
Molecular Evolution of the TET Gene Family in Mammals


Abstract
:
1. Introduction
2. Results
2.1. Mammalian TET Family Gene Sequences
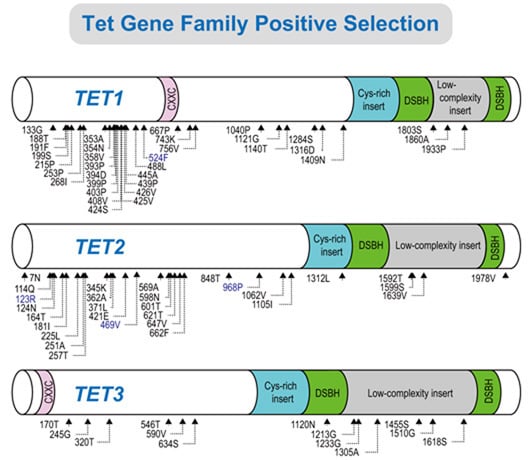
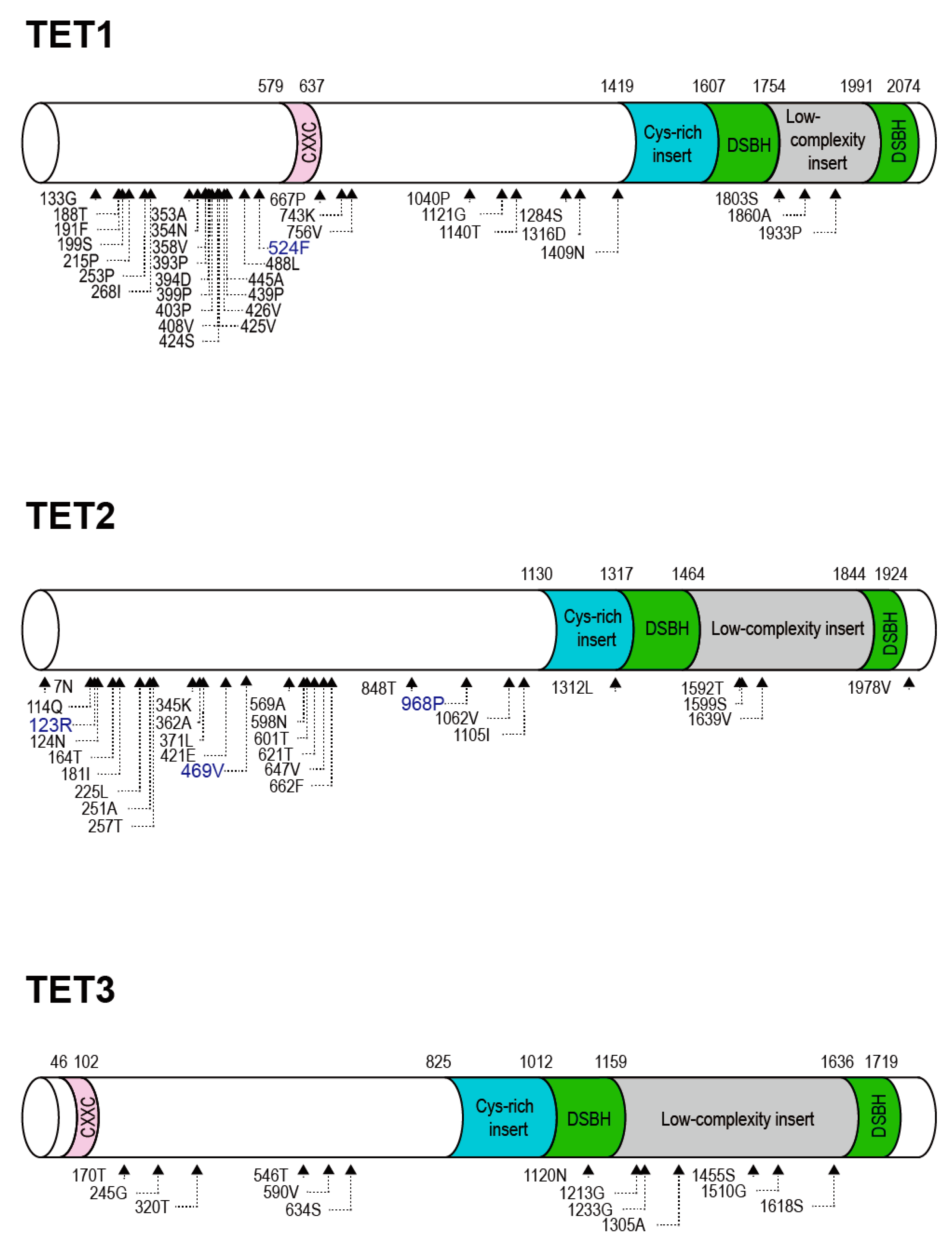
2.2. Positive Selection and the Diversification of TET Family Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Parameter Estimates a | LnL | 2ΔlnL (p-Value) | Positively Selected Sites b,c |
|---|---|---|---|---|
| TET1 | ||||
| M0 | ω = 0.385 | −87,143.158462 | None | |
| M1a | ω0 = 0.205, p0 = 0.623, ω1 = 1.000, p1 = 0.377 | −86,007.766228 | Not allowed | |
| M2a | ω0 = 0.205, p0 = 0.623, ω1 = 1.000, p1 = 0.298, ω2 = 1.000, p2 = 0.078 | −86,007.766228 | M2a vs. M1a | None |
| 0 (p = 1) | ||||
| M7 | p = 0.679, q = 0.983 | −85,718.609725 | Not allowed | |
| M8 | p0 = 0.962, p = 0.744, q = 1.197, p1 = 0.038, ω1 = 1.294 | −85,707.99818 | M8 vs. M7 | 524F, 811M |
| 21.22309 (p < 0.001) | ||||
| TET2 | ||||
| M0 | ω = 0.346 | −75,677.716183 | None | |
| M1a | ω0 = 0.184, p0 = 0.664, ω1 = 1.000, p1 = 0.336 | −74,593.700860 | Not allowed | |
| M2a | ω0 = 0.184, p0 = 0.664, ω1 = 1.000, p1 = 0.292, ω2 = 1.000, p2 = 0.044 | −74,593.700860 | M2a vs. M1a | None |
| 0 (p = 1) | ||||
| M7 | p = 0.617, q = 1.064 | −74,286.497185 | Not allowed | |
| M8 | p0 = 0.978, p = 0.671, q = 1.250, p1 = 0.022, ω1 = 1.558 | −74,265.863135 | M8 vs. M7 | 123R*, 469V, 673V, 968P, 1002T, 1205S, 1767S |
| 41.2681 (p < 0.001) | ||||
| TET3 | ||||
| M0 | ω = 0.115 | −48,719.653509 | None | |
| M1a | ω0 = 0.056, p0 = 0.860, ω1 = 1.000, p1 = 0.140 | −47,597.069946 | Not allowed | |
| M2a | ω0 = 0.056, p0 = 0.859, ω1 = 1.000, p1 = 0.139, ω2 = 999.000, p2 = 0.002 | −47,599.818203 | M2a vs. M1a | None |
| −5.496514 (p = 1) | ||||
| M7 | p = 0.237, q = 1.380 | −47,268.934744 | Not allowed | |
| M8 | p0 = 0.999, p = 0.237, q = 1.380, p1 = 0.001, ω1 = 999.000 | −47,270.552239 | M8 vs. M7 | None |
| −3.23499 (p = 1) | ||||
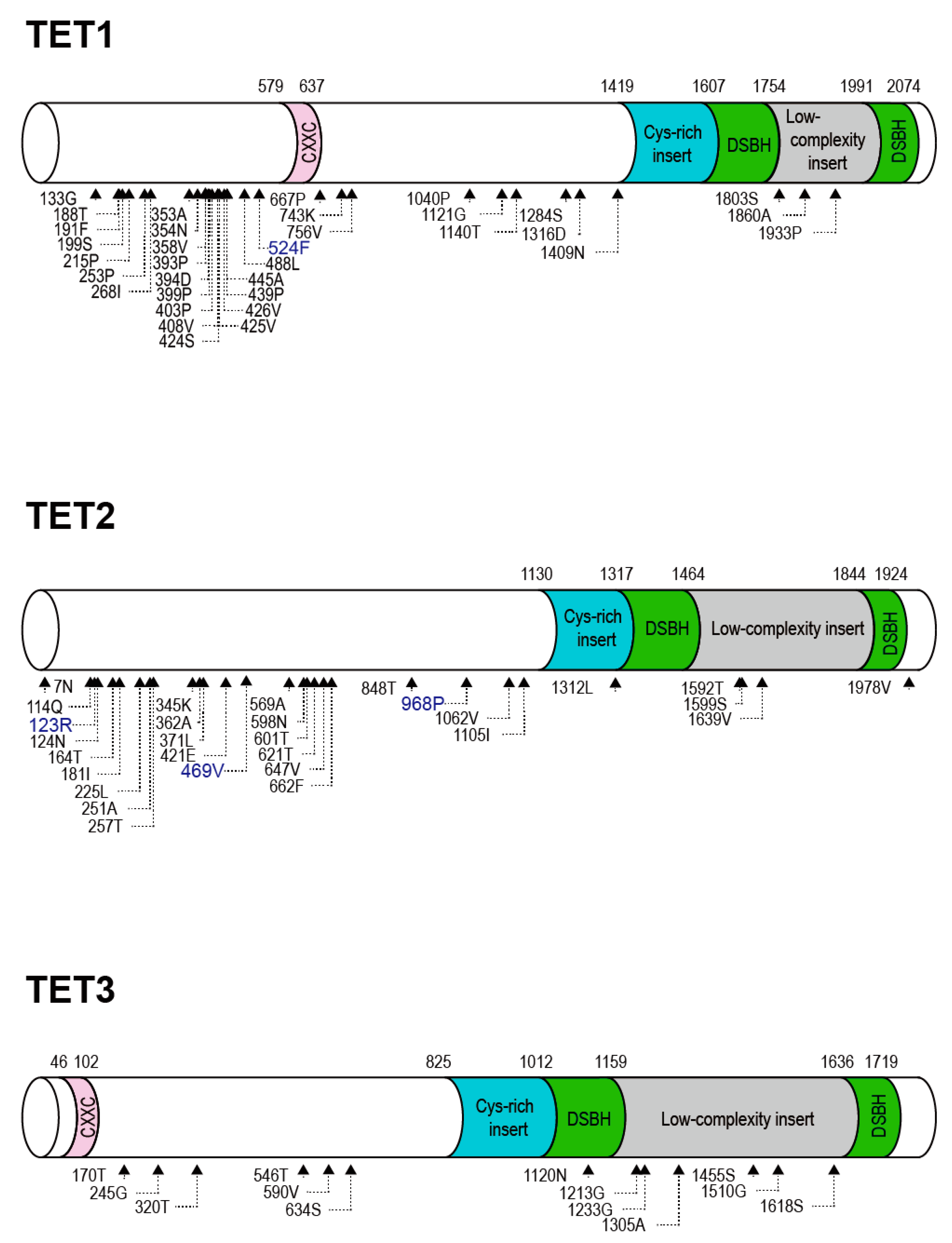
2.3. Identification of Positively Selected Sites
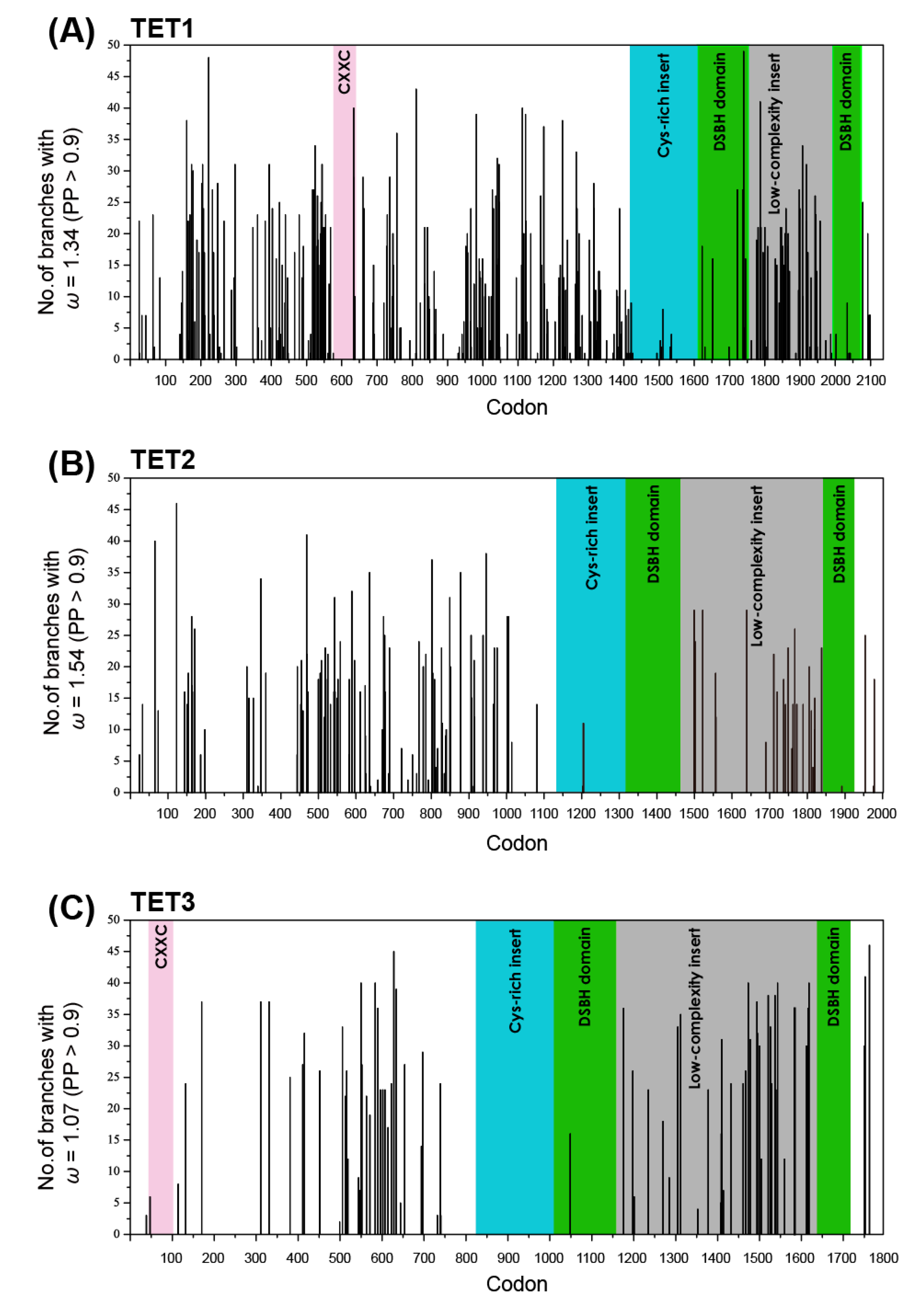
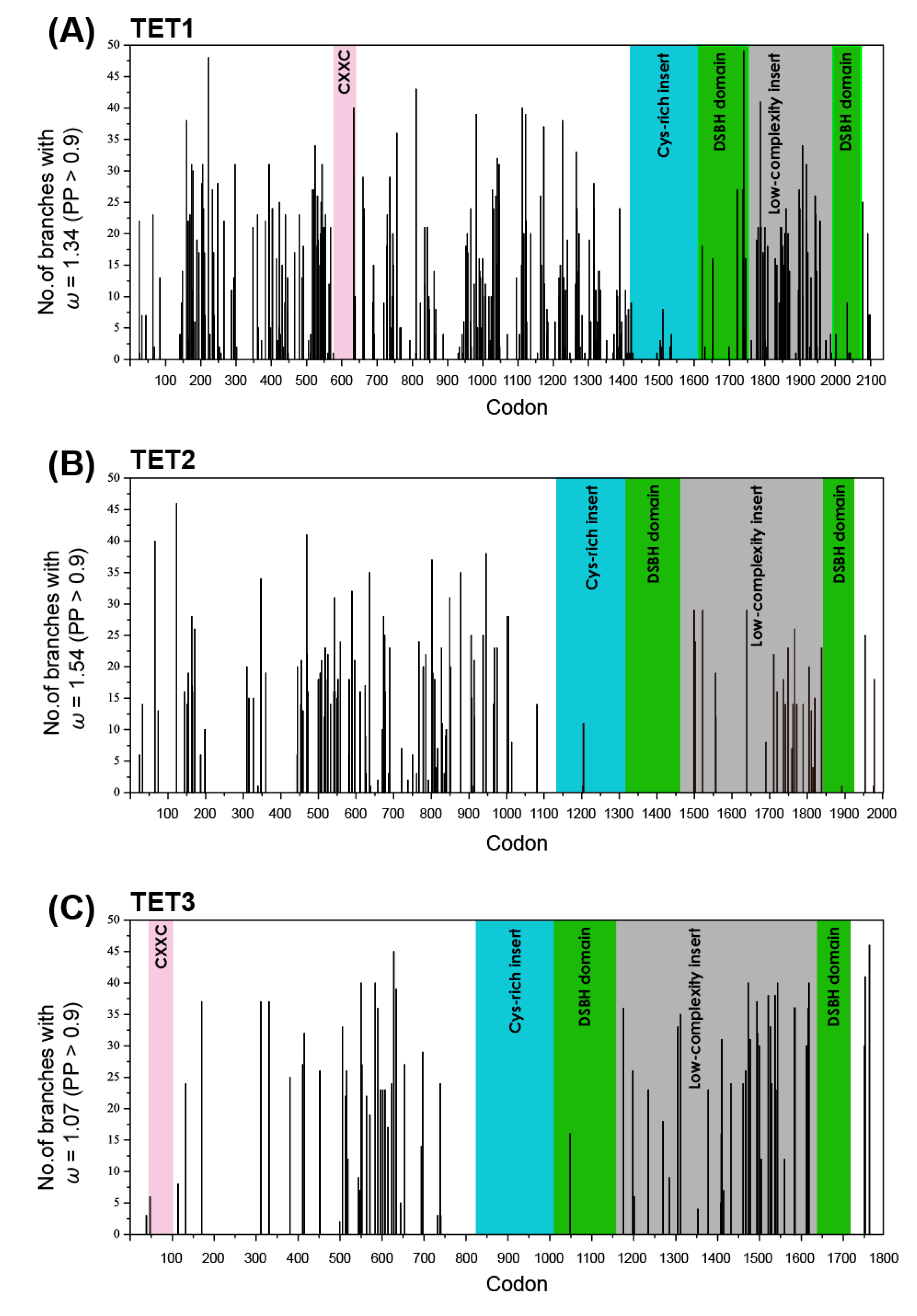
2.4. Shifts in the Site-Specific Selection Process along Lineages
| Gene | M0 vs. M3 | M3 vs. M3 + S1 | M3 + S1 vs. M3 + S2 | |||
|---|---|---|---|---|---|---|
| 2ΔlnL | p-Value | 2ΔlnL | p-Value | 2ΔlnL | p-Value | |
| TET1 | 2858.77 | <0.001 | 376.92 | <0.001 | 71.11 | <0.001 |
| TET2 | 2803.21 | <0.001 | 345.29 | <0.001 | 63.69 | <0.001 |
| TET3 | 2926.57 | <0.001 | 160.22 | <0.001 | 12.59 | <0.002 |
| Parameter | M0 | M3 | M3 + S1 | M3 + S2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TET1 | ||||||||||
| LnL | −87,135.73 | −85,706.34 | −85,517.88 | −85,482.33 | ||||||
| ω1 ω2 ω3 | 0.39 | 0.04 | 0.35 | 0.90 | 0.00 | 0.42 | 1.21 | 0.01 | 0.23 | 1.34 |
| p1 p2 p3 | 0.25 | 0.48 | 0.26 | 0.37 | 0.41 | 0.22 | 0.28 | 0.46 | 0.25 | |
| R12 R13 R23 | 1.54 | 1.54 | 1.54 | 0.71 | 0.2 | 3.34 | ||||
| TET2 | ||||||||||
| LnL | −75,677.72 | −74,276.11 | −74,103.47 | −74,071.62 | ||||||
| ω1 ω2 ω3 | 0.35 | 0.04 | 0.36 | 0.94 | 0.00 | 0.42 | 1.24 | 0.005 | 0.30 | 1.54 |
| p1 p2 p3 | 0.31 | 0.50 | 0.19 | 0.39 | 0.44 | 0.17 | 0.35 | 0.48 | 0.17 | |
| R12 R13 R23 | 1.6 | 1.6 | 1.6 | 0.74 | 0.33 | 4.34 | ||||
| TET3 | ||||||||||
| LnL | −48,719.65 | −47,256.37 | −47,176.26 | −47,169.96 | ||||||
| ω1 ω2 ω3 | 0.11 | 0.01 | 0.19 | 0.75 | 0.00 | 0.28 | 0.98 | 0.003 | 0.22 | 1.07 |
| p1 p2 p3 | 0.62 | 0.28 | 0.10 | 0.69 | 0.23 | 0.08 | 0.68 | 0.23 | 0.09 | |
| R12 R13 R23 | 2.14 | 2.14 | 2.14 | 1.04 | 2.27 | 9.96 | ||||
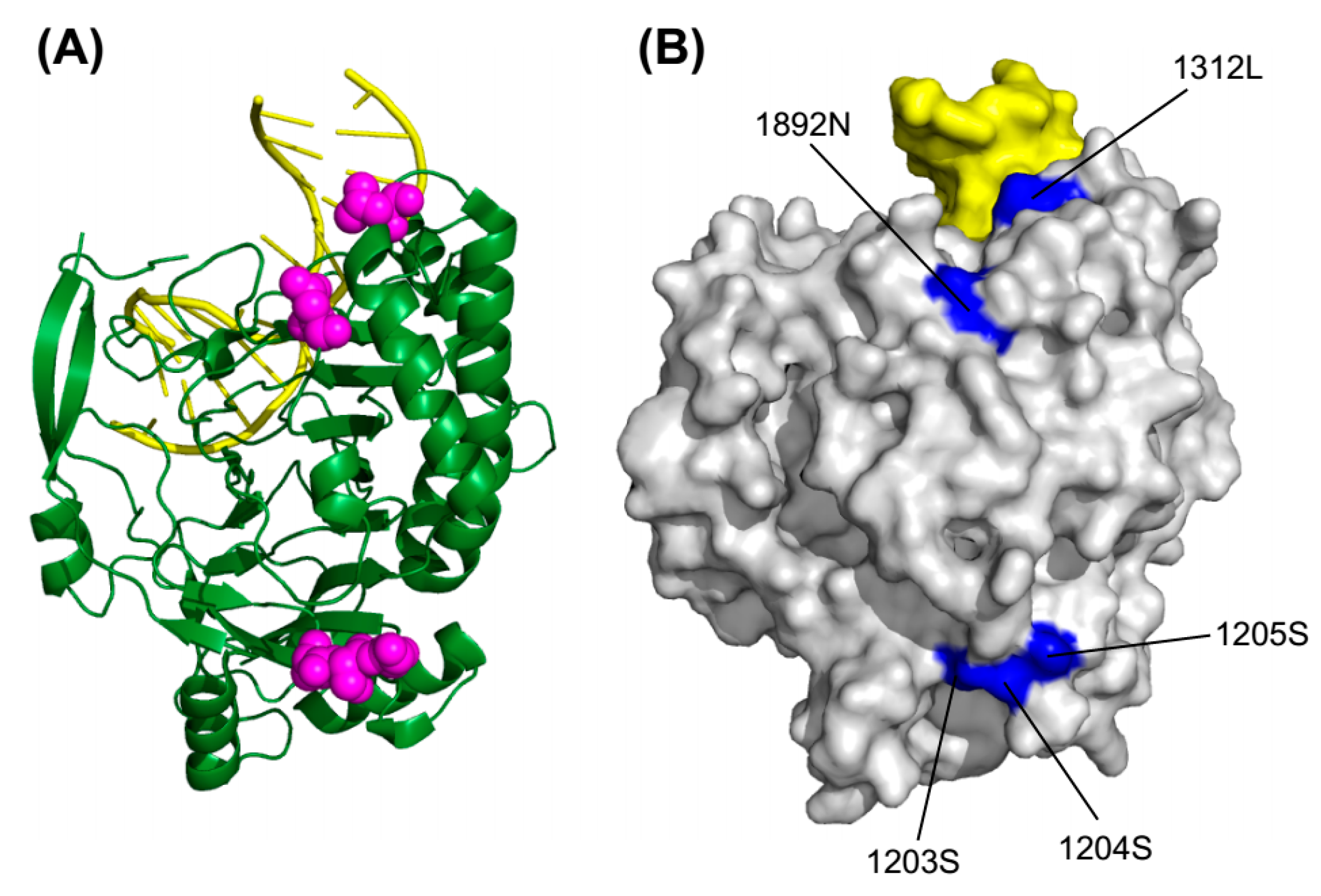
2.5. 3D Structure Mapping
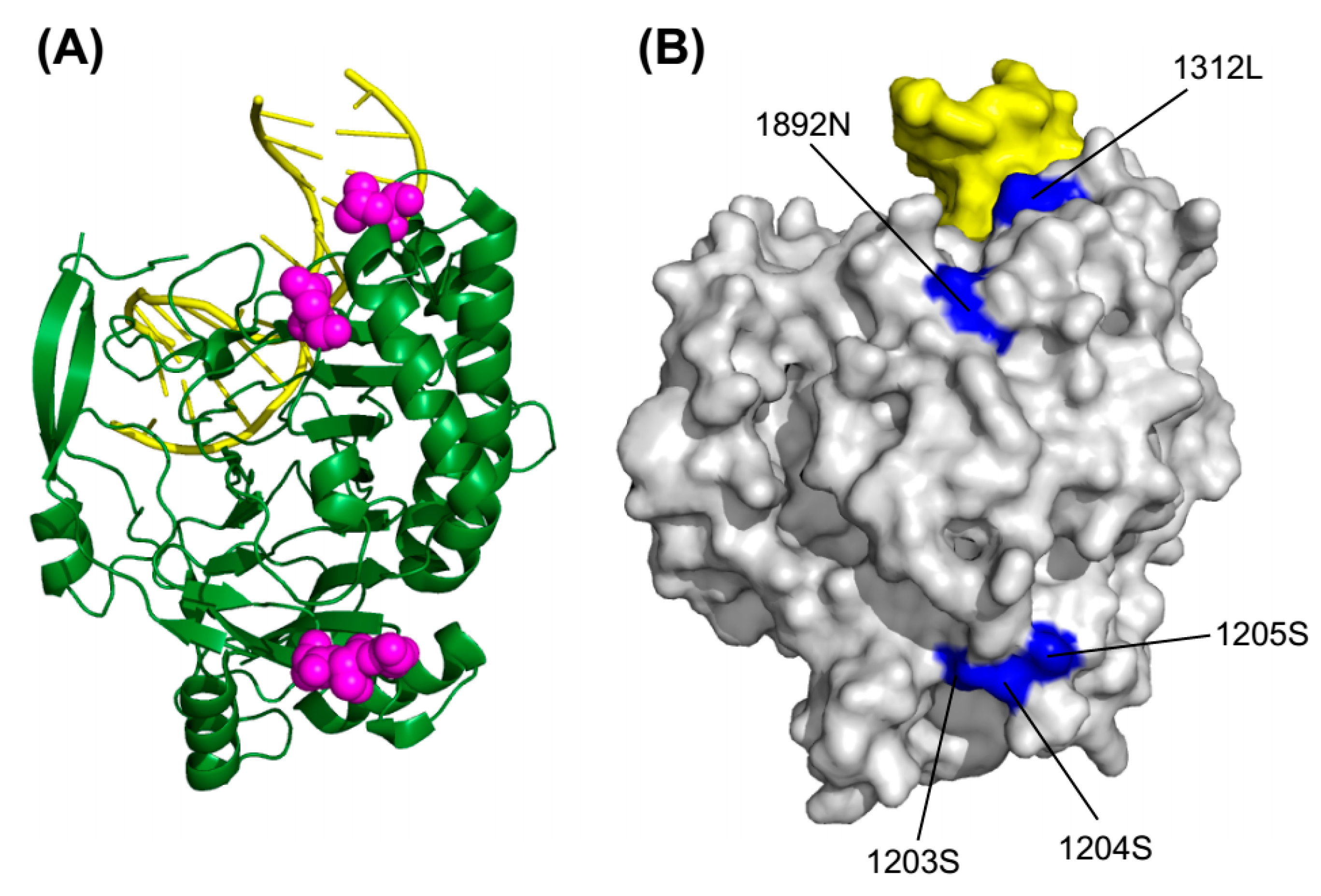
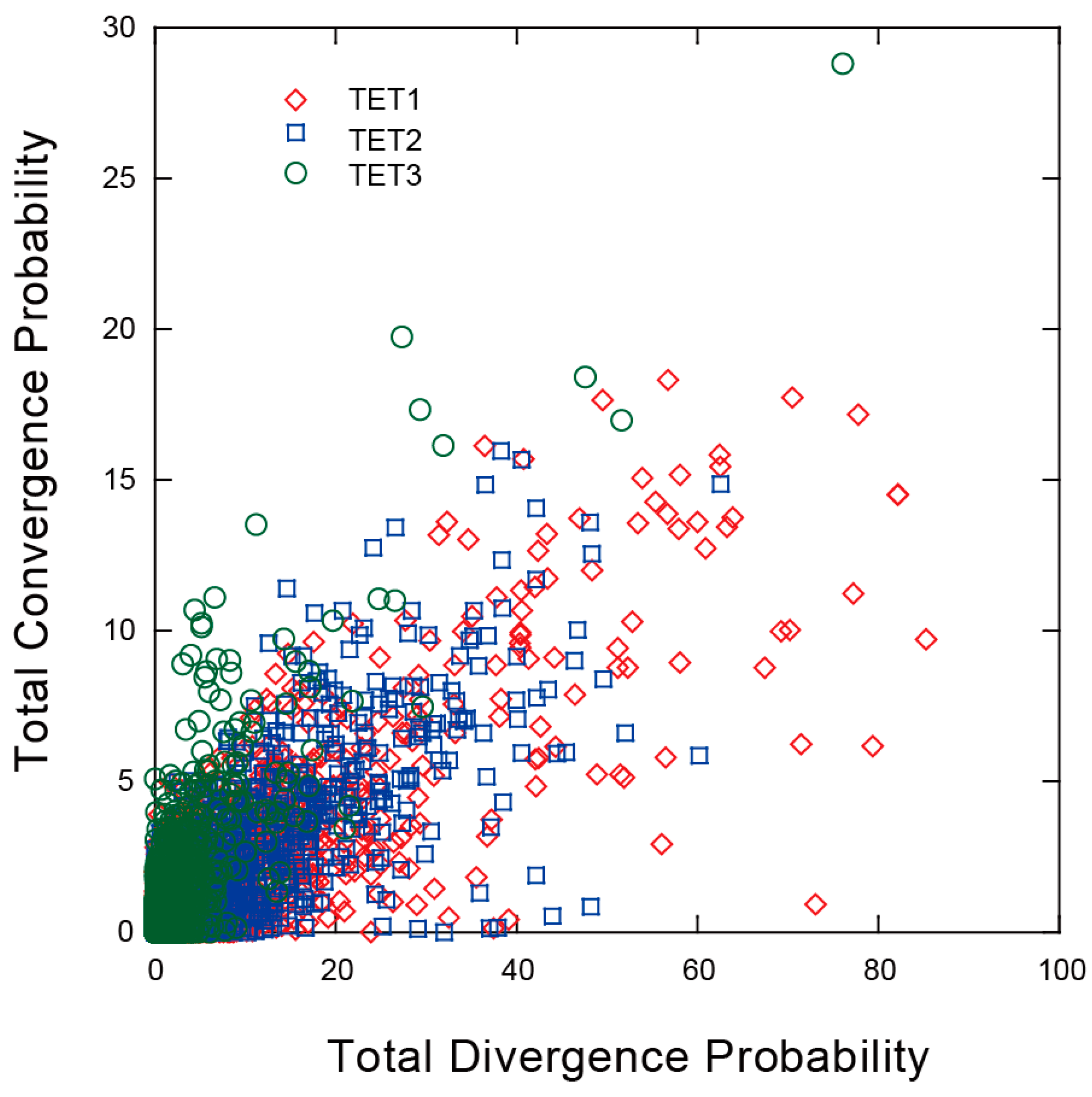
2.6. Convergent and Divergent Changes

3. Discussion
4. Experimental Section
4.1. Sequence Retrieval and Phylogenetic Reconstruction
4.2. Recombination Detection
4.3. Molecular Evolutionary Analysis
4.4. Quantifying the Convergent Amino Acid Substitutions and Divergent Amino Acid Substitutions
4.5. Protein Tertiary Structure Visualizations
5. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Baubec, T.; Schubeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, F.M.; Fisher, A.G. Getting rid of DNA methylation. Trends Cell Biol. 2014, 24, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 2011, 101, 25–104. [Google Scholar] [PubMed]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rao, A. New functions for DNA modifications by TET-JBP. Nat. Struct. Mol. Biol. 2012, 19, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Helin, K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2012, 13, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, Z.; Cheng, J.; Rao, Q.; Gong, W.; Liu, M.; Shi, Y.G.; Zhu, J.; Wang, P.; Xu, Y. Crystal structure of TET2-DNA complex: Insight into TET-mediated 5mC oxidation. Cell 2013, 155, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; An, J.; Bandukwala, H.S.; Chavez, L.; Aijo, T.; Pastor, W.A.; Segal, M.F.; Li, H.; Koh, K.P.; Lahdesmaki, H.; et al. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 2013, 497, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38, e181. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chen, T. Tet family of 5-methylcytosine dioxygenases in mammalian development. J. Hum. Genet. 2013, 58, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky-Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky-Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 2000, 155, 431–449. [Google Scholar] [PubMed]
- Guindon, S.; Rodrigo, A.G.; Dyer, K.A.; Huelsenbeck, J.P. Modeling the site-specific variation of selection patterns along lineages. Proc. Natl. Acad. Sci. USA 2004, 101, 12957–12962. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Nielsen, R.; Yang, Z. Effect of recombination on the accuracy of the likelihood method for detecting positive selection at amino acid sites. Genetics 2003, 164, 1229–1236. [Google Scholar] [PubMed]
- Kosakovsky-Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky-Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- De Matos, A.L.; McFadden, G.; Esteves, P.J. Positive evolutionary selection on the RIG-I-like receptor genes in mammals. PLoS ONE 2013, 8, e81864. [Google Scholar] [CrossRef] [PubMed]
- Wlasiuk, G.; Nachman, M.W. Adaptation and constraint at Toll-like receptors in primates. Mol. Biol. Evol. 2010, 27, 2172–2186. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Guindon, S.; Umemura, T.; Kohidai, L.; Ueda, H. Adaptive evolution of formyl peptide receptors in mammals. J. Mol. Evol. 2015, 80, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Guindon, S. Performance of standard and stochastic branch-site models for detecting positive selection among coding sequences. Mol. Biol. Evol. 2014, 31, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Castoe, T.A.; de Koning, A.P.; Kim, H.M.; Gu, W.; Noonan, B.P.; Naylor, G.; Jiang, Z.J.; Parkinson, C.L.; Pollock, D.D. Evidence for an ancient adaptive episode of convergent molecular evolution. Proc. Natl. Acad. Sci. USA 2009, 106, 8986–8991. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.T.; Tsagkogeorga, G.; Bennett, N.C.; Davalos, L.M.; Faulkes, C.G.; Rossiter, S.J. Molecular evolution of growth hormone and insulin-like growth factor 1 receptors in long-lived, small-bodied mammals. Gene 2014, 549, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Meletis, K.; Fu, D.; Jhaveri, S.; Jaenisch, R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev. Dyn. 2007, 236, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Sehgal, M.; Singh, T.R. Hydroxymethylation and its potential implication in DNA repair system: A review and future perspectives. Gene 2015, 564, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Ikeo, K.; Gojobori, T. Large-scale search for genes on which positive selection may operate. Mol. Biol. Evol. 1996, 13, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, A.M.; Szpurka, H.; Tiu, R.V.; Makishima, H.; Afable, M.; Huh, J.; O’Keefe, C.L.; Ganetzky, R.; McDevitt, M.A.; Maciejewski, J.P. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood 2009, 113, 6403–6410. [Google Scholar] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [PubMed]
- Stabenau, A.; McVicker, G.; Melsopp, C.; Proctor, G.; Clamp, M.; Birney, E. The Ensembl core software libraries. Genome Res. 2004, 14, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, S.; Umemura, T.; Tanaka, K.; Watanabe, K.; Hayashi, M.; Muto, Y. Genome-wide evidence of positive selection in Bacteroides fragilis. Comput. Biol. Chem. 2014, 52, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky-Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akahori, H.; Guindon, S.; Yoshizaki, S.; Muto, Y. Molecular Evolution of the TET Gene Family in Mammals. Int. J. Mol. Sci. 2015, 16, 28472-28485. https://doi.org/10.3390/ijms161226110
Akahori H, Guindon S, Yoshizaki S, Muto Y. Molecular Evolution of the TET Gene Family in Mammals. International Journal of Molecular Sciences. 2015; 16(12):28472-28485. https://doi.org/10.3390/ijms161226110
Chicago/Turabian StyleAkahori, Hiromichi, Stéphane Guindon, Sumio Yoshizaki, and Yoshinori Muto. 2015. "Molecular Evolution of the TET Gene Family in Mammals" International Journal of Molecular Sciences 16, no. 12: 28472-28485. https://doi.org/10.3390/ijms161226110
APA StyleAkahori, H., Guindon, S., Yoshizaki, S., & Muto, Y. (2015). Molecular Evolution of the TET Gene Family in Mammals. International Journal of Molecular Sciences, 16(12), 28472-28485. https://doi.org/10.3390/ijms161226110