
Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
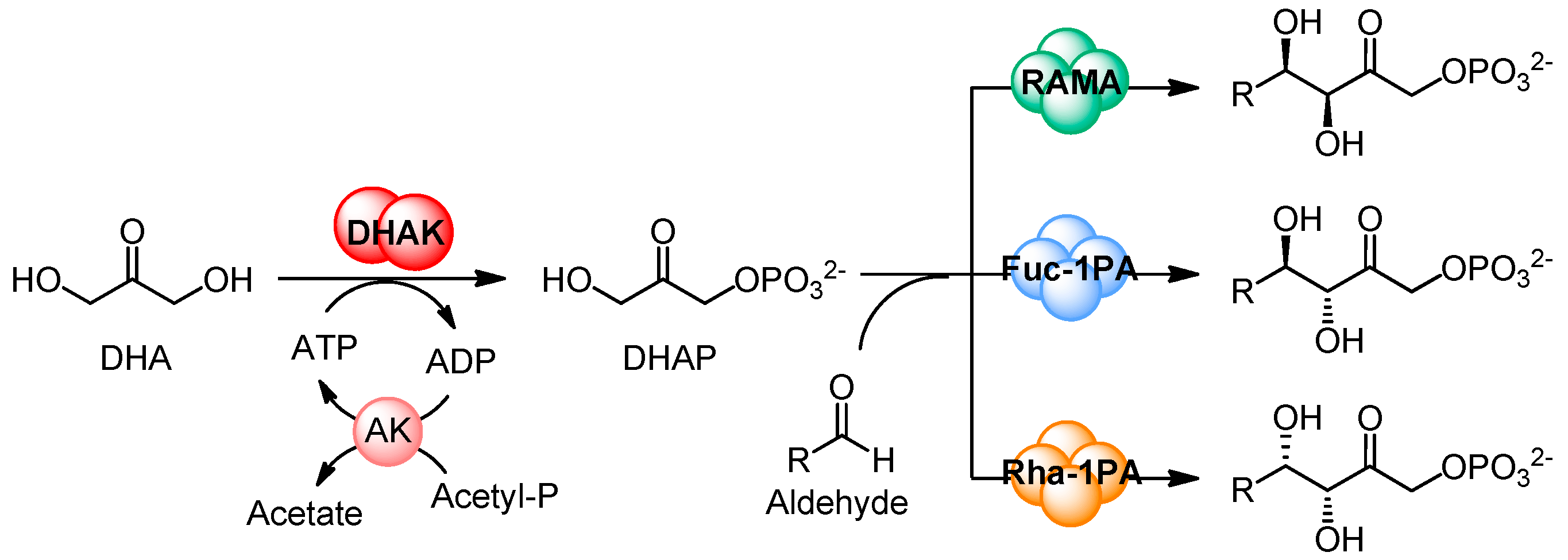
2. Results and Discussion
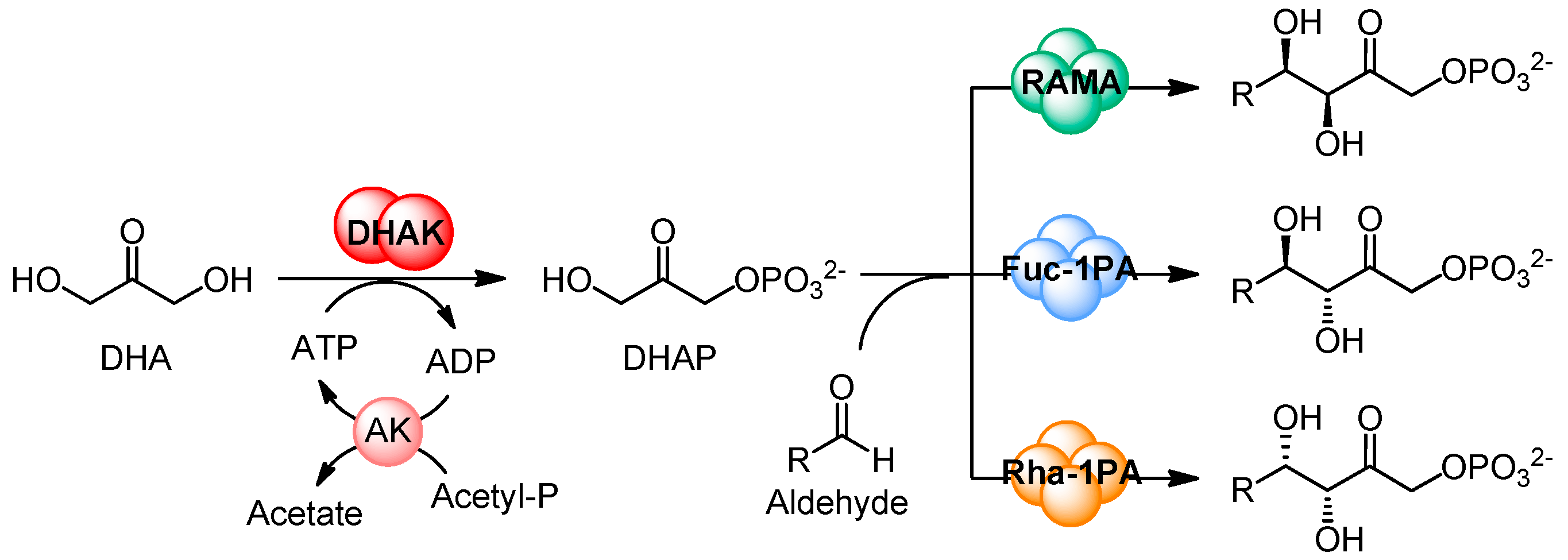
2.1. Substrate Specificity of the Wild-Type DHAK
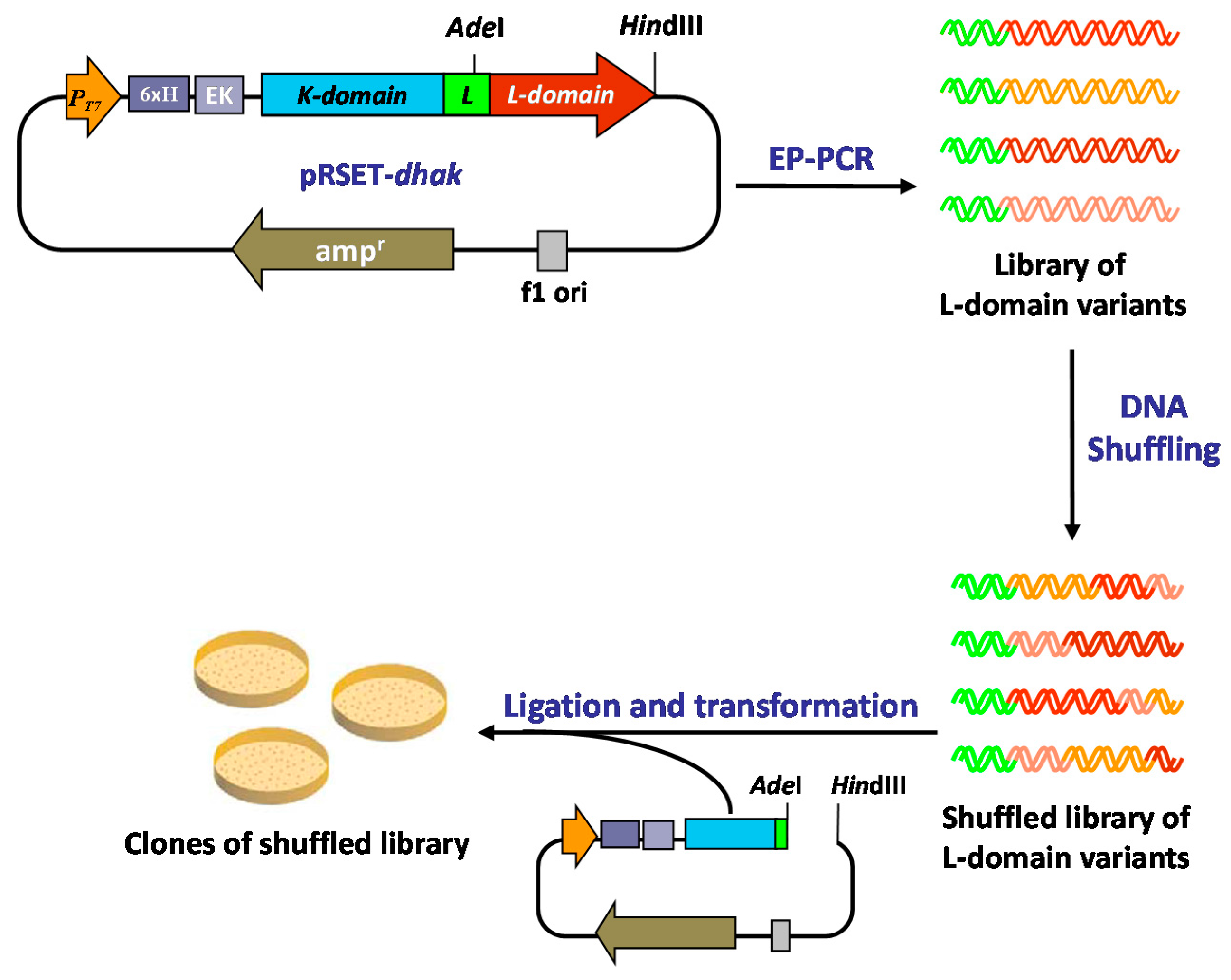
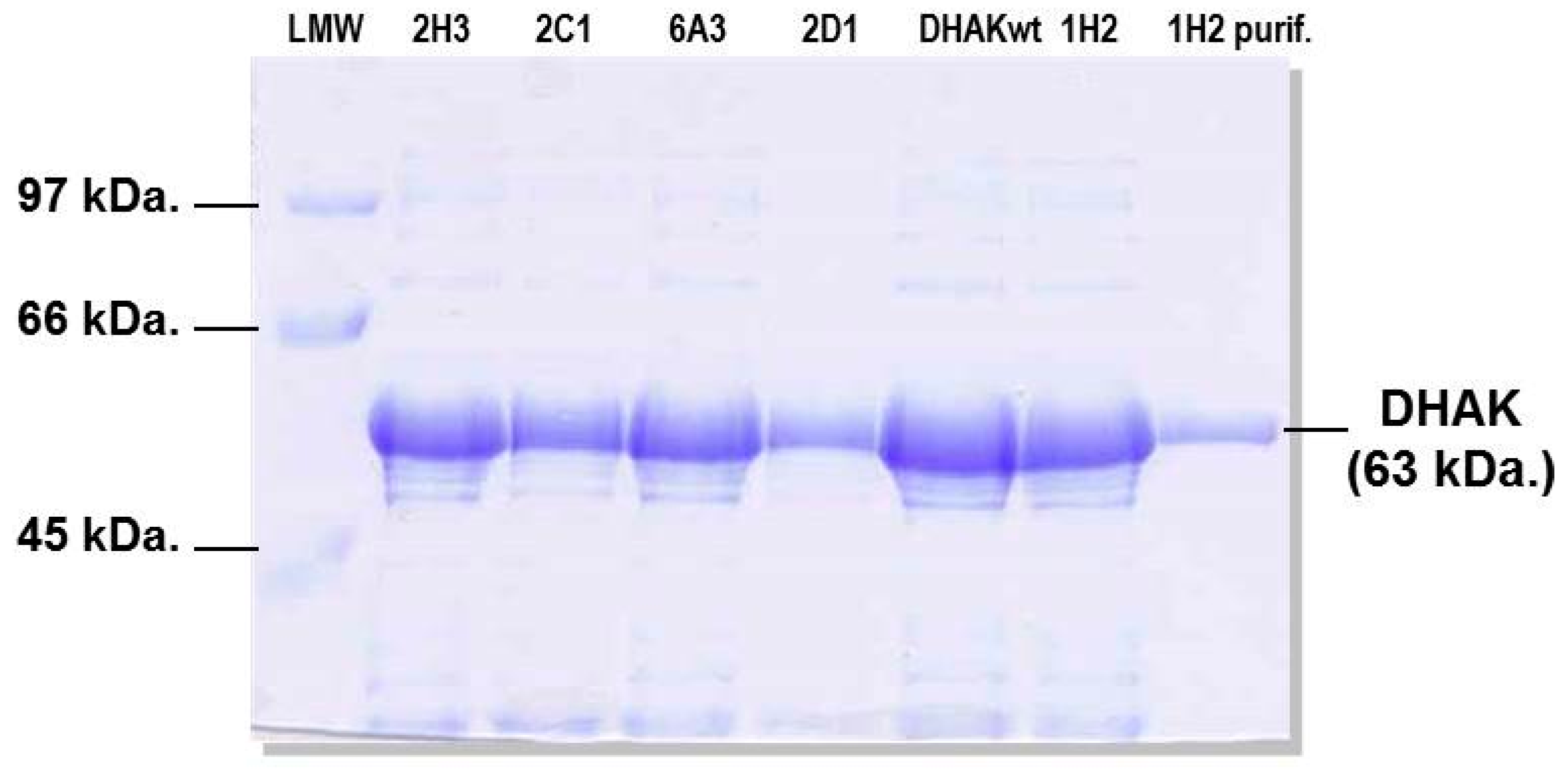
2.2. Generation of a DHAK Mutant Library
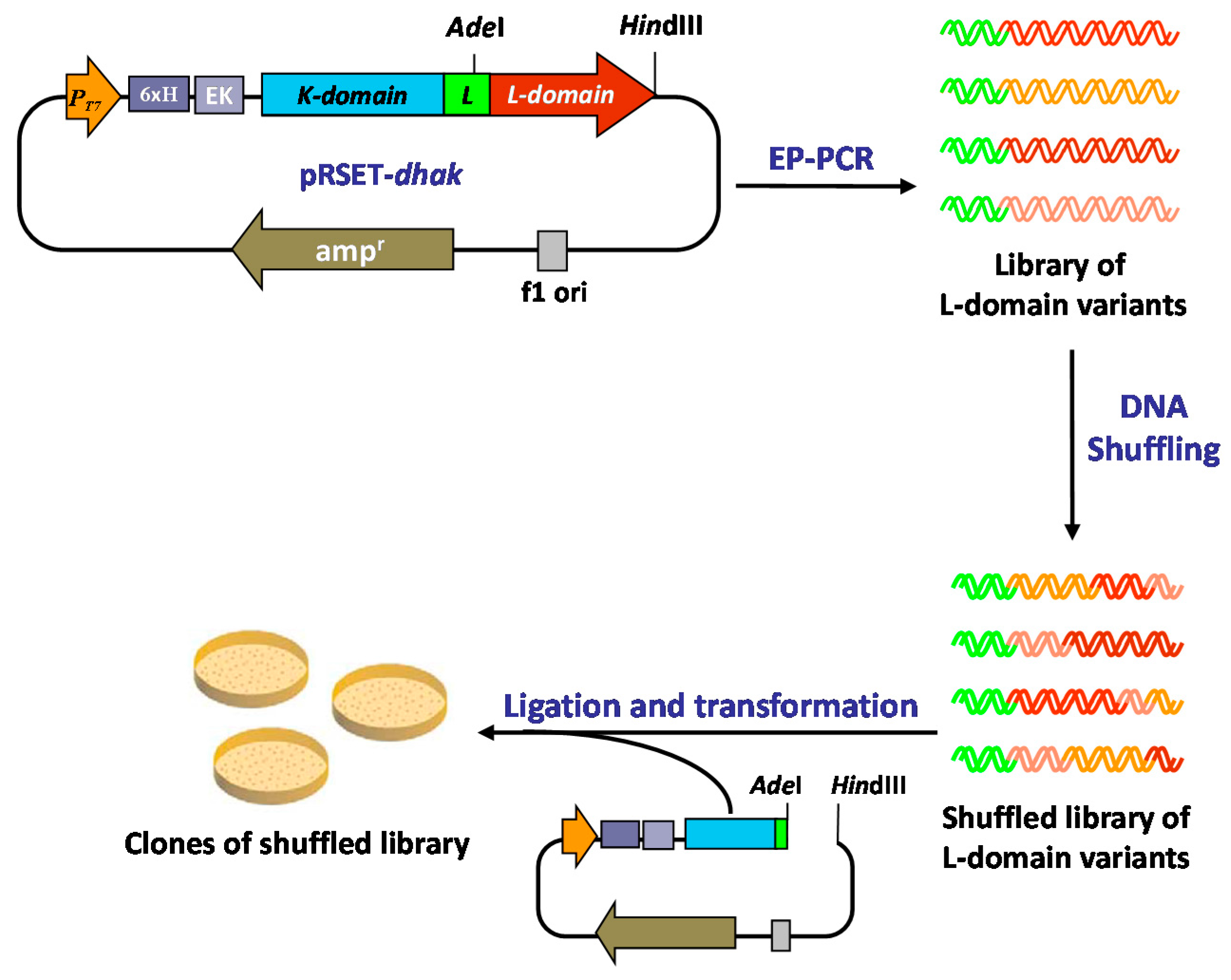
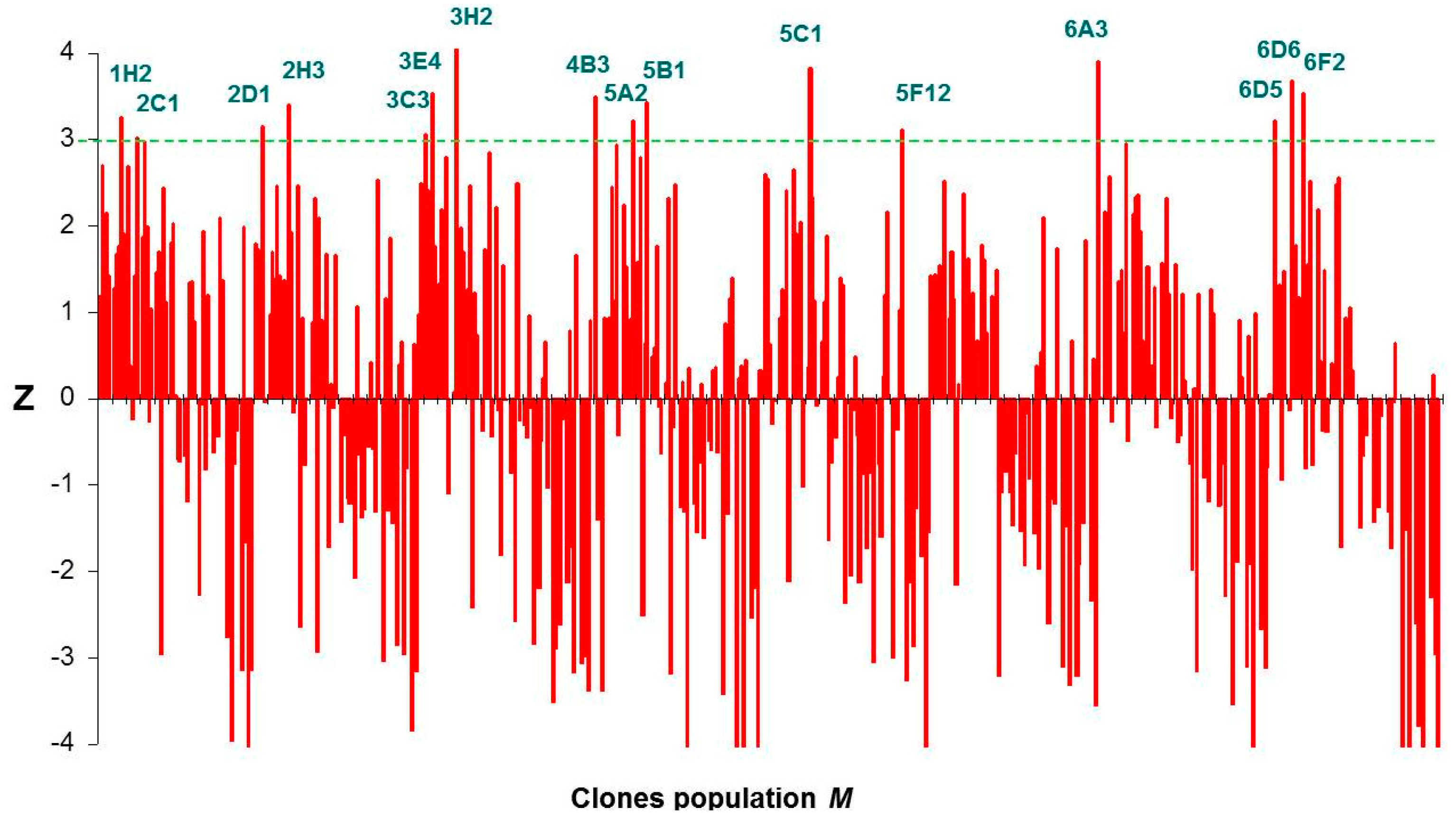
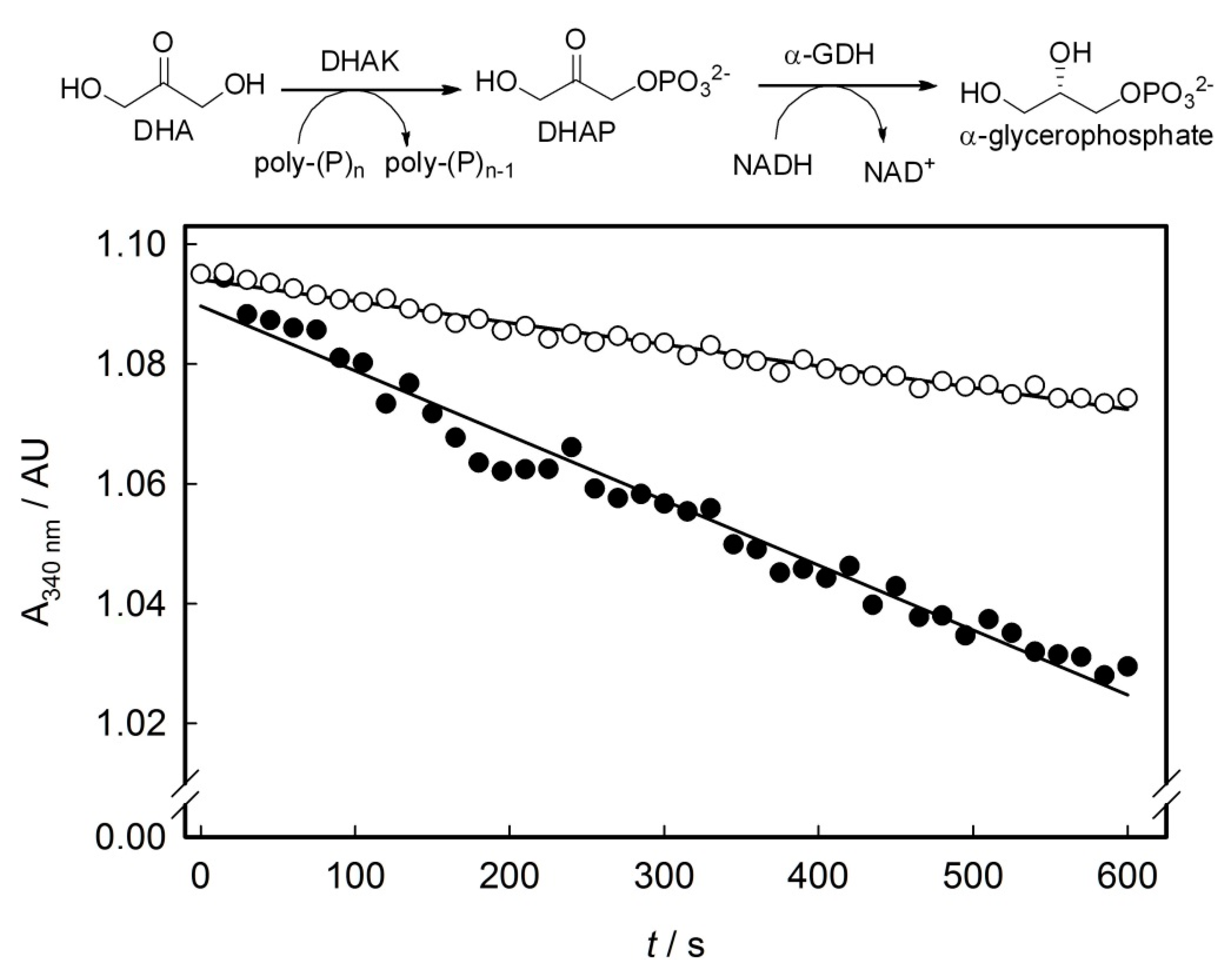
2.3. Screening of the DHAK Mutant Library

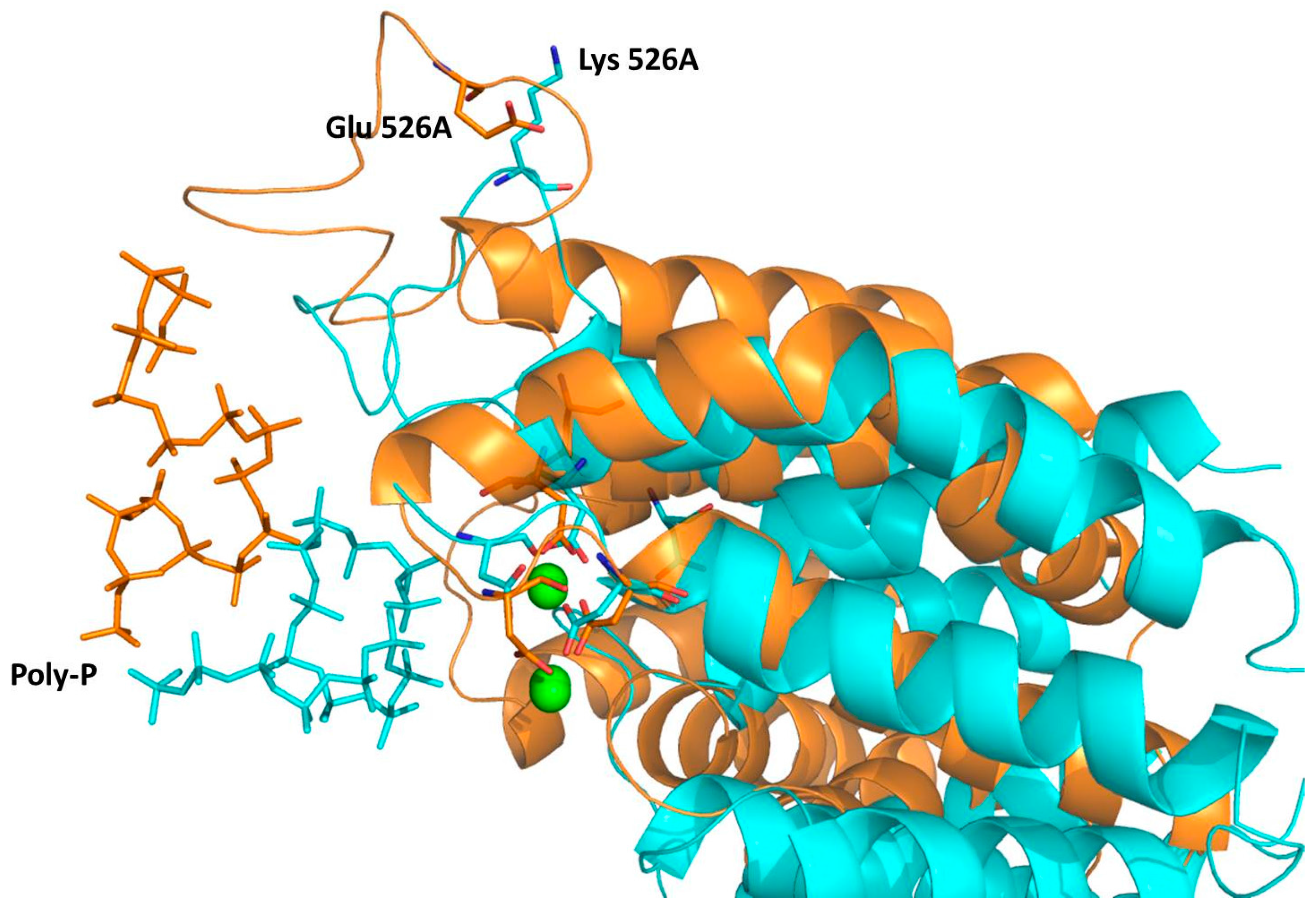
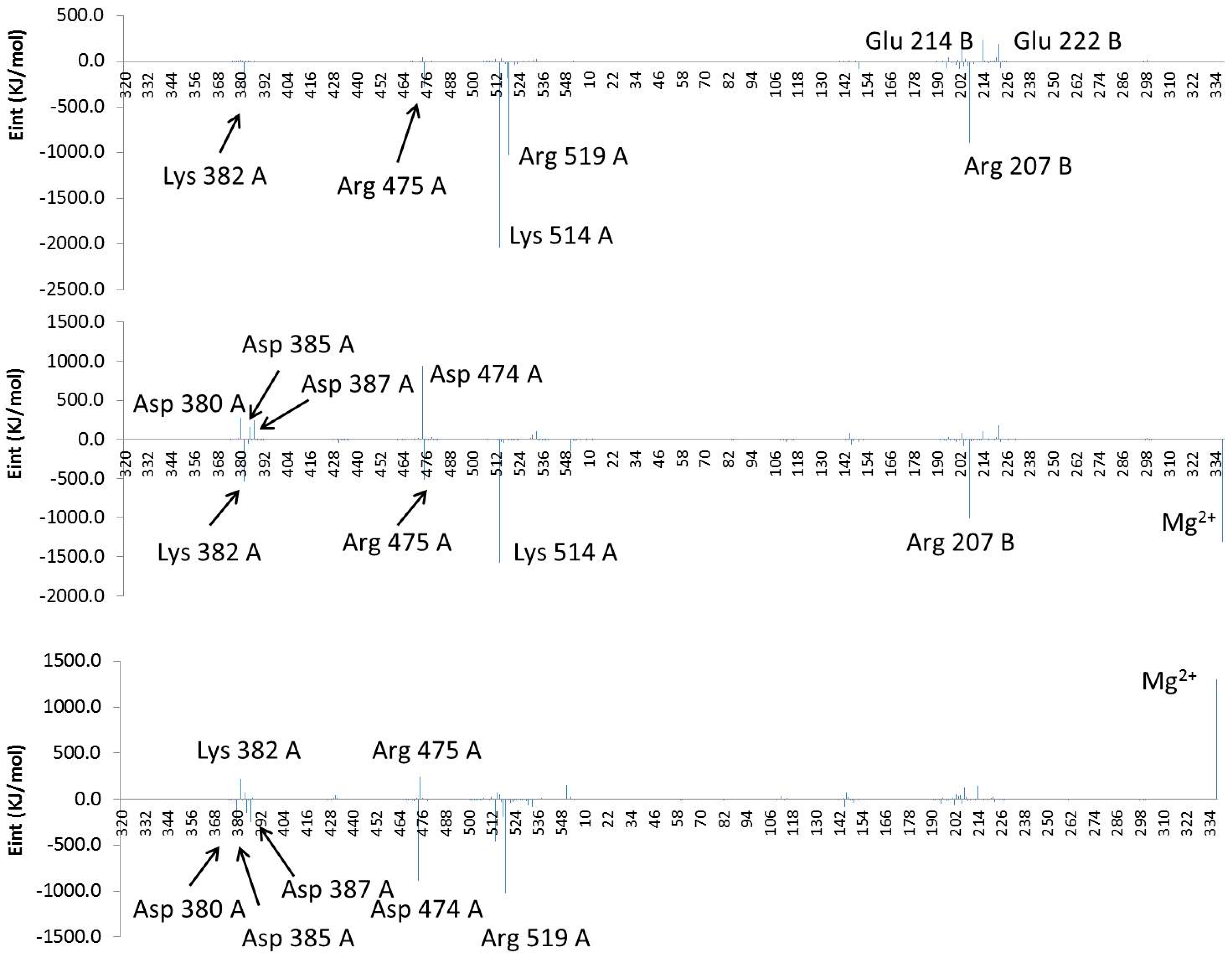
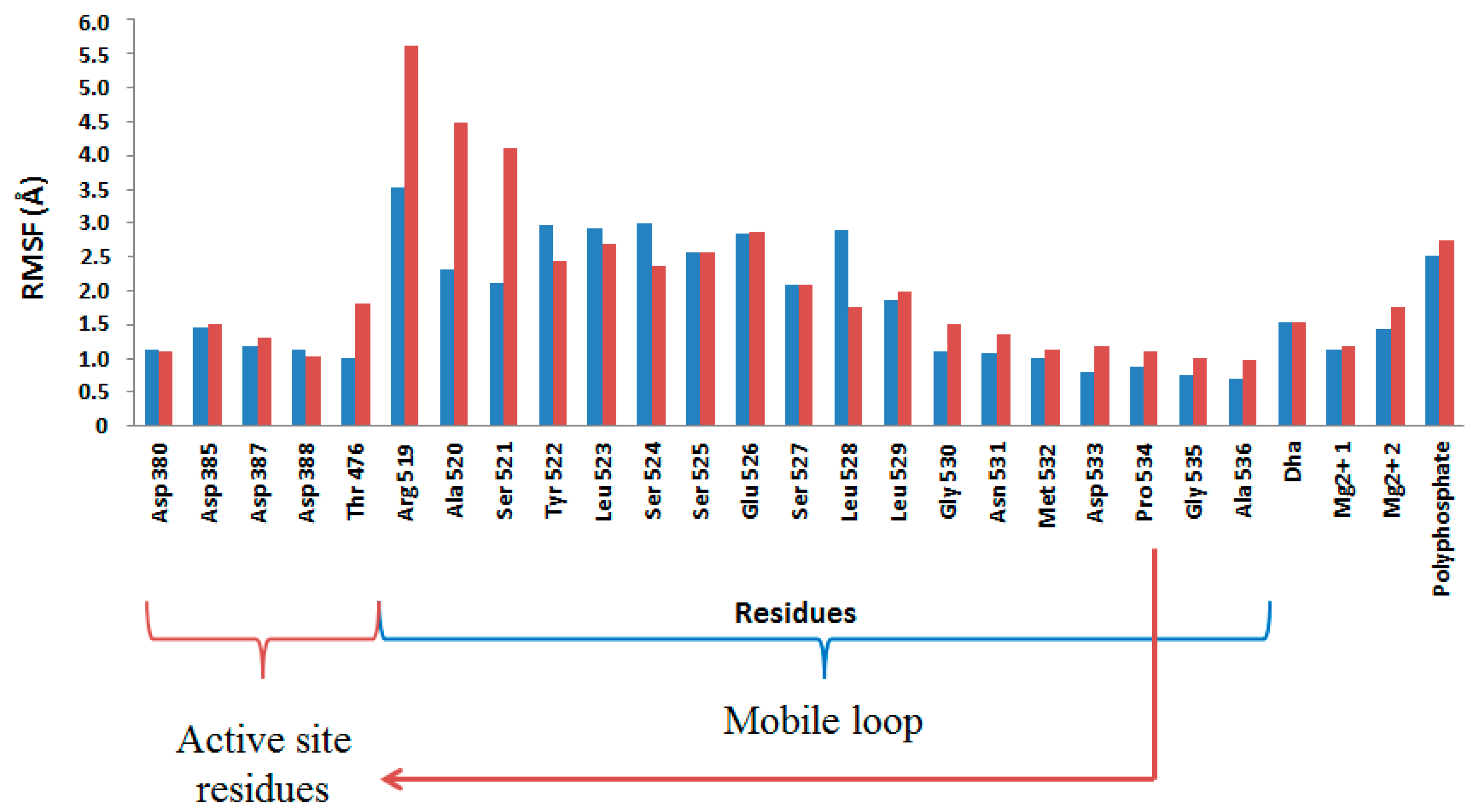
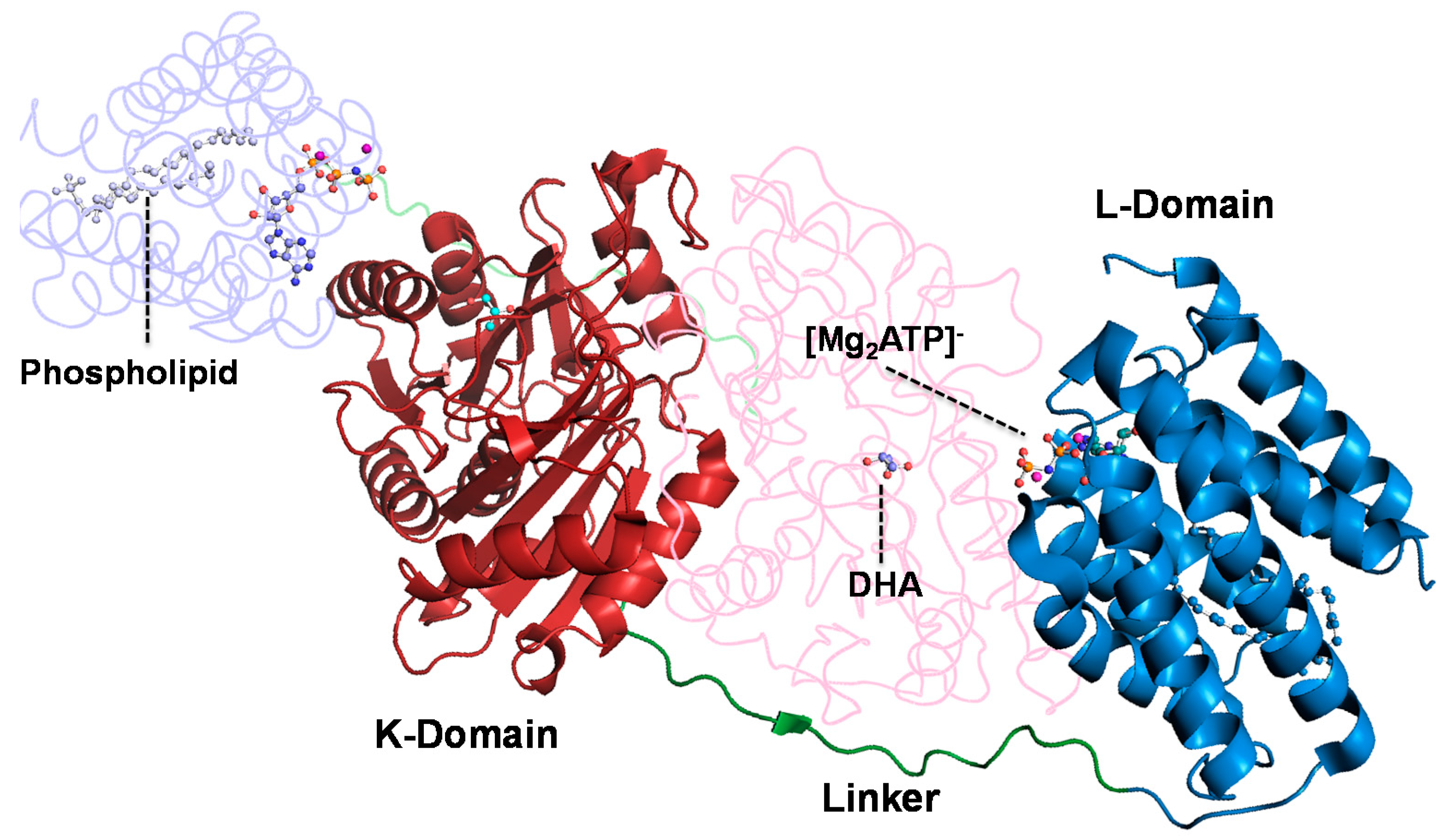
2.4. Computational Studies of the Wild-Type and the Mutant 1H2
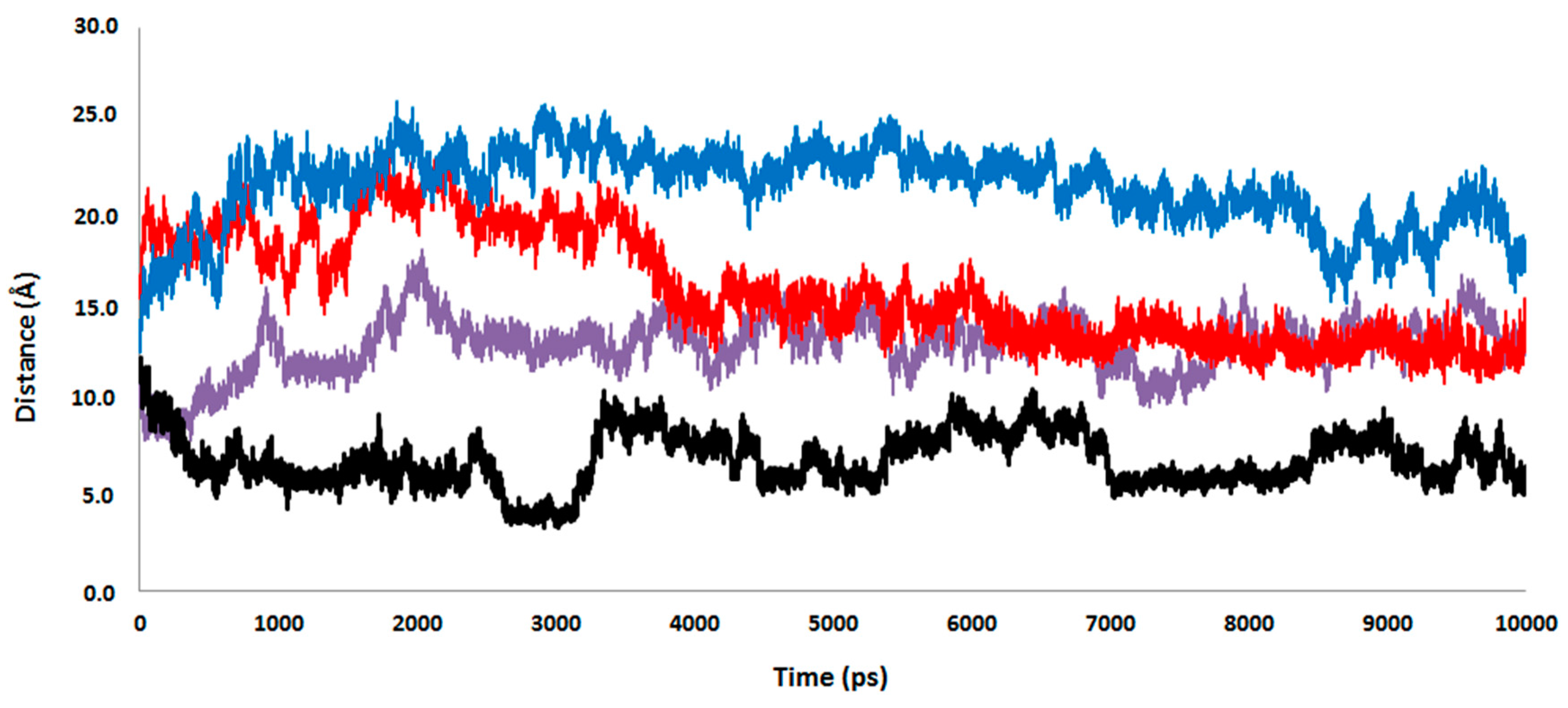
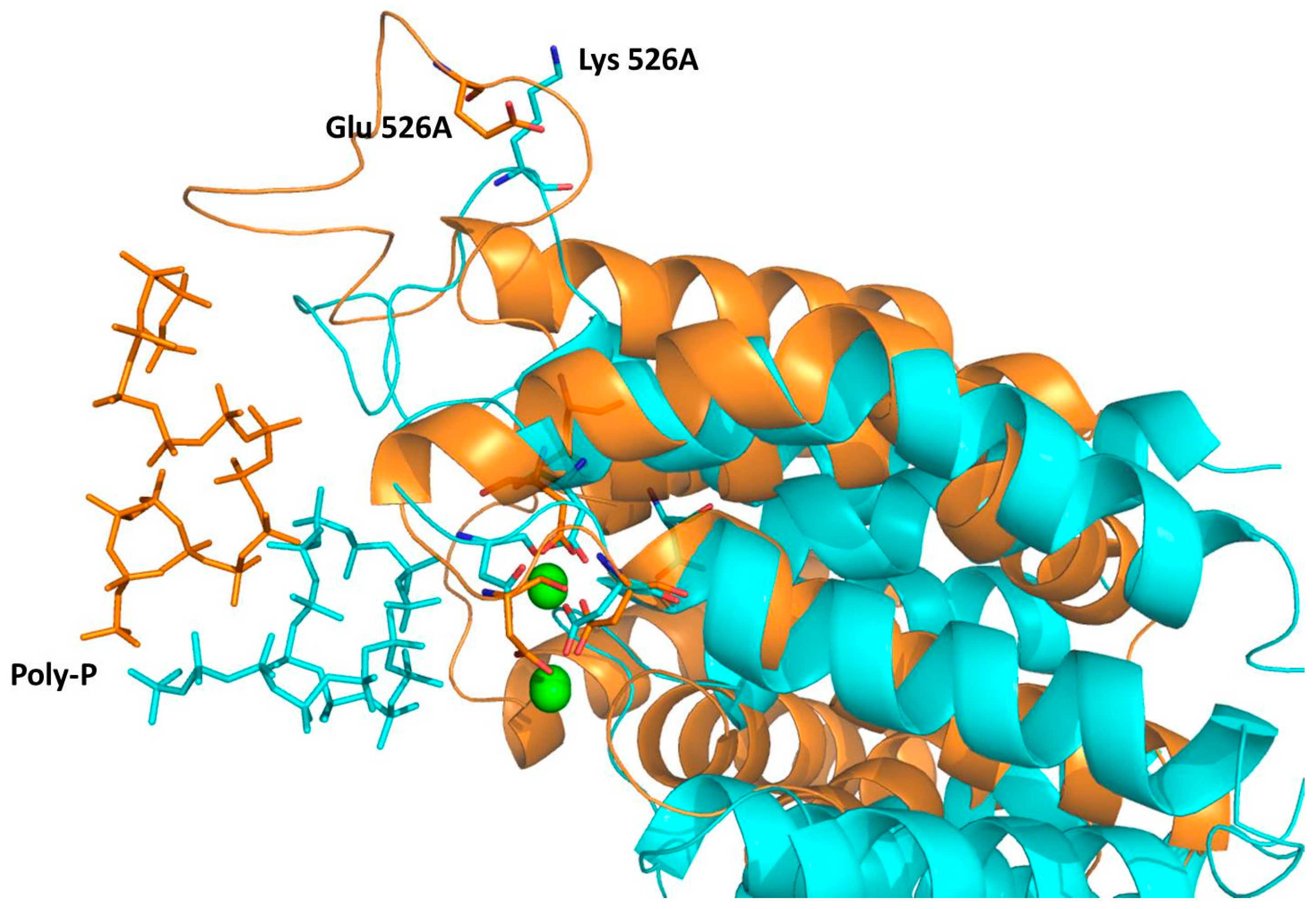
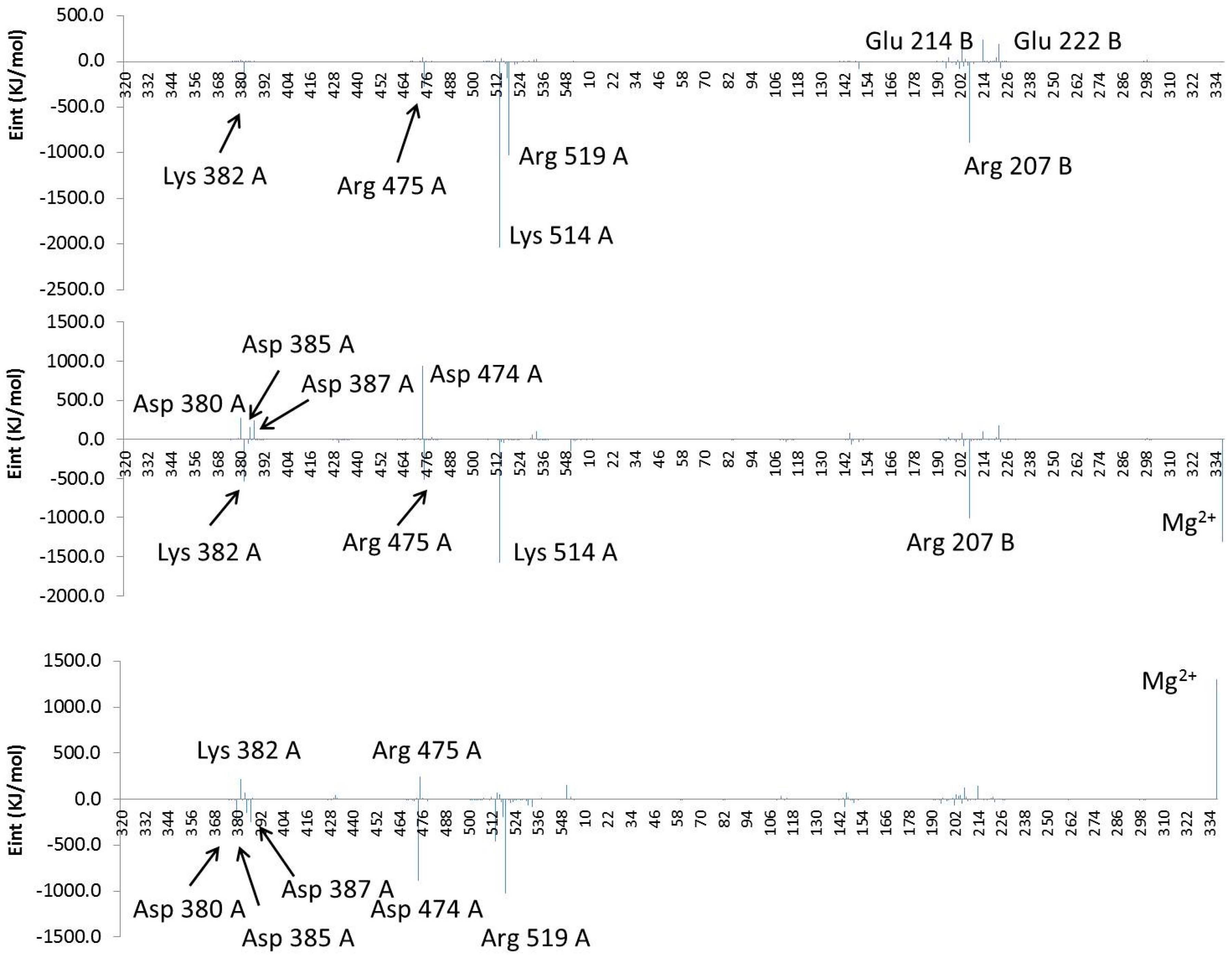
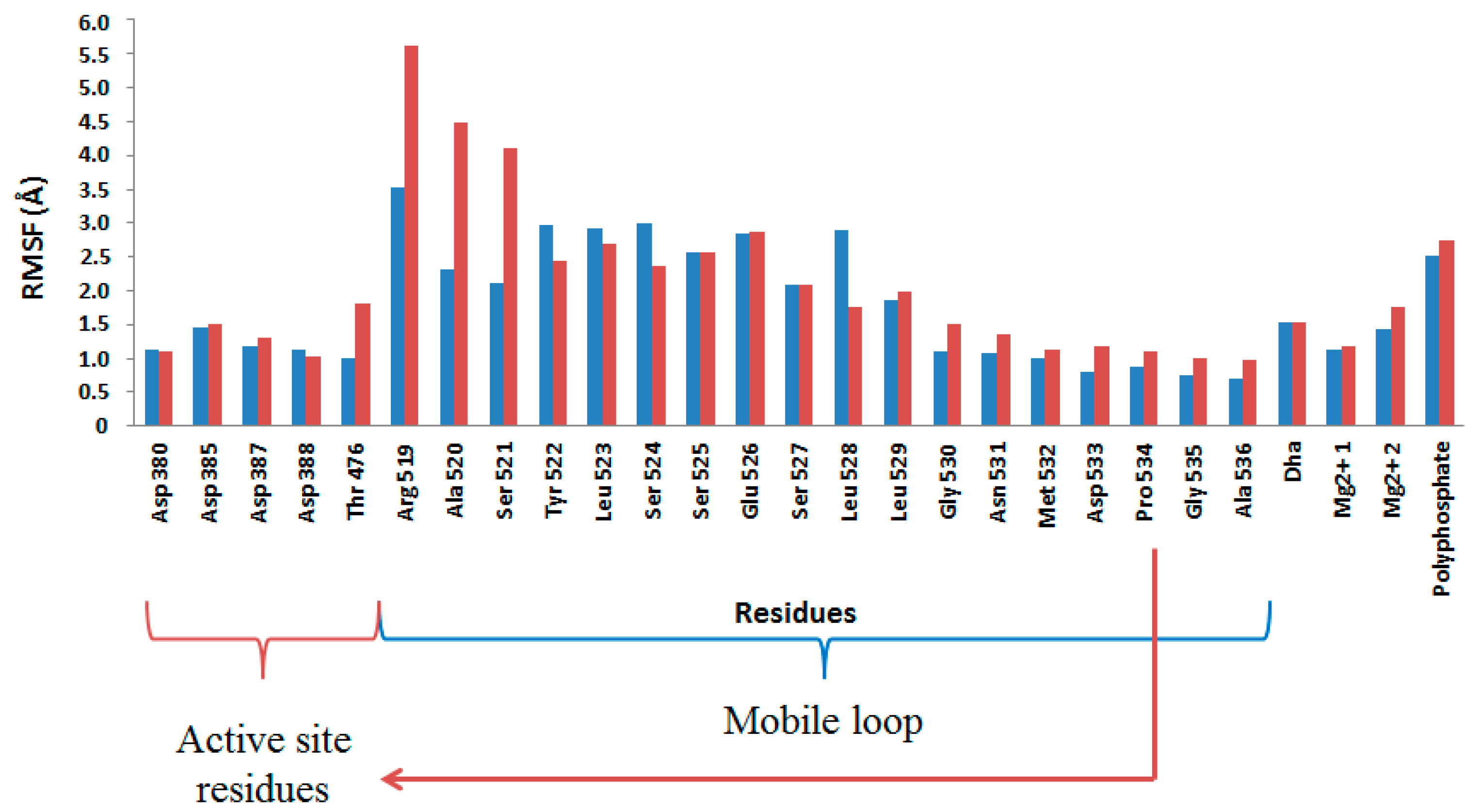
3. Experimental Section
3.1. Materials and General Procedures
3.2. Error-Prone PCR Generated Library
3.3. In Vitro Recombination by DNA Shuffling
3.4. Screening of DHAK Mutant Library
3.5. Expression and Purification of Selected Mutants

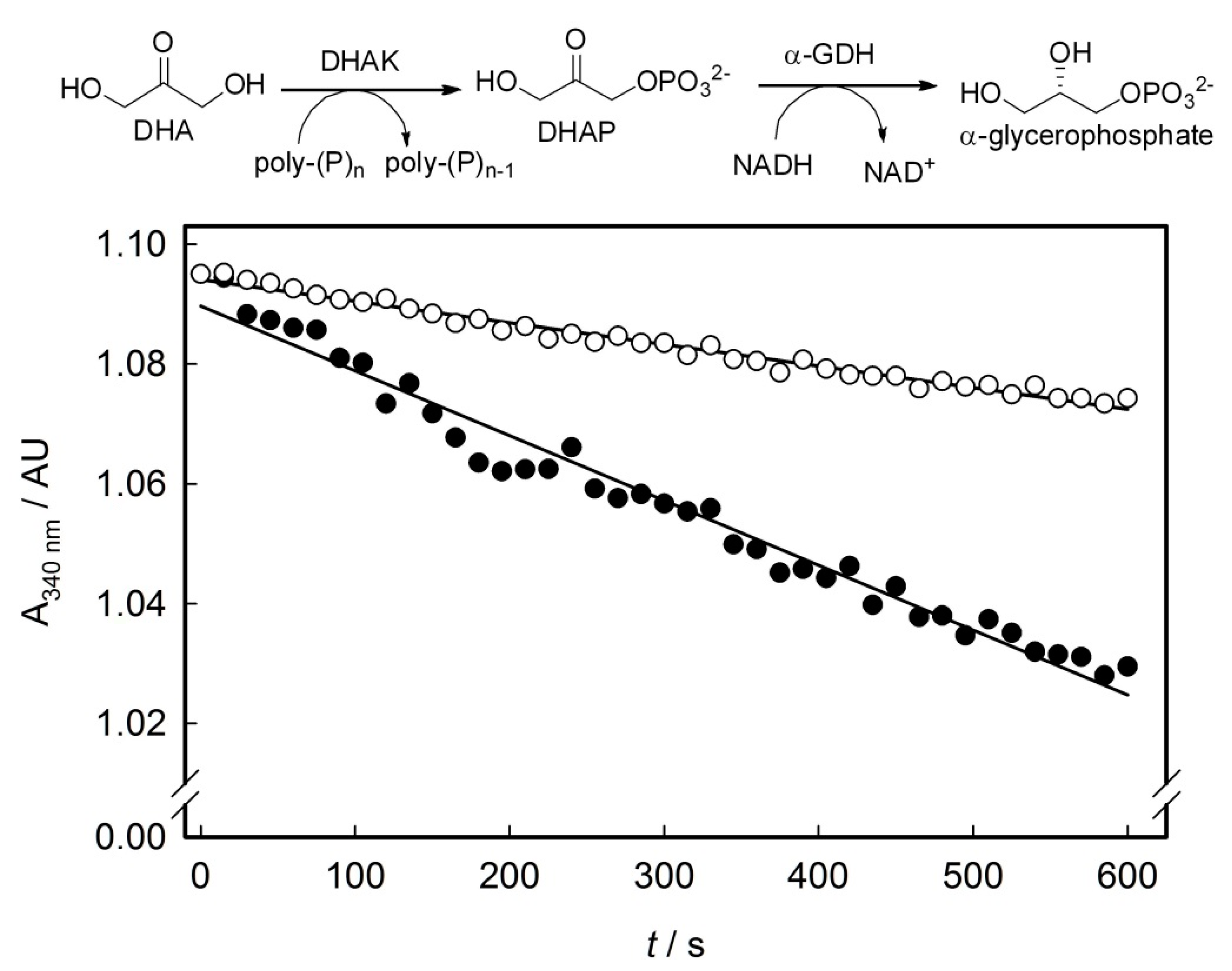
3.6. Enzyme Activity Assays of Purified Mutants and wtDHAK
3.7. Computational Model and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Enders, D.; Voith, M.; Lenzen, A. The dihydroxyacetone unit—A versatile C3 building block in organic synthesis. Angew. Chem. Int. Ed. 2005, 44, 1304–1325. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J. Organic reactions in aqueous media with a focus on carbon-carbon bond formations: A decade update. Chem. Rev. 2005, 105, 3095–3166. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Oiarbide, M.; García, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P. The direct catalytic asymmetric cross-aldol reaction of aldehydes. Angew. Chem. Int. Ed. 2003, 42, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Clapés, P.; Joglar, J. Enzyme-catalyzed aldol additions. In Modern Methods in Stereoselective Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 475–527. [Google Scholar]
- Fesko, K.; Gruber-Khadjawi, M. Biocatalytic methods for C–C bond formation. ChemCatChem 2013, 5, 1248–1272. [Google Scholar] [CrossRef]
- Müller, M. Recent developments in enzymatic asymmetric C–C bond formation. Adv. Synth. Catal. 2012, 354, 3161–3174. [Google Scholar] [CrossRef]
- Brovetto, M.; Gamenara, D.; Saenz Méndez, P.; Seoane, G.A. C–C bond-forming lyases in organic synthesis. Chem. Rev. 2011, 111, 4346–4403. [Google Scholar] [CrossRef] [PubMed]
- Clapés, P.; Fessner, W.-D.; Sprenger, G.A.; Samland, A.K. Recent progress in stereoselective synthesis with aldolases. Curr. Opin. Chem. Biol. 2010, 14, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Iturrate, L.; García-Junceda, E. DHAP-dependent aldolases in the core of multistep processes. In Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; García-Junceda, E., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 61–81. [Google Scholar]
- Schümperli, M.; Pellaux, R.; Panke, S. Chemical and enzymatic routes to dihydroxyacetone phosphate. Appl. Microbiol. Biotechnol. 2007, 75, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Moreno, I.; García-García, J.F.; Bastida, A.; García-Junceda, E. Multienzyme system for dihydroxyacetone phosphate-dependent aldolase catalyzed C–C bond formation from dihydroxyacetone. Chem. Commun. 2004, 14, 1634–1635. [Google Scholar]
- Sánchez-Moreno, I.; Iturrate, L.; Doyagüez, E.G.; Martínez, J.A.; Fernández-Mayoralas, A.; García-Junceda, E. Activated α,β-unsaturated aldehydes as substrate of dihydroxyacetone phosphate (DHAP)-dependent aldolases in the context of a multienzyme system. Adv. Synth. Catal. 2009, 351, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Camps Bres, F.; Guérard-Hélaine, C.; Hélaine, V.; Fernandes, C.; Sánchez-Moreno, I.; Traïkia, M.; García-Junceda, E.; Lemaire, M. l-Rhamnulose-1-phosphate and l-fuculose-1-phosphate aldolase mediated multi-enzyme cascade systems for nitrocyclitol synthesis. J. Mol. Catal. B: Enzym. 2015, 114, 50–57. [Google Scholar] [CrossRef]
- Oroz-Guinea, I.; Hernández, K.; Camps Bres, F.; Guérard-Hélaine, C.; Lemaire, M.; Clapés, P.; García-Junceda, E. l-Rhamnulose-1-phosphate aldolase from Thermotoga maritima in organic synthesis: One-pot multistep reactions for the preparation of imino- and nitrocyclitols. Adv. Synth. Catal. 2015, 357, 1951–1960. [Google Scholar] [CrossRef]
- Itoh, N.; Tujibata, Y.; Liu, J.Q. Cloning and overexpression in Escherichia coli of the gene encoding dihydroxyacetone kinase isoenzyme I from Schizosaccharomyces pombe, and its application to dihydroxyacetone phosphate production. Appl. Microbiol. Biotechnol. 1999, 51, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Iturrate, L.; Sanchez-Moreno, I.; Doyaguez, E.G.; Garcia-Junceda, E. Substrate channelling in an engineered bifunctional aldolase/kinase enzyme confers catalytic advantage for C–C bond formation. Chem. Commun. 2009, 13, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Iturrate, L.; Sánchez-Moreno, I.; Oroz-Guinea, I.; Pérez-Gil, J.; García-Junceda, E. Preparation and characterization of a bifunctional aldolase/kinase enzyme: A more efficient biocatalyst for C–C bond formation. Chem. Eur. J. 2010, 16, 4018–4030. [Google Scholar] [CrossRef] [PubMed]
- Siebold, C.; Arnold, I.; García-Alles, L.F.; Baumann, U.; Erni, B. Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded α-helical barrel ATP-binding domain. J. Biol. Chem. 2003, 278, 48236–48244. [Google Scholar] [CrossRef] [PubMed]
- Cheek, S.; Ginalski, K.; Zhang, H.; Grishin, N.V. A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 2005, 5. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Moreno, I.; Iturrate, L.; Martín-Hoyos, R.; Jimeno, M.L.; Mena, M.; Bastida, A.; García-Junceda, E. From Kinase to Cyclase: An unusual example of catalytic promiscuity modulated by metal switching. ChemBioChem 2009, 10, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A. Inorganic polyphosphate: Toward making a forgotten polymer unforgettable. J. Bacteriol. 1995, 177, 491–496. [Google Scholar] [PubMed]
- Kornberg, A.; Rao, N.N.; Ault-Riche, D. Inorganic polyphosphate: A molecule of many functions. Annu. Rev. Biochem. 1999, 68, 89–125. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Kornberg, A. Inorganic polyphosphate in the origin and survival of species. Proc. Natl. Acad. Sci. USA 2004, 101, 16085–16087. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.R.; Kornberg, A. The long and short of it—polyphosphate, PPK and bacterial survival. Trends Biochem. Sci. 2008, 33, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.N.; Gómez-García, M.R.; Kornberg, A. Inorganic polyphosphate: Essential for growth and survival. Annu. Rev. Biochem. 2009, 78, 605–647. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, P.A.; Dieter, L.; Rittle, K.J.; Shepler, L.S.; Paszkowskiy, A.L.; Moe, O.A. Catalytic properties of Escherichia coli polyphosphate kinase: An enzyme for ATP regeneration. Biotechnol. Appl. Biochem. 1992, 15, 125–133. [Google Scholar] [PubMed]
- Ferrer, S.; Martí, S.; Andrés, J.; Moliner, V.; Tuñón, I.; Bertrán, J. Molecular mechanism of promiscuous MbtI. Improving the secondary activity by a computational design. Theor. Chem. Acc. 2011, 128, 601–607. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef] [PubMed]
- Martí, S.; Andrés, J.; Moliner, V.; Silla, E.; Tuñón, I.; Bertrán, J. Computer-aided rational design of catalytic antibodies: The 1F7 case. Angew. Chem. Int. Ed. 2007, 46, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Martí, S.; Andrés, J.; Moliner, V.; Silla, E.; Tuñón, I.; Bertrán, J. Predicting an improvement of secondary catalytic activity of promiscuous isochorismate pyruvate lyase by computational design. J. Am. Chem. Soc. 2008, 130, 2894–2895. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Chen, E.; Goeddel, D.V. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique 1989, 1, 11–15. [Google Scholar]
- Cirino, P.C.; Mayer, K.M.; Umeno, D. Generating mutant libraries using error-prone PCR. In Methods in Molecular Biology, Volume 231: Directed Evolution Library Creation; Arnold, F.H., Georgiou, G., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2003; pp. 3–9. [Google Scholar]
- McCullum, E.O.; Williams, B.A.R.; Zhang, J.; Chaput, J.C. Random mutagenesis by error-prone PCR. In Methods in Molecular Biology, Volume 634: In Vitro Mutagenesis Protocols, 3rd ed.; Braman, J., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2010; pp. 103–109. [Google Scholar]
- Rasila, T.S.; Pajunen, M.I.; Savilahti, H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal. Biochem. 2009, 388, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kvam, P.H.; Vidakovic, B. Nonparametric Statistics with Applications to Science and Engineering; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Stemmer, W.P.C. DNA shuffling by random fragmentation and reassembly: In vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 10747–10751. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, W.P.C. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput.-Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins: Struct. Funct. Bioinf. 2008, 73, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct. Funct. Bioinf. 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Field, M.J. A Practical Introduction to the Simulation of Molecular Systems; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comp. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tiradorives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Pranata, J.; Wierschke, S.G.; Jorgensen, W.L. OPLS potential functions for nucleotide bases. Relative association constants of hydrogen-bonded base pairs in chloroform. J. Am. Chem. Soc. 1991, 113, 2810–2819. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Moreno, I.; Bordes, I.; Castillo, R.; Ruiz-Pernía, J.J.; Moliner, V.; García-Junceda, E. Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies. Int. J. Mol. Sci. 2015, 16, 27835-27849. https://doi.org/10.3390/ijms161126073
Sánchez-Moreno I, Bordes I, Castillo R, Ruiz-Pernía JJ, Moliner V, García-Junceda E. Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies. International Journal of Molecular Sciences. 2015; 16(11):27835-27849. https://doi.org/10.3390/ijms161126073
Chicago/Turabian StyleSánchez-Moreno, Israel, Isabel Bordes, Raquel Castillo, José Javier Ruiz-Pernía, Vicent Moliner, and Eduardo García-Junceda. 2015. "Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies" International Journal of Molecular Sciences 16, no. 11: 27835-27849. https://doi.org/10.3390/ijms161126073
APA StyleSánchez-Moreno, I., Bordes, I., Castillo, R., Ruiz-Pernía, J. J., Moliner, V., & García-Junceda, E. (2015). Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies. International Journal of Molecular Sciences, 16(11), 27835-27849. https://doi.org/10.3390/ijms161126073