
DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia


Abstract
:
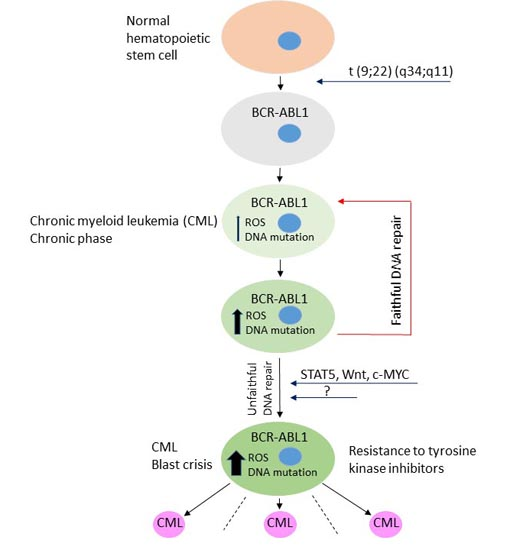{kind=link}
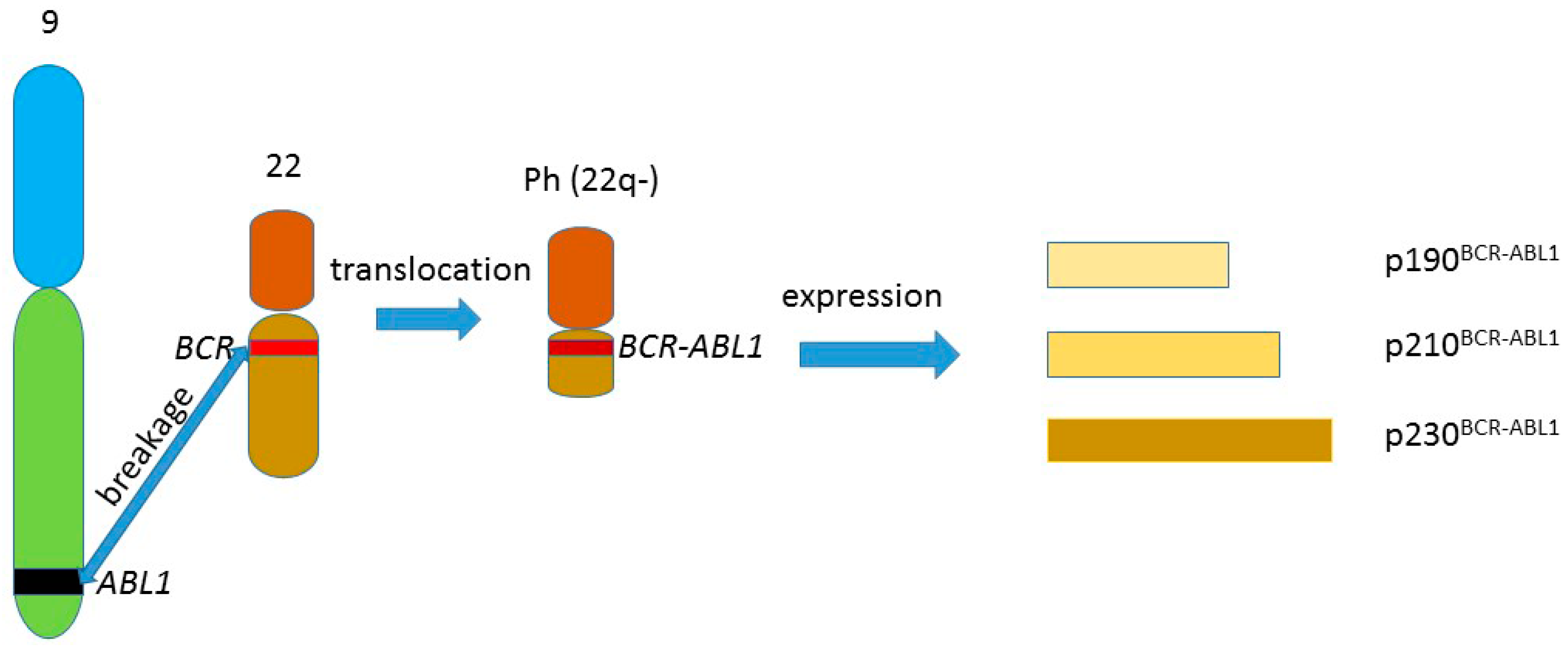{kind=link}
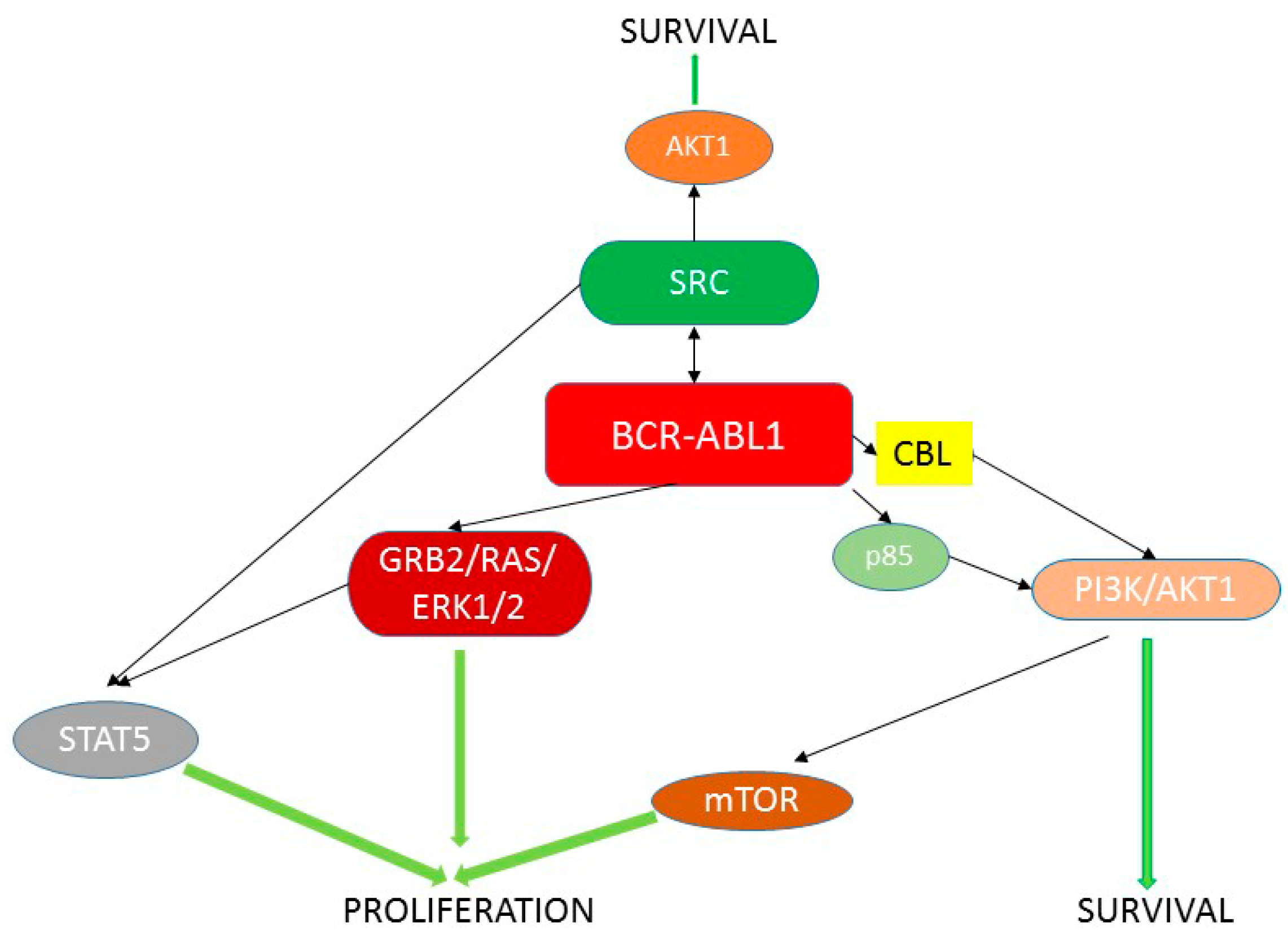{kind=link}
{kind=link}
{kind=link}
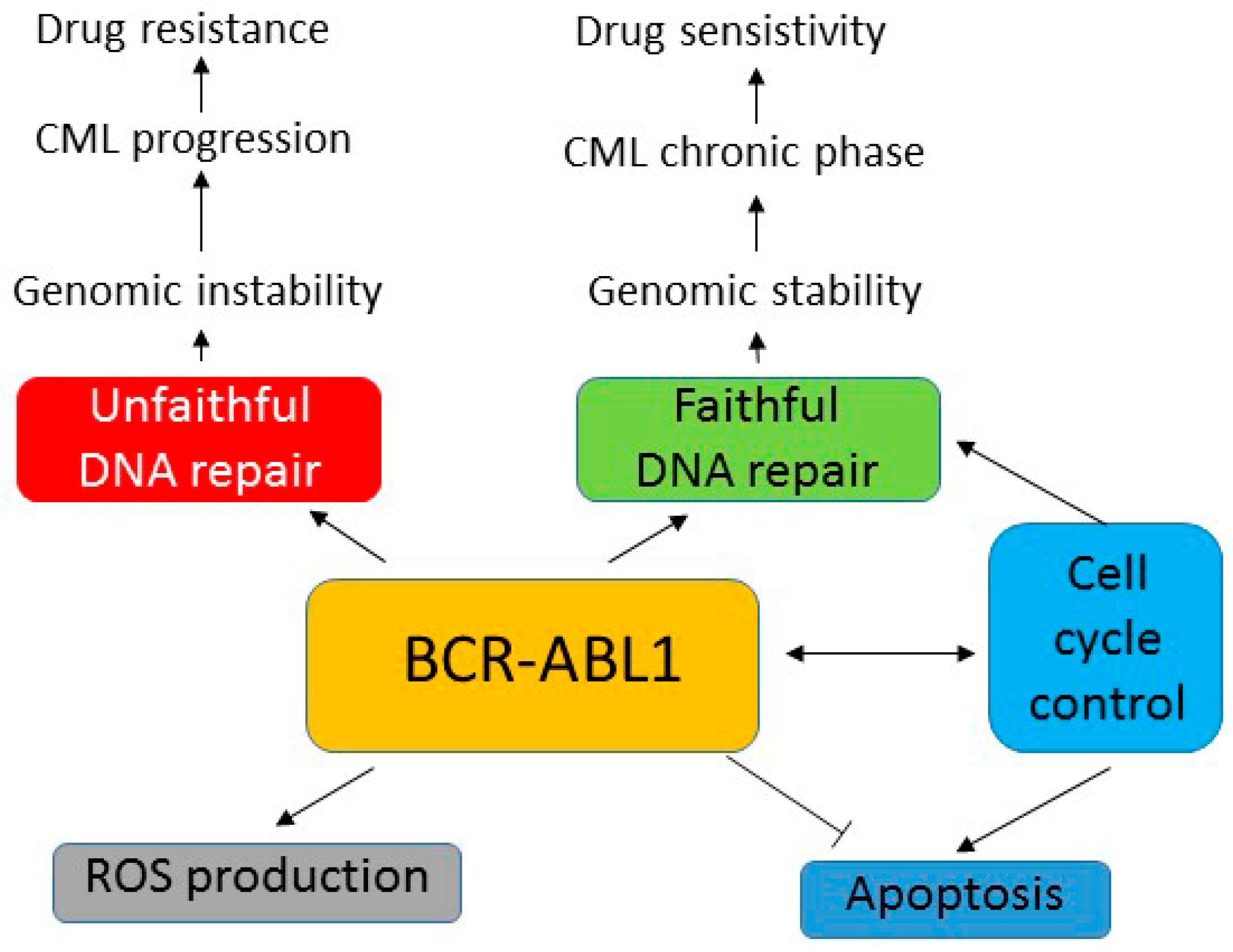{kind=link}
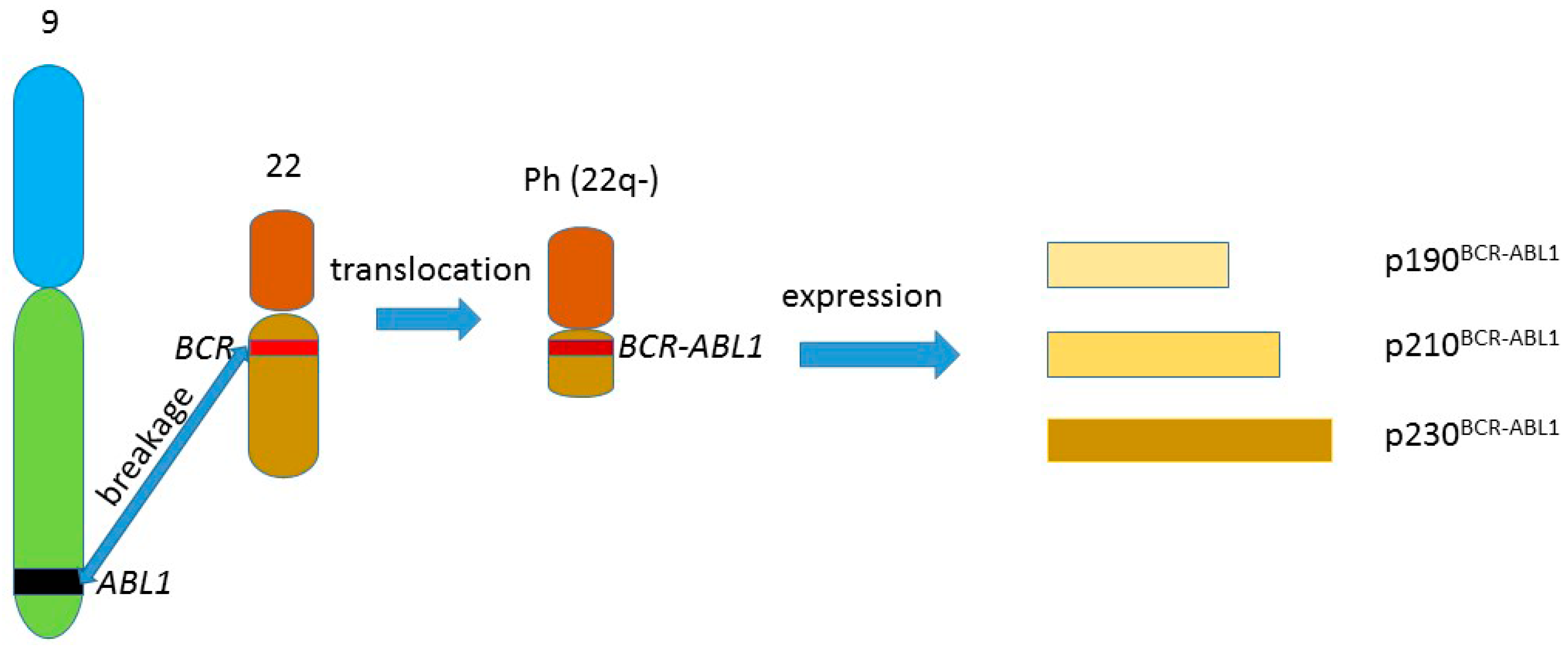
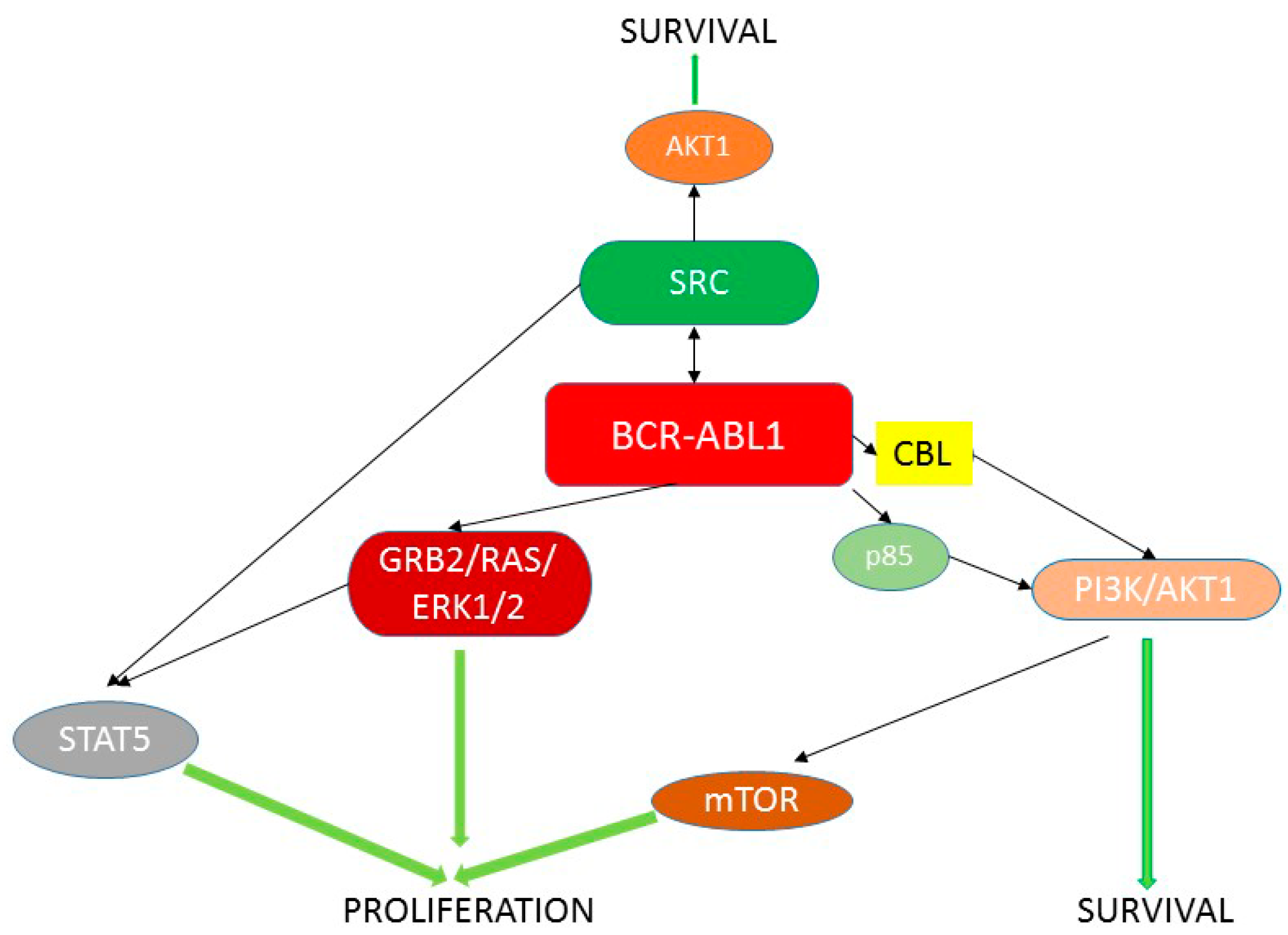
1. BCR-ABL1 and Chronic Myeloid Leukemia
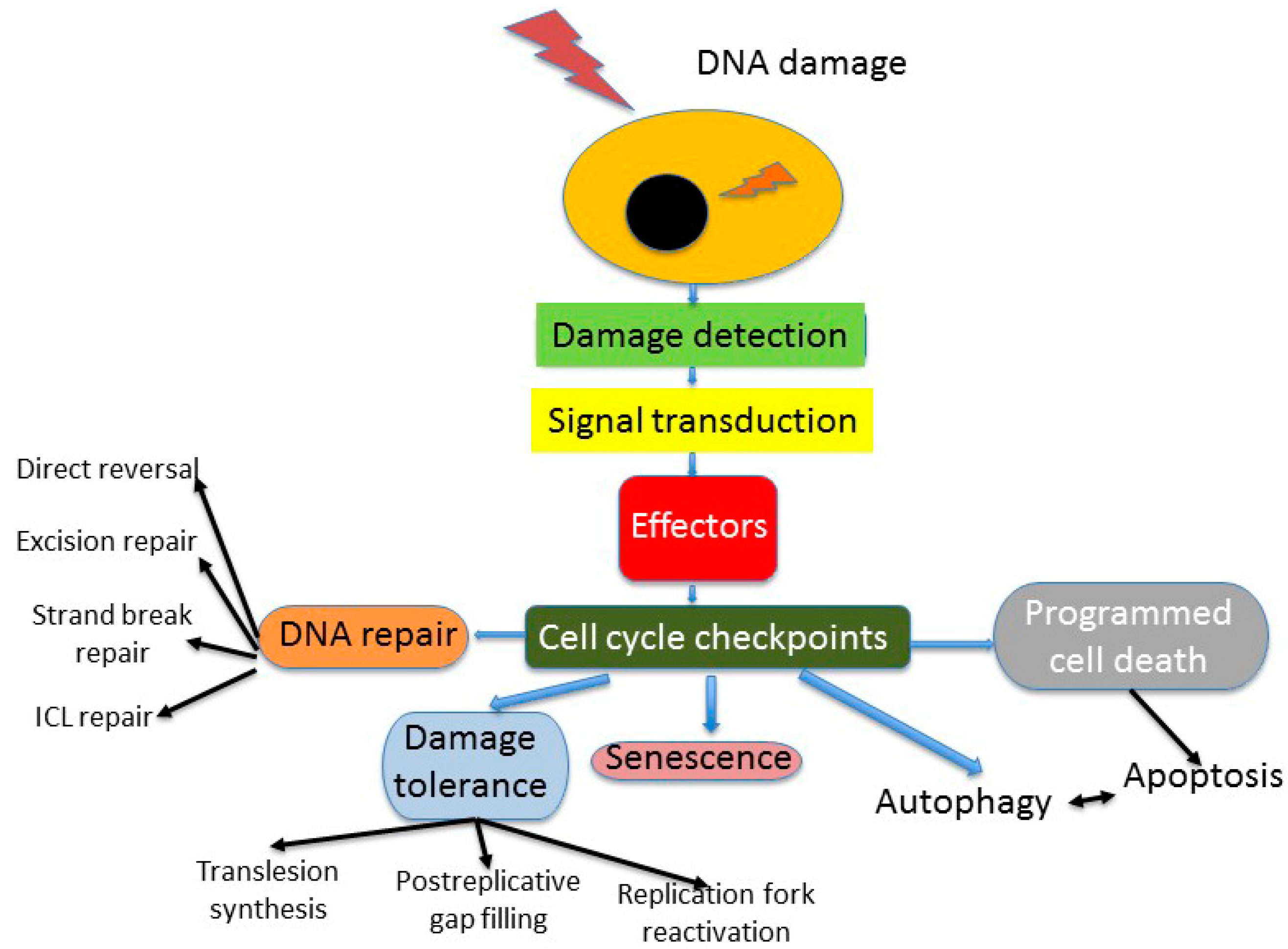
2. DNA Repair in Cellular DNA Damage Response
3. Genomic Instability—A Common Feature of Cancer Cells

4. DNA Repair in Cancer Cells
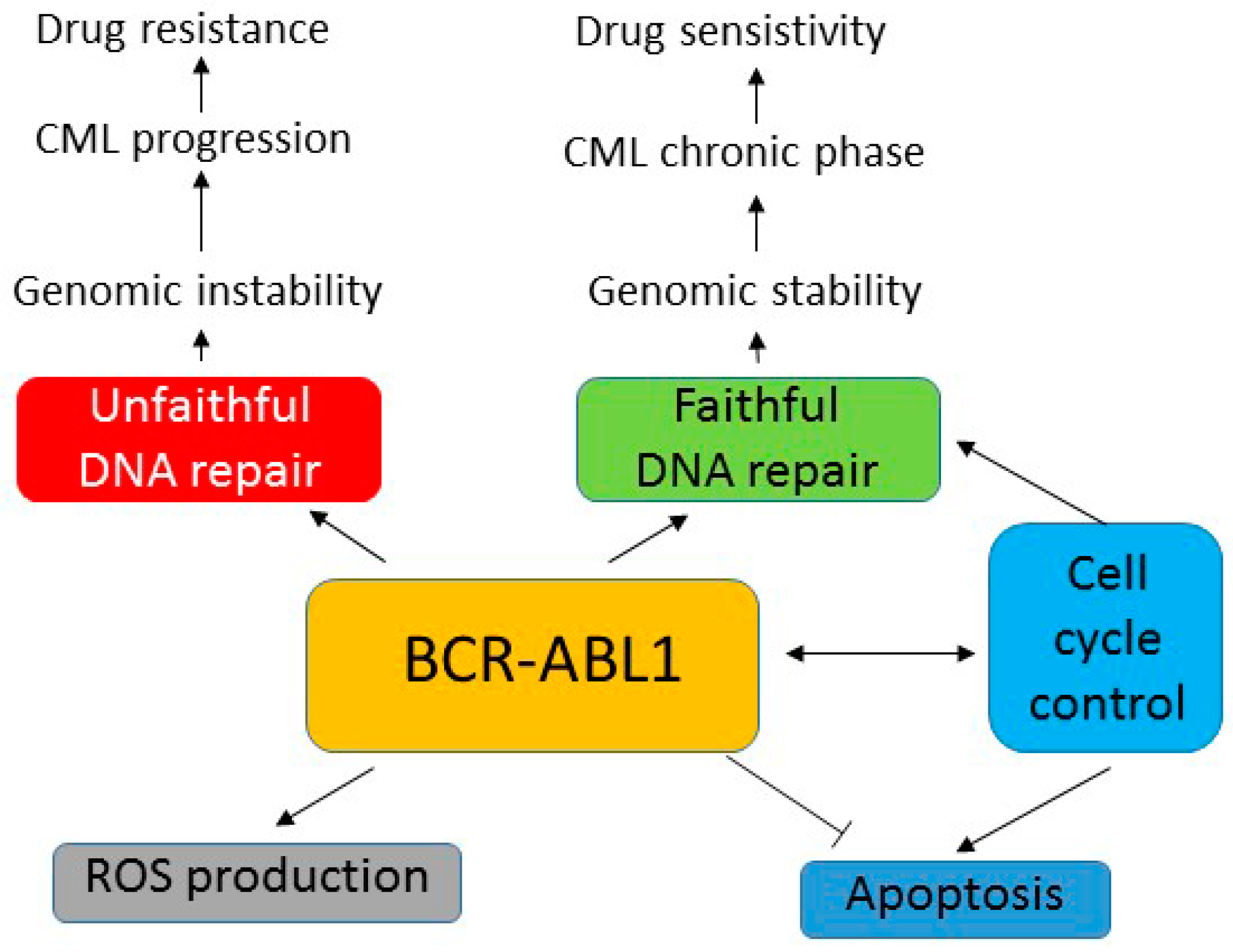
5. BCR-ABL1 Can Induce Genomic Instability Independently of Its Leukemogenic Effect
6. Role of BCR-ABL1 in Mutagenesis—ROS Production and Unfaithful DNA Repair
7. Aberrant DNA Repair in BCR-ABL1-Expressing Cells
8. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deininger, M.W.; Vieira, S.; Mendiola, R.; Schultheis, B.; Goldman, J.M.; Melo, J.V. BCR-ABL tyrosine kinase activity regulates the expression of multiple genes implicated in the pathogenesis of chronic myeloid leukemia. Cancer Res. 2000, 60, 2049–2055. [Google Scholar] [PubMed]
- Quentmeier, H.; Eberth, S.; Romani, J.; Zaborski, M.; Dexler, G.H. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J. Hematol. Oncol. 2011, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Maino, E.; Sancetta, R.; Viero, P.; Imbergamo, S.; Scattolin, A.M.; Vespignani, M.; Bassan, R. Current and future management of Ph/BCR-ABL positive ALL. Expert Rev. Anticancer Ther. 2014, 14, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Al-Barrak, J.; Cheung, W.Y. Adherence to imatinib therapy in gastrointestinal stromal tumors and chronic myeloid leukemia. Support. Care Cancer 2013, 21, 2351–2357. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Keating, M.J.; Talpaz, M.; Walters, R.S.; Smith, T.L.; Cork, A.; McCredie, K.B.; Freireich, E.J. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am. J. Med. 1987, 83, 445–454. [Google Scholar] [CrossRef]
- Glowacki, S.; Synowiec, E.; Blasiak, J. The role of mitochondrial DNA damage and repair in the resistance of BCR/ABL-expressing cells to tyrosine kinase inhibitors. Int. J. Mol. Sci. 2013, 14, 16348–16364. [Google Scholar] [CrossRef] [PubMed]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Bixby, D.; Talpaz, M. Seeking the causes and solutions to imatinib-resistance in chronic myeloid leukemia. Leukemia 2011, 25, 7–22. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, T.; Eide, C.A.; Deininger, M.W. BCR-ABL kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Mauro, M.J.; Martinelli, G.; Kim, D.W.; Soverini, S.; Müller, M.C.; Hochhaus, A.; Cortes, J.; Chuah, C.; Dufva, I.H.; et al. Epidemiologic study on survival of chronic myeloid leukemia and Ph(+) acute lymphoblastic leukemia patients with BCR-ABL T315I mutation. Blood 2009, 114, 5271–5278. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, N.; Frank, D.A.; Griffin, J.D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med. 1996, 183, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Halpern, J.; ten Hoeve, J.; Rao, X.; Sawyers, C.L. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996, 13, 247–254. [Google Scholar] [PubMed]
- Ilaria, R.L., Jr.; van Etten, R.A. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 1996, 271, 31704–31710. [Google Scholar] [CrossRef] [PubMed]
- Wrasch, W.; Grudschober, E.; berger, A.; Gille, L.; Cerny-Reiterer, S.; Tigan, A.S.; Hoelbl-Kovacic, A.; Valent, P.; Moriggi, R.; Sexl, V. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget 2012, 3, 1669–1687. [Google Scholar] [PubMed]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol. Allergy Clin. N. Am. 2014, 34, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, Y.; Fang, S.; Wang, L.; Qi, H.; Zhang, Y.; Zhang, J.; Li, W. BCR/ABL oncogene-induced PI3K signaling pathway leads to chronic myeloid leukemia pathogenesis by impairing immuno-modulatory function of hemangioblasts. Cancer Gene Ther. 2015, 22, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Scheller, M.; Schönheit, J.; Zimmermann, K.; Leser, U.; Rosenbauer, F.; Leutz, A. Cross talk between Wnt/β-catenin and Irf8 in leukemia progression and drug resistance. J. Exp. Med. 2013, 210, 2239–2256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Baydoun, H.H.; Pancewicz, J.; Nicot, C. Human T-lymphotropic type 1 virus p30 inhibits homologous recombination and favors unfaithful DNA repair. Blood 2011, 17, 5897–5906. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Soulas-Sprauel, P.; Rivera-Munoz, P.; Malivert, L.; le Guyader, G.; Abramowski, V.; Revy, P.; de Villartay, J.P. V(D)J and immunoglobulin class switch recombinations: A paradigm to study the regulation of DNA end-joining. Oncogene 2007, 26, 7780–7791. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Atsumi, Y.; Nakagama, H.; Teraoka, H. Development of cancer-initiating cells and immortalized cells with genomic instability. World J. Stem Cells 2015, 7, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Recovery of antediluvian DNA. Nature 1993, 365, 700. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Biol. 2011, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yu, H. The Smc complexes in DNA damage response. Cell Biosci. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.A.; Dhanaraman, T.; D’Amours, D. The Smc5–Smc6 heterodimer associates with DNA through several independent binding domains. Sci. Rep. 2015, 5, 9797. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; van der Linden, E.; Sanchez, H.; Wyman, C. RAD50, an Smc family member with multiple roles in DNA break repair: How does ATP affect function? Chromosome Res. 2009, 17, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Nasmyth, K. Segregating sister genomes: The molecular biology of chromosome separation. Science 2002, 297, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Smc proteins and chromosome mechanics: From bacteria to humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmidt, C.; Woodcock, M.; Stevens, D.L.; Hill, M.A.; Rothkamm, K.; Helleday, T. Cohesin phosphorylation and mobility of Smc1 at ionizing radiation-induced DNA double-strand breaks in human cells. Exp. Cell Res. 2011, 317, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human Smc5/6 complex promotes sister chromatid homologous recombination by recruiting the Smc1/3 cohesin complex to double-strand breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef] [PubMed]
- Schär, P.; Fäsi, M.; Jessberger, R. Smc1 coordinates DNA double-strand break repair pathways. Nucleic Acids Res. 2004, 32, 3921–3929. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Li, Y.; Zhang, J.; Xi, Y.; Li, Y.; Yang, T.; Jung, S.Y.; Pan, X.; Chen, R.; Li, W.; et al. Genome-wide reinforcement of cohesin binding at pre-existing cohesin sites in response to ionizing radiation in human cells. J. Biol. Chem. 2010, 285, 22784–22792. [Google Scholar] [CrossRef] [PubMed]
- Gligoris, T.G.; Scheinost, J.C.; Bürmann, F.; Petela, N.; Chan, K.L.; Uluocak, P.; Beckouët, F.; Gruber, S.; Nasmyth, K.; Löwe, J. Closing the cohesin ring: Structure and function of its Smc3-kleisin interface. Science 2014, 346, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.D.; Song, Q.; Li, C.W.; Chen, J.; Tang, H.M.; Peng, Z.H.; Wang, X.C. Structural maintenance of chromosomes 4 is a predictor of survival and a novel therapeutic target in colorectal cancer. Asian Pac. J. Cancer Prev. 2014, 15, 9459–9465. [Google Scholar] [CrossRef] [PubMed]
- Morawiec, Z.; Janik, K.; Kowalski, M.; Stetkiewicz, T.; Szaflik, J.; Morawiec-Bajda, A.; Sobczuk, A.; Blasiak, J. DNA damage and repair in children with Down’s syndrome. Mutat. Res. 2008, 637, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Ayed, W.; Gouas, L.; Penault-Llorca, F.; Amouri, A.; Tchirkov, A.; Vago, P. Trisomy 21 and cancers. Morphologie 2012, 96, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Weckselblatt, B.; Rudd, M.K. Human structural variation: Mechanism of chromosome rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Niu, H.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.A.; Strande, N.T.; Wyatt, D.W.; Pryor, J.M.; Ramsden, D.A. Nonhomologous end joining: A good solution for bad ends. DNA Repair 2014, 17, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Martín-López, J.V.; Fishel, R. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam. Cancer 2013, 12, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers 2015, 7, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.J.; Lehmann, P.Z.; Li, W. Nuclear PI3K signaling in cell growth and tumorigenesis. Front. Cell Dev. Biol. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Sipos, F.; Fűri, I.; Constantinovits, M.; Tulassay, Z.; Műzes, G. Contribution of TLR signaling to the pathogenesis of colitis-associated cancer in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 12713–12721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, S.; Kakizaki, M.; Hirose, Y.; Ishikawa, Y.; Funato, H.; Yanagisawa, M.; Yu, Y.; Liu, Q. Orexin/hypocretin activates mTOR complex 1 (mTORC1) via an Erk/Akt-independent and calcium–stimulated lysosome v-ATPase pathway. J. Biol. Chem. 2014, 289, 31950–31959. [Google Scholar] [CrossRef] [PubMed]
- Salloukh, H.F.; Laneuville, P. Increase in mutant frequencies in mice expressing the BCR-ABL activated tyrosine kinase. Leukemia 2000, 14, 1401–1404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brain, J.M.; Saksena, A.; Laneuville, P. The kinase inhibitor STI571 reverses the BCR/ABL induced point mutation frequencies observed in pre-leukemic P190BCR/ABL transgenic mice. Leuk. Res. 2001, 26, 1011–1016. [Google Scholar] [CrossRef]
- Kharbanda, S.; Ren, R.; Pandey, P.; Shafman, T.D.; Feller, S.M.; Weichselbaum, R.R.; Kufe, D.W. Activation of the c-ABL tyrosine kinase in the stress response to DNA-damaging agents. Nature 1995, 376, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.M.; Huang, Y.; Fan, M.M.; Sawyers, C.; Kharbanda, S.; Kufe, D. Genotoxic drugs induce interaction of the c-ABL tyrosine kinase and the tumor suppressor protein p53. J. Biol. Chem. 1996, 271, 26457–26460. [Google Scholar] [PubMed]
- Shafman, T.; Khanna, K.K.; Kedar, P.; Spring, K.; Kozlov, S.; Yen, T.; Hobson, K.; Gatei, M.; Zhang, N.; Watters, D.; et al. Interaction between ATM protein and c-ABL in response to DNA damage. Nature 1997, 387, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.M.; Huang, Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Kharbanda, S.; Wang, R.; Sung, P.; Shinohara, A.; Weichselbaum, R.; et al. Regulation of Rad51 function by c-ABL in response to DNA damage. J. Biol. Chem. 1998, 273, 3799–3802. [Google Scholar] [CrossRef] [PubMed]
- Gesbert, F.; Sellers, W.R.; Signoretti, S.; Loda, M.; Griffin, J.D. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J. Biol. Chem. 2000, 275, 39223–39230. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.; Sun, G.; Doorly, M.; Wang, J.Y.; Schwartz, M.A. Modulation of DNA damage-induced apoptosis by cell adhesion is independently mediated by p53 and c-ABL. Proc. Natl. Acad. Sci. USA 2003, 100, 10281–10286. [Google Scholar] [CrossRef] [PubMed]
- Canitrot, Y.; Lautier, D.; Laurent, G.; Fréchet, M.; Ahmed, A.; Turhan, A.G.; Salles, B.; Cazaux, C.; Hoffmann, J.S. Mutator phenotype of BCR-ABL transfected Ba/F3 cell lines and its association with enhanced expression of DNA polymerase β. Oncogene 1999, 18, 2676–2680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Canitrot, Y.; Laurent, G.; Astarie-Dequeker, C.; Bordier, C.; Cazaux, C.; Hoffmann, J.S. Enhanced expression and activity of DNA polymerase β in chronic myelogenous leukemia. Anticancer Res. 2006, 26, 523–525. [Google Scholar] [PubMed]
- Nieborowska-Skorska, M.; Flis, S.; Skorski, T. AKT-induced reactive oxygen species generate imatinib-resistant clones emerging from chronic myeloid leukemia progenitor cells. Leukemia 2014, 28, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Hoser, G.; Hochhaus, A.; Stoklosa, T.; Skorski, T. Anti-oxidant vitamin E prevents accumulation of imatinib-resistant BCR-ABL1 kinase mutations in CML-CP xenografts in NSG mice. Leukemia 2013, 27, 2253–2254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slupianek, A.; Falinski, R.; Znojek, P.; Stoklosa, T.; Flis, S.; Doneddu, V.; Pytel, D.; Synowiec, E.; Blasiak, J.; Bellacosa, A.; et al. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia 2013, 27, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Kopinski, P.K.; Ray, R.; Hoser, G.; Ngaba, D.; Flis, S.; Cramer, K.; Reddy, M.M.; Koptyra, M.; Penserga, T.; et al. Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors. Blood 2012, 119, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef] [PubMed]
- Flis, K.; Irvine, D.; Copland, M.; Bhatia, R.; Skorski, T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk. Lymphoma 2012, 53, 2474–2478. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sorel, N.; Bonnet, M.L.; Guillier, M.; Guilhot, F.; Brizard, A.; Turhan, A.G. Evidence of ABL-kinase domain mutations in highly purified primitive stem cell populations of patients with chronic myelogenous leukemia. Biochem. Biophys. Res. Commun. 2004, 323, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Singh, A.; Khosravi-Far, R. FoxO tumor suppressors and BCR-ABL-induced leukemia: A matter of evasion of apoptosis. Biochim. Biophys. Acta 2008, 1785, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Gloc, E.; Mlynarski, W.; Drzewoski, J.; Skorski, T. Amifostine differentially modulates DNA damage evoked by idarubicin in normal and leukemic cells. Leuk. Res. 2002, 26, 1093–1096. [Google Scholar] [CrossRef]
- Hoser, G.; Majsterek, I.; Romana, D.E.; Slupianek, A.; Blasiak, J.; Skorski, T. Fusion oncogenic tyrosine kinases alter DNA damage and repair after genotoxic treatment: Role in drug resistance. Leuk. Res. 2003, 27, 267–273. [Google Scholar] [CrossRef]
- Majsterek, I.; Slupianek, A.; Hoser, G.; Skorski, T.; Blasiak, J. ABL-fusion oncoproteins activate multi-pathway of DNA repair: Role in drug resistance? Biochimie 2004, 86, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Hoser, G.; Majsterek, I.; Bronisz, A.; Malecki, M.; Blasiak, J.; Fishel, R.; Skorski, T. Fusion tyrosine kinases induce therapeutic drug resistance by stimulation of homology-dependent recombination repair, prolongation of G2/M phase and protection from apoptosis. Mol. Cell. Biol. 2002, 22, 4189–4201. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Stoklosa, T.; Datta, M.; Czechowska, A.; Rink, L.; Słupianek, A.; Koptyra, M.; Seferyncka, I.; Krszyna, K.; Blasiak, J.; et al. ATR-Chk1 axis protects BCR/ABL leukemia cells from the lethal effect of DNA double-strand breaks. Cell Cycle 2006, 5, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Wood, L.D.; Whitaker, L.L.; Canman, C.E.; Morgan, S.E.; Xu, Y.; Barlow, C.; Baltimore, D.; Wynshaw-Boris, A.; Kastan, M.B.; et al. Ataxia telangiectasia mutant protein activates c-ABL tyrosine kinase in response to ionizing radiation. Nature 1997, 387, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Sato, M.; Piao, J.; Miyamoto, S.; Isoda, T.; Kitagawa, M.; Honda, H.; Mizutani, S. ATM-dependent DNA damage-response pathway as a determinant in chronic myelogenous leukemia. DNA Repair 2013, 12, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Sears, D.; Luong, P.; Yuan, M.; Nteliopoulos, G.; Man, Y.K.; Melo, J.V.; Basu, S. Functional phosphoproteomic analysis reveals cold-shock domain protein A to be a BCR-ABL effector-regulating proliferation and transformation in chronic myeloid leukemia. Cell. Death Dis. 2010, 1, e93. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, S.; Yuan, Z.M.; Weichselbaum, R.; Kufe, D. Determination of cell fate by c-ABL activation in response to DNA damage. Oncogene 1998, 17, 3309–3318. [Google Scholar] [CrossRef] [PubMed]
- Brady, N.; Gaymes, T.J.; Cheung, M.; Mufti, G.J.; Rassool, F.V. Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res. 2003, 63, 1798–1805. [Google Scholar] [PubMed]
- Nowicki, M.O.; Falinski, R.; Koptyra, M.; Slupianek, A.; Stoklosa, T.; Gloc, E.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood 2004, 104, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar] [PubMed]
- Shibata, A.; Jeggo, P.A. DNA double-strand break repair in a cellular context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Johnston, C.; Reeves, W.H.; Carter, T.; Wyche, J.H.; Hendrickson, E.A. Characterization of a Ku86 variant protein that results in altered DNA binding and diminished DNA-dependent protein kinase activity. J. Biol. Chem. 1996, 271, 14098–14104. [Google Scholar] [PubMed]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [PubMed]
- Kabotyanski, E.B.; Gomelsky, L.; Han, J.O.; Stamato, T.D.; Roth, D.B. Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Res. 1998, 26, 5333–5342. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Fan, J.; Rassool, F.V. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008, 270, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Dugray, A.; AbdulKarim, B.; Marangoni, E.; Maggiorella, L.; Vaganay, S.; M’Kacher, R.; Rasy, S.D.; Eschwege, F.; Vainchenker, W.; et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood 2001, 97, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Slupianek, A.; Schmutte, C.; Tombline, G.; Nieborowska-Skorska, M.; Hoser, G.; Nowicki, M.O.; Pierce, A.J.; Fishel, R.; Skorski, T. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol. Cell 2001, 8, 795–806. [Google Scholar] [CrossRef]
- Pastwa, E.; Poplawski, T.; Czechowska, A.; Malinowski, M.; Blasiak, J. Non-homologous DNA end joining repair in normal and leukemic cells depends on the substrate ends. Z. Naturforsch. C 2005, 60, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Muvarak, N.; Kelley, S.; Robert, C.; Baer, M.R.; Perrotti, D.; Gambacorti-Passerini, C.; Civin, C.; Scheibner, K.; Rassool, F.V. c-MYC generates repair errors via increased transcription of alternative-NHEJ factors, LIG3 and PARP1, in tyrosine kinase-activated leukemias. Mol. Cancer Res. 2015, 13, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlowska, E.; Blasiak, J. DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2015, 16, 27535-27549. https://doi.org/10.3390/ijms161126049
Pawlowska E, Blasiak J. DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2015; 16(11):27535-27549. https://doi.org/10.3390/ijms161126049
Chicago/Turabian StylePawlowska, Elzbieta, and Janusz Blasiak. 2015. "DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia" International Journal of Molecular Sciences 16, no. 11: 27535-27549. https://doi.org/10.3390/ijms161126049
APA StylePawlowska, E., & Blasiak, J. (2015). DNA Repair—A Double-Edged Sword in the Genomic Stability of Cancer Cells—The Case of Chronic Myeloid Leukemia. International Journal of Molecular Sciences, 16(11), 27535-27549. https://doi.org/10.3390/ijms161126049