKM | Human fibroblasts | [32] | ~1% | |
| Non-integrating DNA adenoviral | OSKM | Tail tip fibroblasts, hepatocytes, fatal liver cells | [34] | 0.0001%–0.001% | |
| Excisable lentiviral | OSK | Mouse fibroblasts, human fibroblasts | [36] | Not reported | |
| PiggyBAC transposon | OSKM or OSKML | Mouse fibroblasts | [38] | ~1% | |
| Synthetic mRNA | OSKM | Human fibroblasts | [39] | 2% | |
| Polyarginine-tagged polypeptides | OSKM | Human fibroblasts | [42] | 0.001% | |
| Magnet-based nanofection of polypeptides | OSKM | Mouse fibroblasts | [43] | 0.001%–0.003% | |
| MicroRNAs (lentiviral) | miR302/367 | Mouse fibroblasts, human fibroblasts | [44] | Up to 10% |
3.3. Cell Types from Which Induced Pluripotent Stem Cells (iPSCs) Have Been Derived
4. Gene Targeting and Disease Modeling
| System | Endogenous System | Advantages | Disadvantages |
|---|---|---|---|
| Homing endonucleases | Restriction enzymes with large restriction sites, encoded by mobile genetic elements [63] |
| |
| Zinc finger nucleases (ZFNs) | Zinc finger domain found in many eukaryotic transcription factors [66,67] |
| |
| Transcriptional activator-like effector nucleases (TALENs) | TAL-effector proteins from Xanthomonas, a plant pathogen [69] |
|
|
| CRISPR/Cas9 | Prokaryotic adaptive immune system [72] |
|
5. CRISPR/Cas9 System and Its Potential in Disease Modeling
6. Gene Editing in Disease Modeling
7. Advantages of iPSCs for Disease Modeling
8. Requirements of iPSCs in Disease Modeling
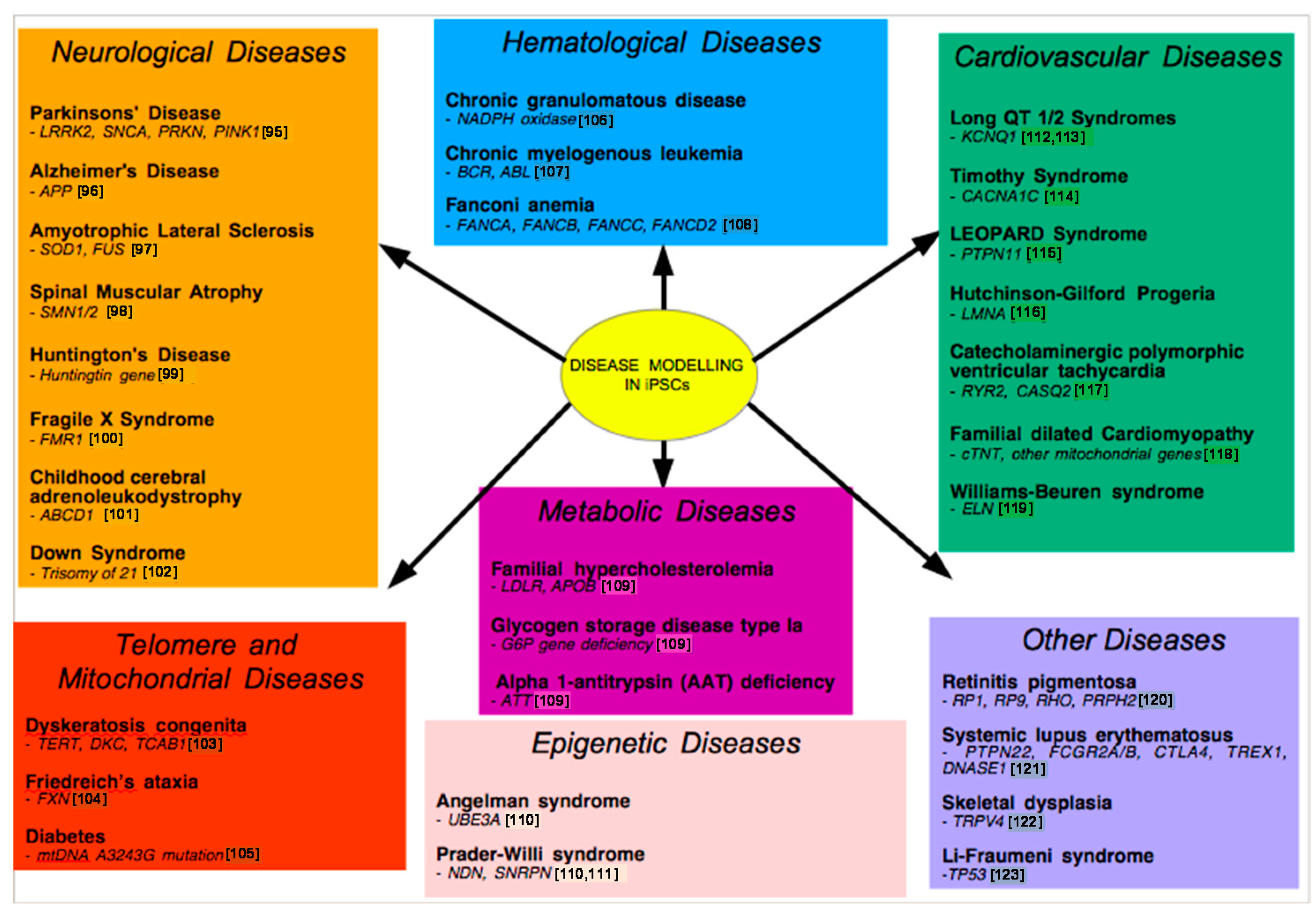
9. Examples of Disease Models
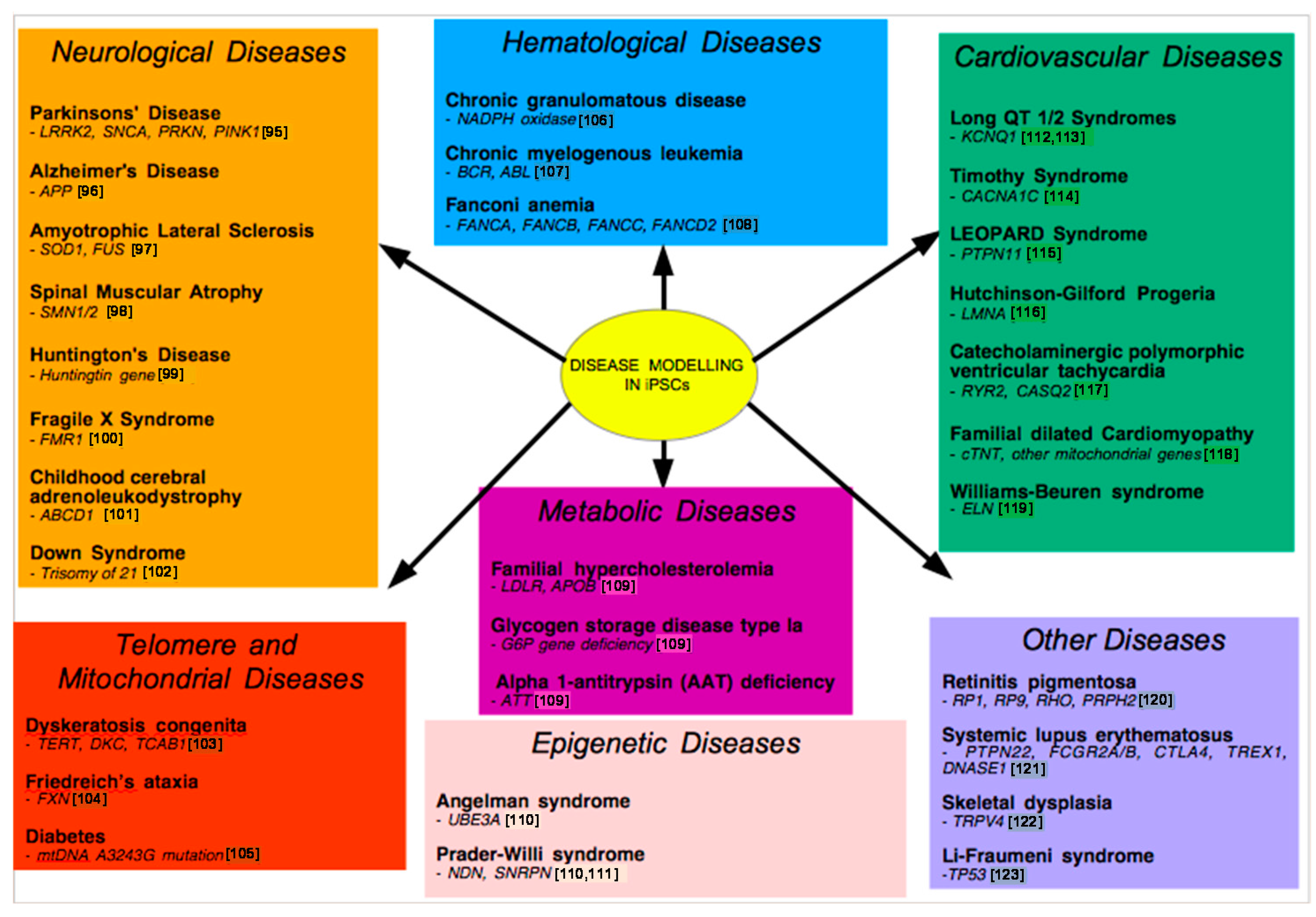
9.1. Neurological Disease Models
9.2. Metabolic Disease Modeling
9.3. Modeling Drug Metabolism
9.4. Cardiovascular Disease Models
9.5. Telomere Disease Models
9.6. Other Disease Models
9.7. Modeling Epigenetic Diseases
9.8. Modeling Infectious Diseases
9.9. Modeling Cancer
10. Limitations of iPSCs in Disease Modeling
11. Diseases That Continue to Be Difficult to Model
| Challenges | Potential Solutions |
|---|---|
| Epigenetic memory [138] | Test various cell types, and use late passage iPSCs |
| Lack of dependable and efficient differentiation protocols, which may result in the generation of a mixture of cell types [139] | Further research into and development of protocols utilise reporter genes to select for the cell type of interest [140] |
| Variable properties independent of genetic background, especially for transgene-based reprogramming | Utilise transgene-free reprogramming utilise more stringent quality controls |
| Modeling of diseases involving the complex interactions between multiple cell types, in a three-dimensional niche [127] | Advances in 3D culture techniques, organoid-growing techniques and tissue engineering strategies [143,144,146,147] |
| Modeling of diseases affected by environmental factors | Use cells cultured in bio-engineered niches and co-culture with primary cells in order to mimic in vivo tissue development |
| Modeling of adult onset diseases | For neurons, exposing cells to oxidative stress [95], progerin [148], or by excessively stimulating neurons with high concentrations of glutamate [149] |
12. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Siminovitch, L.; McCulloch, E.A.; Till, J.E. The distribution of colony-forming cells among spleen colonies. J. Cell. Comp. Physiol. 1963, 62, 327–336. [Google Scholar] [CrossRef]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Nakada, D.; Morrison, S.J. Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol. 2009, 25, 377–406. [Google Scholar] [PubMed]
- Ulloa-Montoya, F.; Verfaillie, C.M.; Hu, W.S. Culture systems for pluripotent stem cells. J. Biosci. Bioeng. 2005, 100, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, O.; Kokaia, Z. Stem cells for the treatment of neurological disorders. Nature 2006, 441, 1094–1096. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.H.; Saha, K.; Jaenisch, R. Pluripotency and cellular reprogramming: Facts, hypotheses, unresolved issues. Cell 2010, 143, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Brock, A.; Goh, H.T.; Yang, B.; Lu, Y.; Li, H.; Loh, Y.H. Cellular reprogramming: A new technology frontier in pharmaceutical research. Pharm. Res. 2012, 29, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Beumer, K.; Trautman, J.K.; Carroll, D. Enhancing gene targeting with designed zinc finger nucleases. Science 2003, 300, 764. [Google Scholar] [CrossRef] [PubMed]
- Grizot, S.; Smith, J.; Daboussi, F.; Prieto, J.; Redondo, P.; Merino, N.; Villate, M.; Thomas, S.; Lemaire, L.; Montoya, G.; et al. Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Res. 2009, 37, 5405–5419. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Gurdon, J.B. The transplantation of nuclei between two species of Xenopus. Dev. Biol. 1962, 5, 68–83. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Maherali, N.; Ahfeldt, T.; Rigamonti, A.; Utikal, J.; Cowan, C.; Hochedlinger, K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 2008, 3, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Merkl, C.; Saalfrank, A.; Riesen, N.; Kühn, R.; Pertek, A.; Eser, S.; Hardt, M.S.; Kind, A.; Saur, D.; Wurst, W.; et al. Efficient generation of rat induced pluripotent stem cells using a non-viral inducible vector. PLoS ONE 2013, 8, e55170. [Google Scholar] [CrossRef] [PubMed]
- Ichida, J.K.; Blanchard, J.; Lam, K.; Son, E.Y.; Chung, J.E.; Egli, D.; Loh, K.M.; Carter, A.C.; di Giorgio, F.P.; Koszka, K.; et al. A small-molecule inhibitor of TGF-β signaling replaces Sox2 in reprogramming by inducing nanog. Cell Stem Cell 2009, 5, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Foreman, R.K.; Staerk, J.; Garcia, M.; Mathur, D.; Markoulaki, S.; Hanna, J.; Lairson, L.L.; Charette, B.D.; Bouchez, L.C.; et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc. Natl. Acad. Sci. USA 2009, 106, 8912–8917. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by Oct4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Schöler, H.R.; Ding, S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Osafune, K.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Chen, S.; Muhlestein, W.; Melton, D.A. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 2008, 26, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Wu, S. Reprogramming with small molecules instead of exogenous transcription factors. Stem Cells Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells Dayt. Ohio 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Pawlik, K.M.; Liu, K.; Sun, C.W.; Li, C.; Schoeb, T.R.; Townes, T.M. Polycistronic lentiviral vector for “hit and run” reprogramming of adult skin fibroblasts to induced pluripotent stem cells. Stem Cells Dayt. Ohio 2009, 27, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.G.; Rozelle, S.S.; Sullivan, S.; Mills, J.A.; Park, S.M.; Smith, B.W.; Iyer, A.M.; French, D.L.; Kotton, D.N.; Gadue, P.; et al. Generation of human induced pluripotent stem cells from peripheral blood using the STEMCCA lentiviral vector. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBAC transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Plews, J.R.; Li, J.; Jones, M.; Moore, H.D.; Mason, C.; Andrews, P.W.; Na, J. Activation of pluripotency genes in human fibroblast cells by a novel mRNA based approach. PLoS ONE 2010, 5, e14397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Kim, J.H.; Lee, H.J.; Jeon, K.; Lim, H.; Choi, H.; Lee, E.R.; Park, S.H.; Park, J.Y.; Hong, S.; et al. The generation of iPS cells using non-viral magnetic nanoparticle based transfection. Biomaterials 2011, 32, 6683–6691. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.; Markoulaki, S.; Schorderet, P.; Carey, B.W.; Beard, C.; Wernig, M.; Creyghton, M.P.; Steine, E.J.; Cassady, J.P.; Foreman, R.; et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 2008, 133, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Eminli, S.; Utikal, J.; Arnold, K.; Jaenisch, R.; Hochedlinger, K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells Dayt. Ohio 2008, 26, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Ohnishi, H.; Oda, Y.; Tadokoro, M.; Sasao, M.; Kato, H.; Hattori, K.; Ohgushi, H. Generation of induced pluripotent stem cells from human adipose-derived stem cells without c-MYC. Tissue Eng. Part A 2010, 16, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Tan, X.; Li, G.; Gao, Y.; Liu, X.; Zhang, L.; Li, Y. Induced pluripotent stem cells from human hair follicle mesenchymal stem cells. Stem Cell Rev. 2013, 9, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Fan, Y.; Sun, X.; Yu, Y. Generation of induced pluripotent stem cells from human amniotic fluid cells by reprogramming with two factors in feeder-free conditions. J. Reprod. Dev. 2013, 59, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Brennand, K.; Panopoulos, A.D.; Herrerías, A.; Gage, F.H.; Izpisua-Belmonte, J.C. High-efficient generation of induced pluripotent stem cells from human astrocytes. PLoS ONE 2010, 5, e15526. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Golic, M.; Golic, K.G.; Carroll, D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 2002, 161, 1169–1175. [Google Scholar] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Carroll, D.; Segal, D.J.; Trautman, J.K.; Smith, J.; Kim, Y.G.; Chandrasegaran, S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001, 21, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Zhu, S.; Xu, Y.; Fu, X. Homologous recombination in human embryonic stem cells using CRISPR/Cas9 nickase and a long DNA donor template. Protein Cell 2014, 5, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Grizot, S.; Arnould, S.; Duclert, A.; Epinat, J.C.; Chames, P.; Prieto, J.; Redondo, P.; Blanco, F.J.; Bravo, J.; et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006, 34, e149. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Stormo, G.D. Context-dependent DNA recognition code for C2H2 zinc-finger transcription factors. Bioinform. Oxf. Engl. 2008, 24, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, B. TAL effector nuclease (TALEN) engineering. Methods Mol. Biol. 2013, 978, 63–72. [Google Scholar] [PubMed]
- Gogarten, J.P.; Hilario, E. Inteins, introns, and homing endonucleases: Recent revelations about the life cycle of parasitic genetic elements. BMC Evol. Biol. 2006, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Poirot, L.; Galetto, R.; Smith, J.; Montoya, G.; Duchateau, P.; Paques, F. Meganucleases and other tools for targeted genome engineering: Perspectives and challenges for gene therapy. Curr. Gene Ther. 2011, 11, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Cathomen, T.; Joung, J.K. Zinc-finger nucleases: The next generation emerges. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Müller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Carlson, D.F.; Fahrenkrug, S.C.; Hackett, P.B. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic Acids 2012, 1, e3. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the IAP gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [PubMed]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; di Carlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Ortiz, O.; Hrabé de Angelis, M.; Wurst, W.; Kühn, R. Modeling disease mutations by gene targeting in one-cell mouse embryos. Proc. Natl. Acad. Sci. USA 2012, 109, 9354–9359. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Casini, S.; Davis, R.P.; Aniello, C.D.; Haas, J.; Ward-van Oostwaard, D.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Egashira, T.; Yuasa, S.; Fukuda, K. Novel insights into disease modeling using induced pluripotent stem cells. Biol. Pharm. Bull. 2013, 36, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M.; Cudkowicz, M.E.; Schoenfeld, D.; Conrad, T.; Taft, J.; Chilton, M.; Urbinelli, L.; Qureshi, M.; Zhang, H.; Pestronk, A.; et al. NEALS Consortium. A clinical trial of creatine in ALS. Neurology 2004, 63, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Gupta, R.S. Species-specific differences in the toxicity and mutagenicity of the anticancer drugs mithramycin, chromomycin A3, and olivomycin. Cancer Res. 1985, 45, 2813–2820. [Google Scholar] [PubMed]
- Chun, Y.S. Applications of patient-specific induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease. Int. J. Biol. Sci. 2010, 796–805. [Google Scholar] [CrossRef]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ohi, Y.; Qin, H.; Hong, C.; Blouin, L.; Polo, J.M.; Guo, T.; Qi, Z.; Downey, S.L.; Manos, P.D.; Rossi, D.J.; et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 2011, 13, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Mekhoubad, S.; Bock, C.; de Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Moad, M.; Pal, D.; Hepburn, A.C.; Williamson, S.C.; Wilson, L.; Lako, M.; Armstrong, L.; Hayward, S.W.; Franco, O.E.; Cates, J.M.; et al. A novel model of urinary tract differentiation, tissue regeneration, and disease: Reprogramming human prostate and bladder cells into induced pluripotent stem cells. Eur. Urol. 2013, 64, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.N.; Byers, B.; Cord, B.; Shcheglovitov, A.; Byrne, J.; Gujar, P.; Kee, K.; Schüle, B.; Dolmetsch, R.E.; Langston, W.; et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011, 8, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS ONE 2011, 6, e26203. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Yik, W.Y.; Zhang, P.; Lu, W.; Dranchak, P.K.; Shibata, D.; Steinberg, S.J.; Hacia, J.G. The gene expression profiles of induced pluripotent stem cells from individuals with childhood cerebral adrenoleukodystrophy are consistent with proposed mechanisms of pathogenesis. Stem Cell Res. Ther. 2012, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.E.; Yang, Y.C.; Chen, S.M.; Su, H.L.; Huang, P.C.; Tsai, M.S.; Wang, T.H.; Tseng, C.P.; Hwang, S.M. Modeling neurogenesis impairment in Down syndrome with induced pluripotent stem cells from Trisomy 21 amniotic fluid cells. Exp. Cell Res. 2013, 319, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.Z.; Pech, M.F.; Zhong, F.L.; Nguyen, H.N.; Xie, K.T.; Zaug, A.J.; Crary, S.M.; Choi, J.; Sebastiano, V.; Cherry, A.; et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 2011, 474, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Soragni, E.; Campau, E.; Thomas, E.A.; Altun, G.; Laurent, L.C.; Loring, J.F.; Napierala, M.; Gottesfeld, J.M. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAATTC triplet repeat instability. Cell Stem Cell 2010, 7, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, J.; Nakao, K.; Sone, M.; Noguchi, M.; Mori, E.; Naito, M.; Taura, D.; Harada-Shiba, M.; Kishimoto, I.; Watanabe, A.; et al. Induced pluripotent stem cells generated from diabetic patients with mitochondrial DNA A3243G mutation. Diabetologia 2012, 55, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cowley, S.A.; Siler, U.; Melguizo, D.; Tilgner, K.; Browne, C.; Dewilton, A.; Przyborski, S.; Saretzki, G.; James, W.S.; et al. Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells Dayt. Ohio 2012, 30, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Arai, S.; Hosoi, M.; Taoka, K.; Takayama, N.; Otsu, M.; Nagae, G.; Ueda, K.; Nakazaki, K.; Kamikubo, Y.; et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood 2012, 119, 6234–6242. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.U.W.; Schlaeger, T.M.; de Vine, A.L.; Williams, D.A. Induced pluripotent stem cells as a tool for gaining new insights into Fanconi anemia. Cell Cycle Georget. Tex 2012, 11, 2985–2990. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cai, J.; Zhang, Y.; Wang, X.; Li, W.; Xu, J.; Li, F.; Guo, X.; Deng, K.; Zhong, M.; et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. J. Biol. Chem. 2010, 285, 40303–40311. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.J.; Chen, P.F.; Ng, K.Y.; Bourgois-Rocha, F.; Lemtiri-Chlieh, F.; Levine, E.S.; Lalande, M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA 2010, 107, 17668–17673. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; Souza, S.L.D.; Ang, Y.S.; Schaniel, C.; Lee, D.F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lian, Q.; Zhu, G.; Zhou, F.; Sui, L.; Tan, C.; Mutalif, R.A.; Navasankari, R.; Zhang, Y.; Tse, H.F.; et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef] [PubMed]
- Kinnear, C.; Chang, W.Y.; Khattak, S.; Hinek, A.; Thompson, T.; de Carvalho Rodrigues, D.; Kennedy, K.; Mahmut, N.; Pasceri, P.; Stanford, W.L.; et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.B.; Okamoto, S.; Osakada, F.; Homma, K.; Assawachananont, J.; Hirami, Y.; Iwata, T.; Takahashi, M. Modeling retinal degeneration using patient-specific induced pluripotent stem cells. PLoS ONE 2011, 6, e17084. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Luo, R.; Xu, Y.; Cai, X.; Li, W.; Tan, K.; Huang, J.; Dai, Y. Generation of systemic lupus erythematosus-specific induced pluripotent stem cells from urine. Rheumatol. Int. 2013, 33, 2127–2134. [Google Scholar] [CrossRef] [PubMed]
- Saitta, B.; Passarini, J.; Sareen, D.; Ornelas, L.; Sahabian, A.; Argade, S.; Krakow, D.; Cohn, D.H.; Svendsen, C.N.; Rimoin, D.L. Patient-derived skeletal dysplasia induced pluripotent stem cells display abnormal chondrogenic marker expression and regulation by BMP2 and TGFβ1. Stem Cells Dev. 2014, 23, 1464–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Su, J.; Kim, H.S.; Chang, B.; Papatsenko, D.; Zhao, R.; Yuan, Y.; Gingold, J.; Xia, W.; Darr, H.; et al. Modeling familial cancer with induced pluripotent stem cells. Cell 2015, 161, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Ghodsizadeh, A.; Taei, A.; Totonchi, M.; Seifinejad, A.; Gourabi, H.; Pournasr, B.; Aghdami, N.; Malekzadeh, R.; Almadani, N.; Salekdeh, G.H.; et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev. 2010, 6, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Kim, Y.; Shim, J.S.; Park, J.T.; Wang, R.H.; Leach, S.D.; Liu, J.O.; Deng, C.; Ye, Z.; Jang, Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013, 57, 2458–2468. [Google Scholar] [CrossRef] [PubMed]
- Irudayam, J.I. Modeling Liver Diseases Using Induced Pluripotent Stem Cell (Ipsc)-Derived Hepatocytes. J. Stem Cell Res. Ther. 2014, 4, 218. [Google Scholar] [CrossRef]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, M.; Sinigaglia, A.; Desole, G.; Berto, A.; Pacenti, M.; Palù, G.; Barzon, L. Modeling Viral Infectious Diseases and Development of Antiviral Therapies Using Human Induced Pluripotent Stem Cell-Derived Systems. Viruses 2015, 7, 3835–3856. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Huang, S.X.L.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Kambal, A.; Mitchell, G.; Cary, W.; Gruenloh, W.; Jung, Y.; Kalomoiris, S.; Nacey, C.; McGee, J.; Lindsey, M.; Fury, B.; et al. Generation of HIV-1 resistant and functional macrophages from hematopoietic stem cell-derived induced pluripotent stem cells. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Rhimm, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.X.; et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013, 3, 2088–2099. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.W.; Barrilleaux, B.L.; Varlakhanova, N.; Bush, K.M.; Chan, V.; Knoepfler, P.S. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013, 22, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Curry, E.L.; Moad, M.; Robson, C.N.; Heer, R. Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J. Stem Cells 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Liu, H.; Ridky, T.W.; Cassarino, D.; Segal, E.; Chang, H.Y. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2008, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Osafune, K.; Caron, L.; Borowiak, M.; Martinez, R.J.; Fitz-Gerald, C.S.; Sato, Y.; Cowan, C.A.; Chien, K.R.; Melton, D.A. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 2008, 26, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Jaenisch, R. iPSC disease modeling. Science 2012, 338, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A. Epigenetic modifications in pluripotent and differentiated cells. Nat. Biotechnol. 2010, 28, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sancho-Martinez, I.; Nivet, E.; Rodriguez Esteban, C.; Campistol, J.M.; Izpisua Belmonte, J.C. The generation of kidney organoids by differentiation of human pluripotent cells to ureteric bud progenitor-like cells. Nat. Protoc. 2014, 9, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seah, Y.F.S.; EL Farran, C.A.; Warrier, T.; Xu, J.; Loh, Y.-H. Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges. Int. J. Mol. Sci. 2015, 16, 28614-28634. https://doi.org/10.3390/ijms161226119
Seah YFS, EL Farran CA, Warrier T, Xu J, Loh Y-H. Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges. International Journal of Molecular Sciences. 2015; 16(12):28614-28634. https://doi.org/10.3390/ijms161226119
Chicago/Turabian StyleSeah, Yu Fen Samantha, Chadi A. EL Farran, Tushar Warrier, Jian Xu, and Yuin-Han Loh. 2015. "Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges" International Journal of Molecular Sciences 16, no. 12: 28614-28634. https://doi.org/10.3390/ijms161226119
APA StyleSeah, Y. F. S., EL Farran, C. A., Warrier, T., Xu, J., & Loh, Y.-H. (2015). Induced Pluripotency and Gene Editing in Disease Modelling: Perspectives and Challenges. International Journal of Molecular Sciences, 16(12), 28614-28634. https://doi.org/10.3390/ijms161226119