
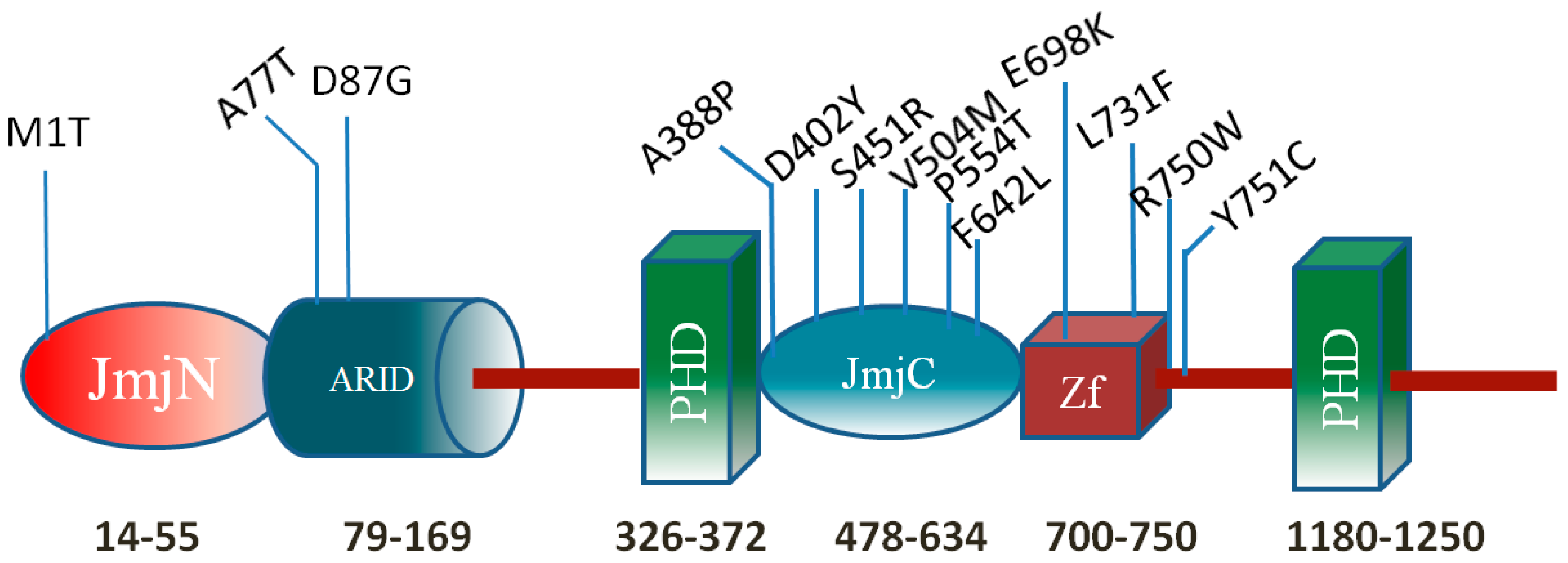
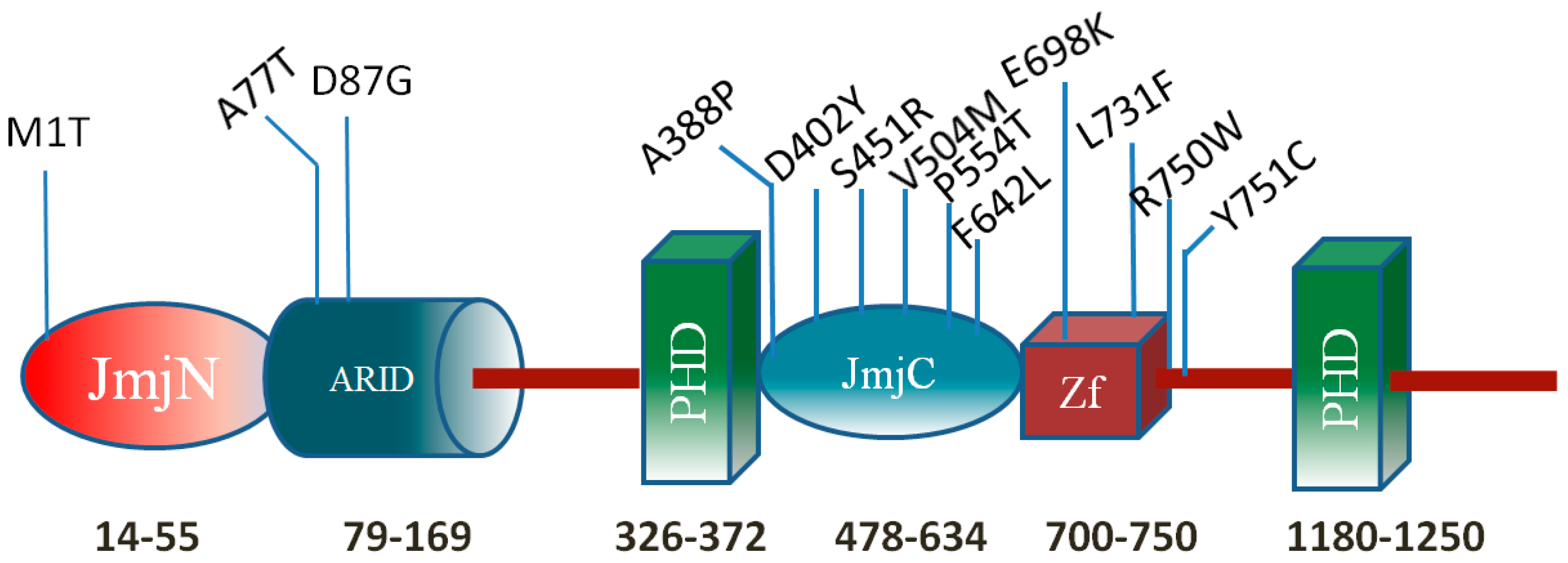
Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease


Abstract
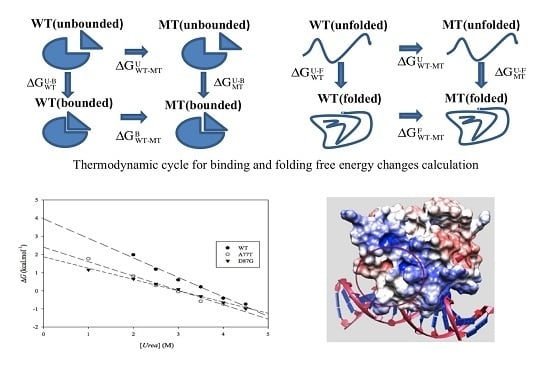
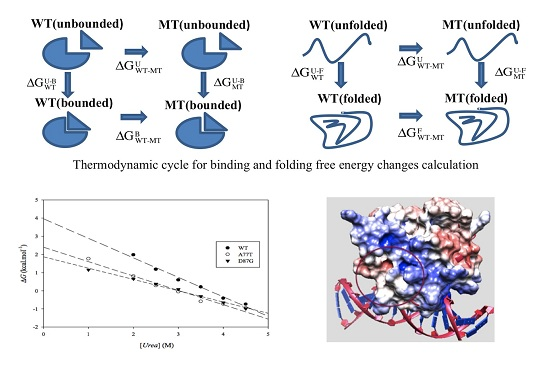
:
1. Introduction
2. Results
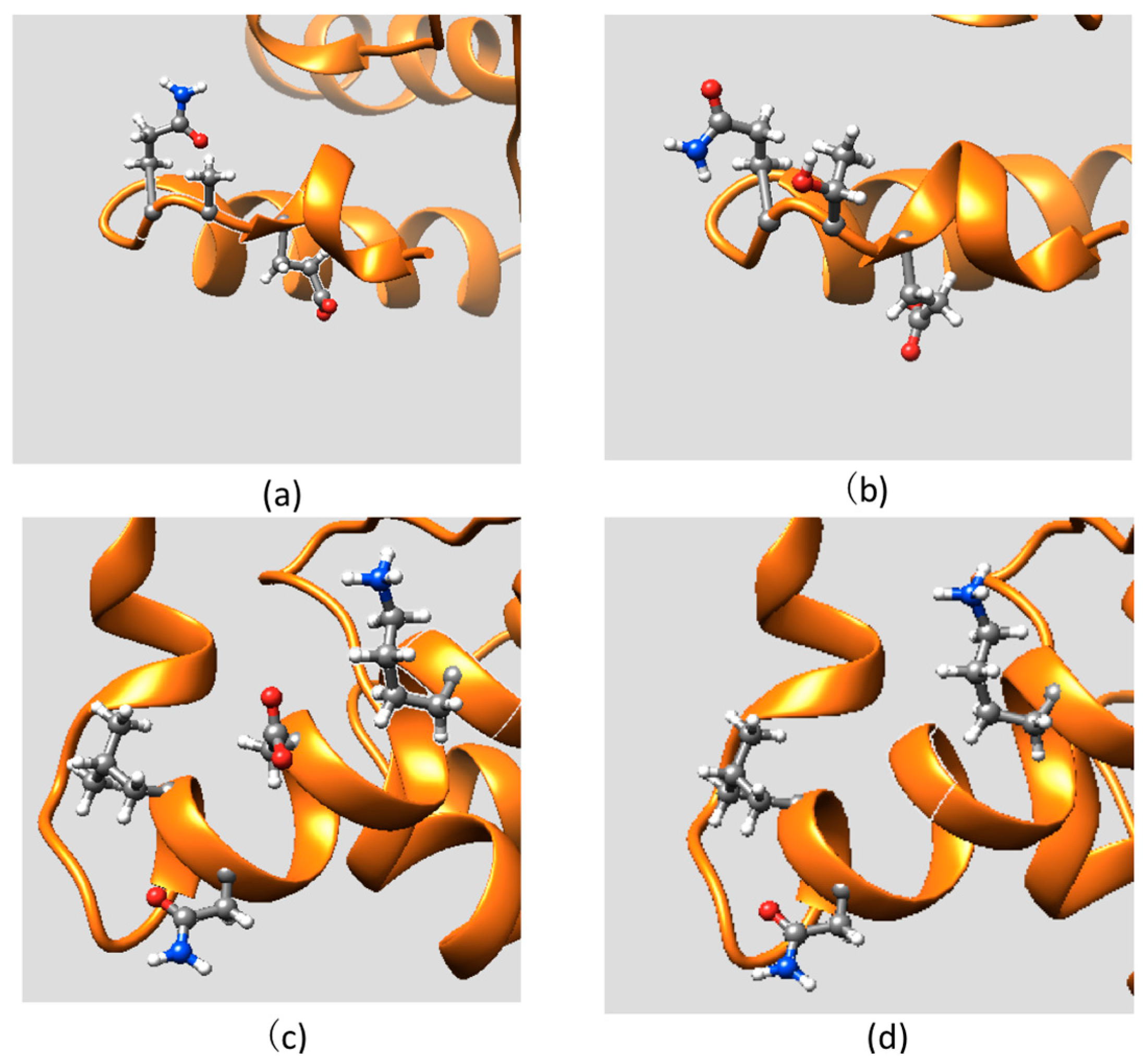
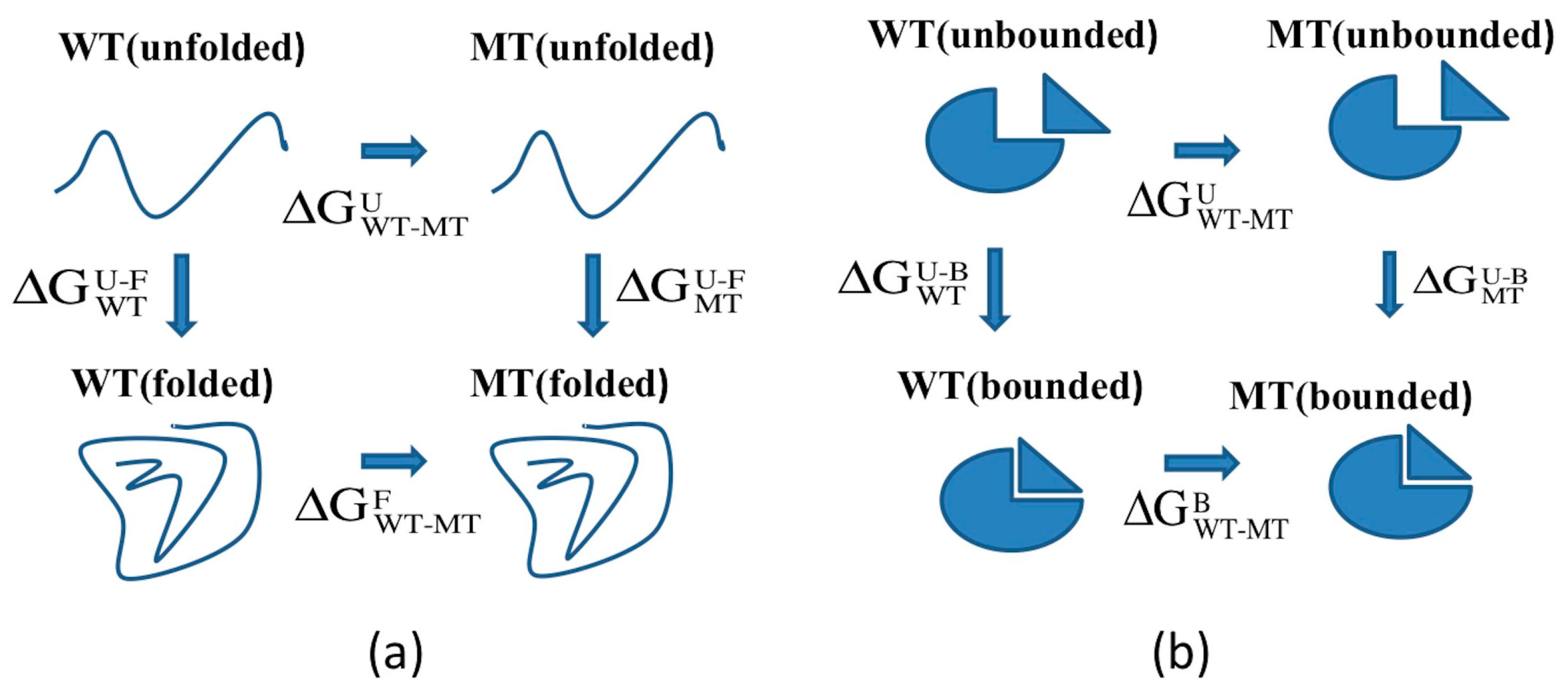
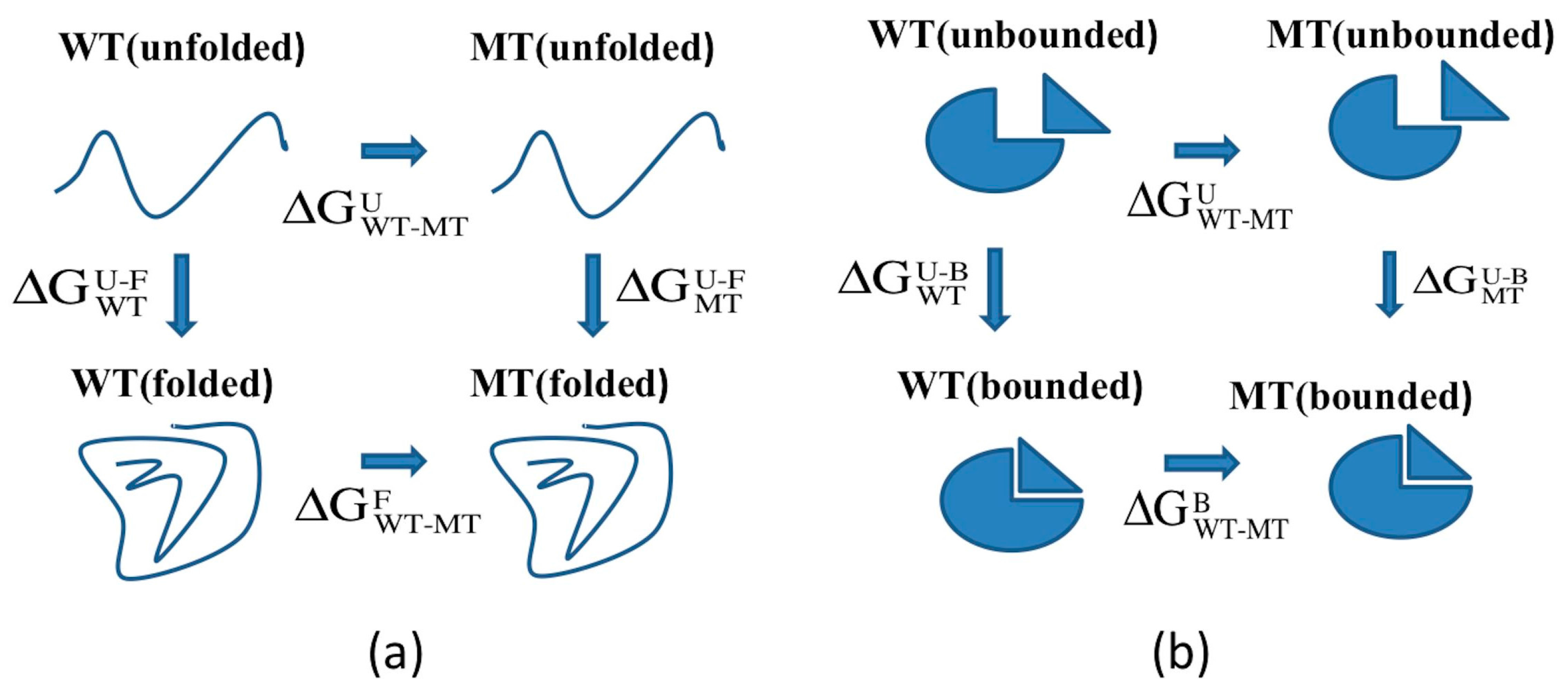
2.1. Protein Stability Changes due to Mutations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | NeEMO (Folding) | PopMusic (Folding) | I-Mutant (Folding) | DUET (Folding) | CUPSAT (Folding) | Foldx (Folding) | FEP (Folding) | Folding (Average) | FEP (Binding) |
|---|---|---|---|---|---|---|---|---|---|
| A77T | −1.03 | −0.22 | −0.75 | −0.76 | −0.29 | −1.40 | 0.13 | −0.74/−0.62 | −0.35 |
| D87G | −0.16 | −0.49 | −0.47 | −0.729 | −0.16 | −0.60 | −0.28 | −0.43/−0.41 | 0.73 |
| R108W | −0.36 | −0.86 | −1.32 | −0.26 | 0.28 | −0.18 | −11.29 | −0.45/−1.99 | −1.44 |
| N142S | −0.21 | −0.27 | −0.09 | −0.02 | 0.17 | −0.3 | −0.98 | −0.12/−0.24 | 0.64 |
| R179H | −0.71 | 0.06 | −0.15 | −0.72 | 0.28 | 0.746 | −8.78 | −0.08/−1.32 | −3.06 |
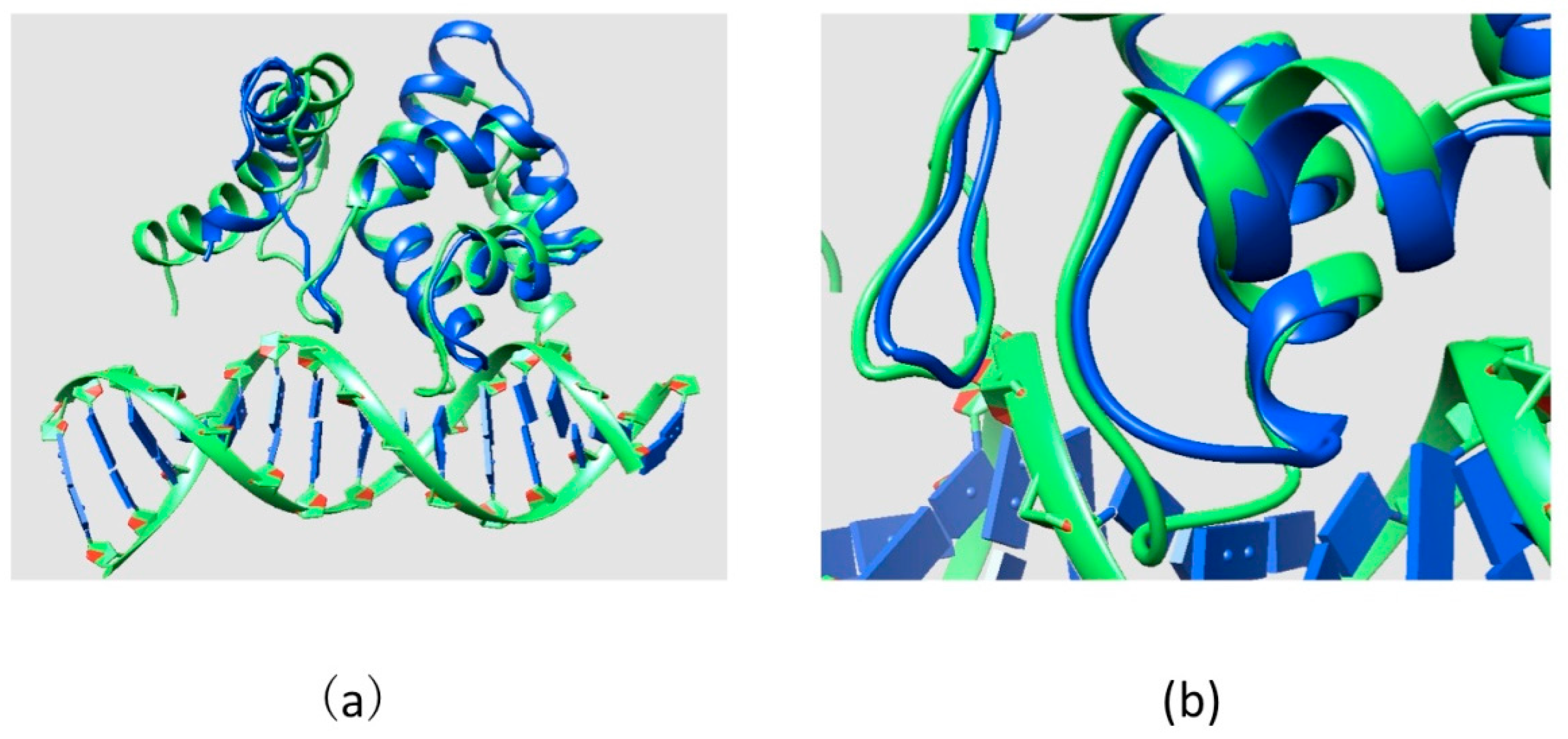
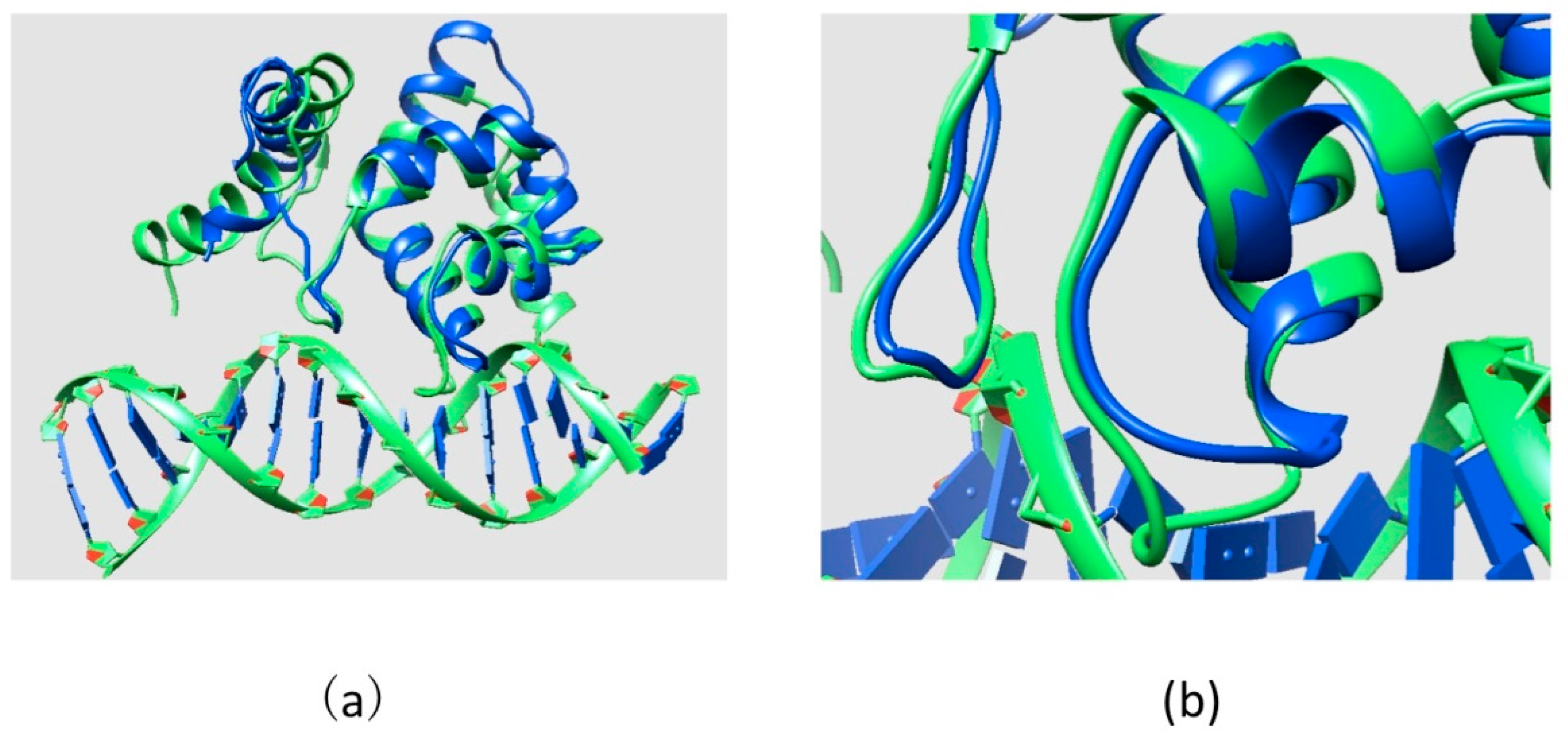
2.2. Effect of Mutations on Protein Structure
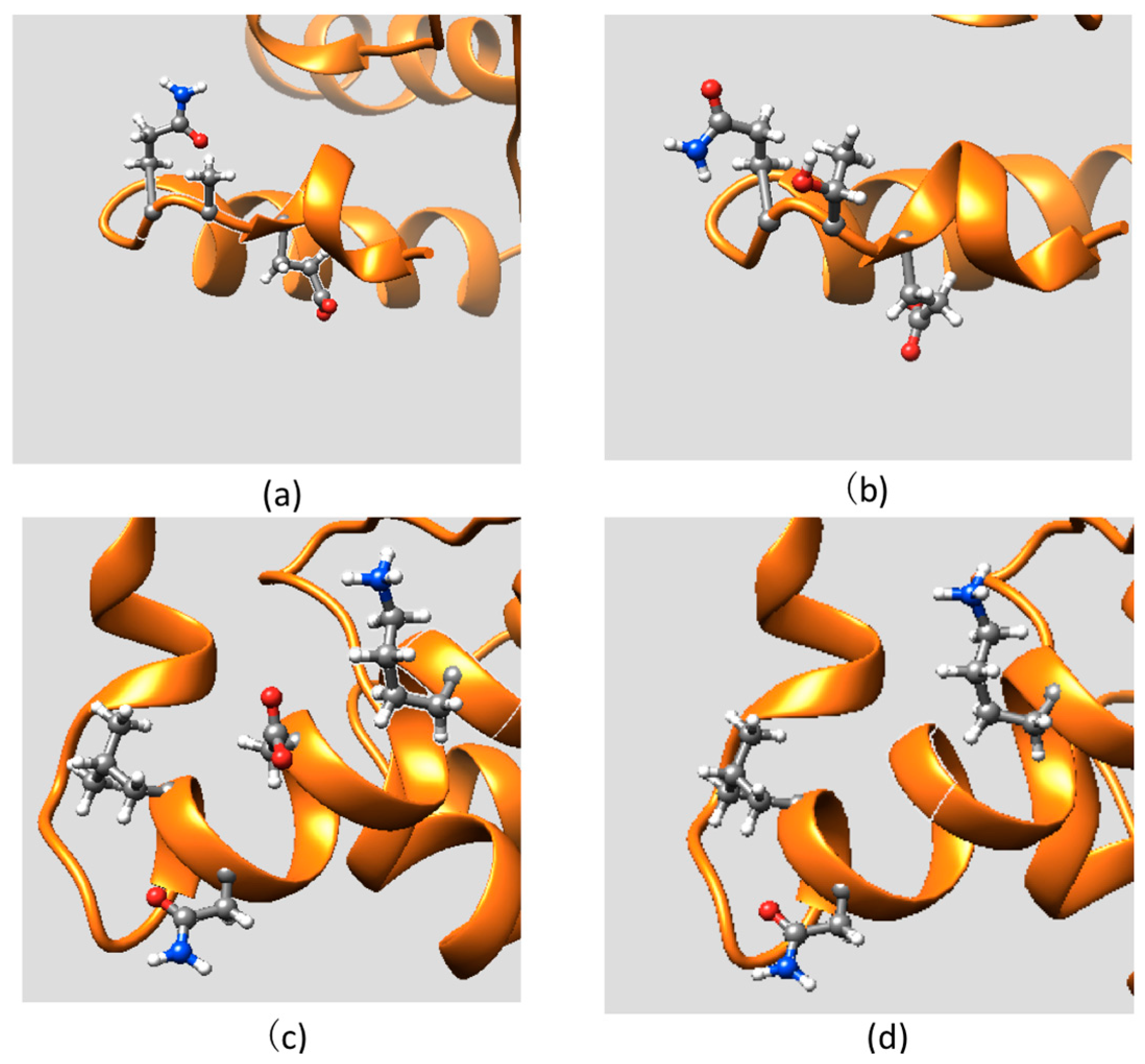
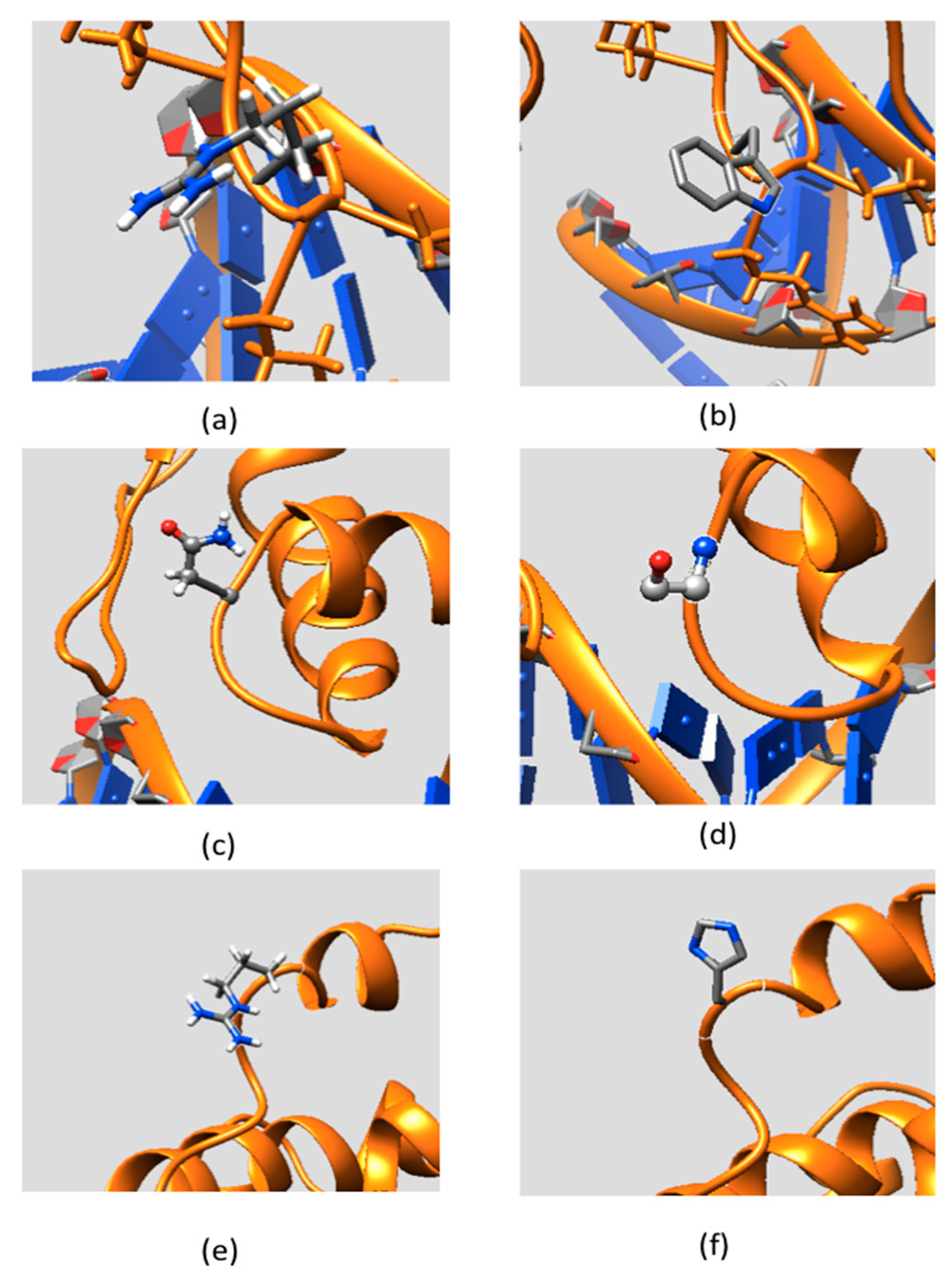
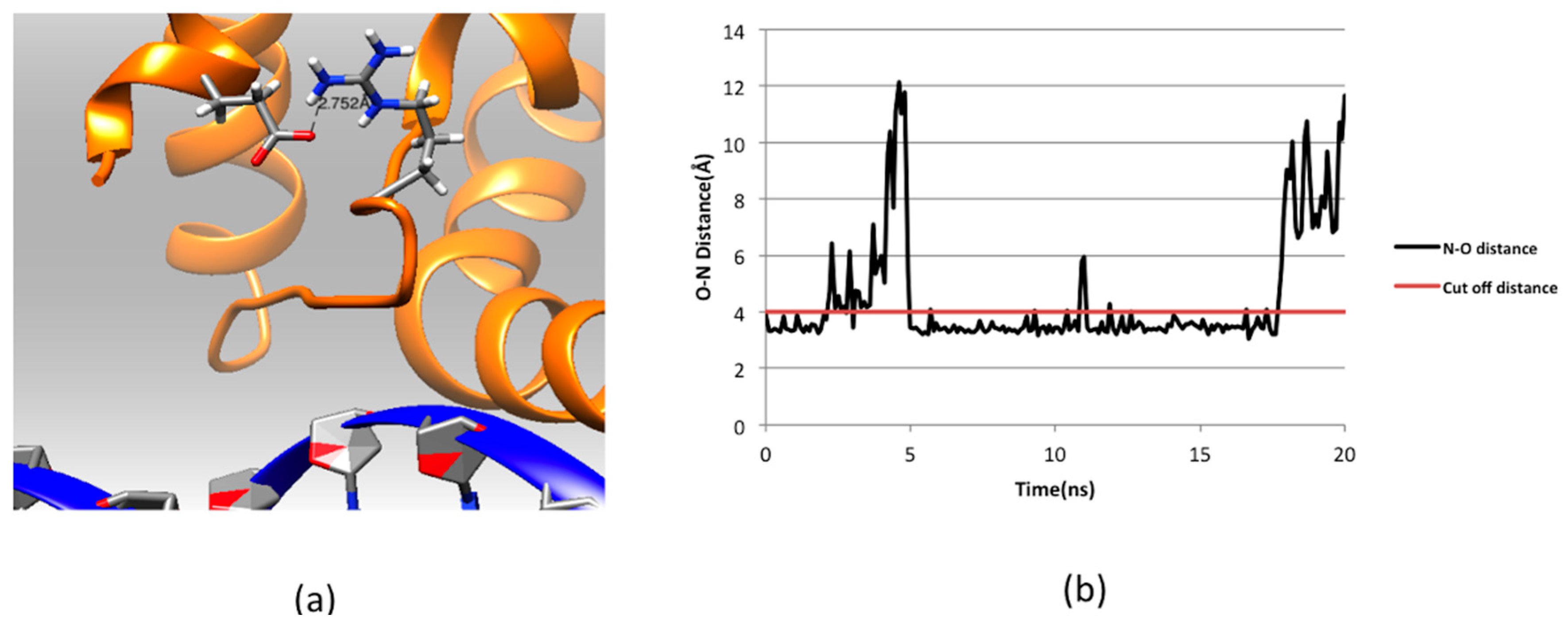
2.3. Effect of Mutations on Protein Dynamics
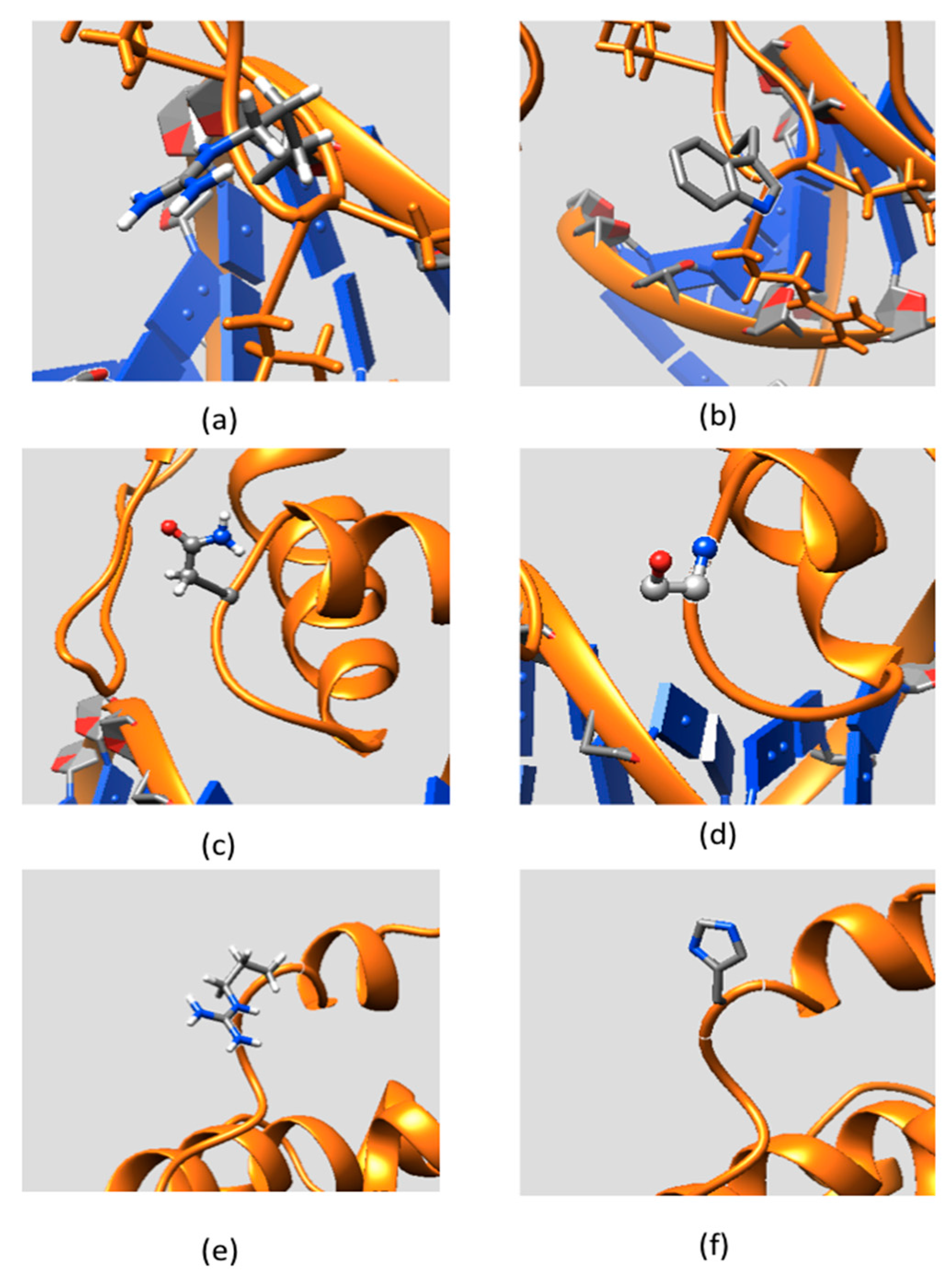
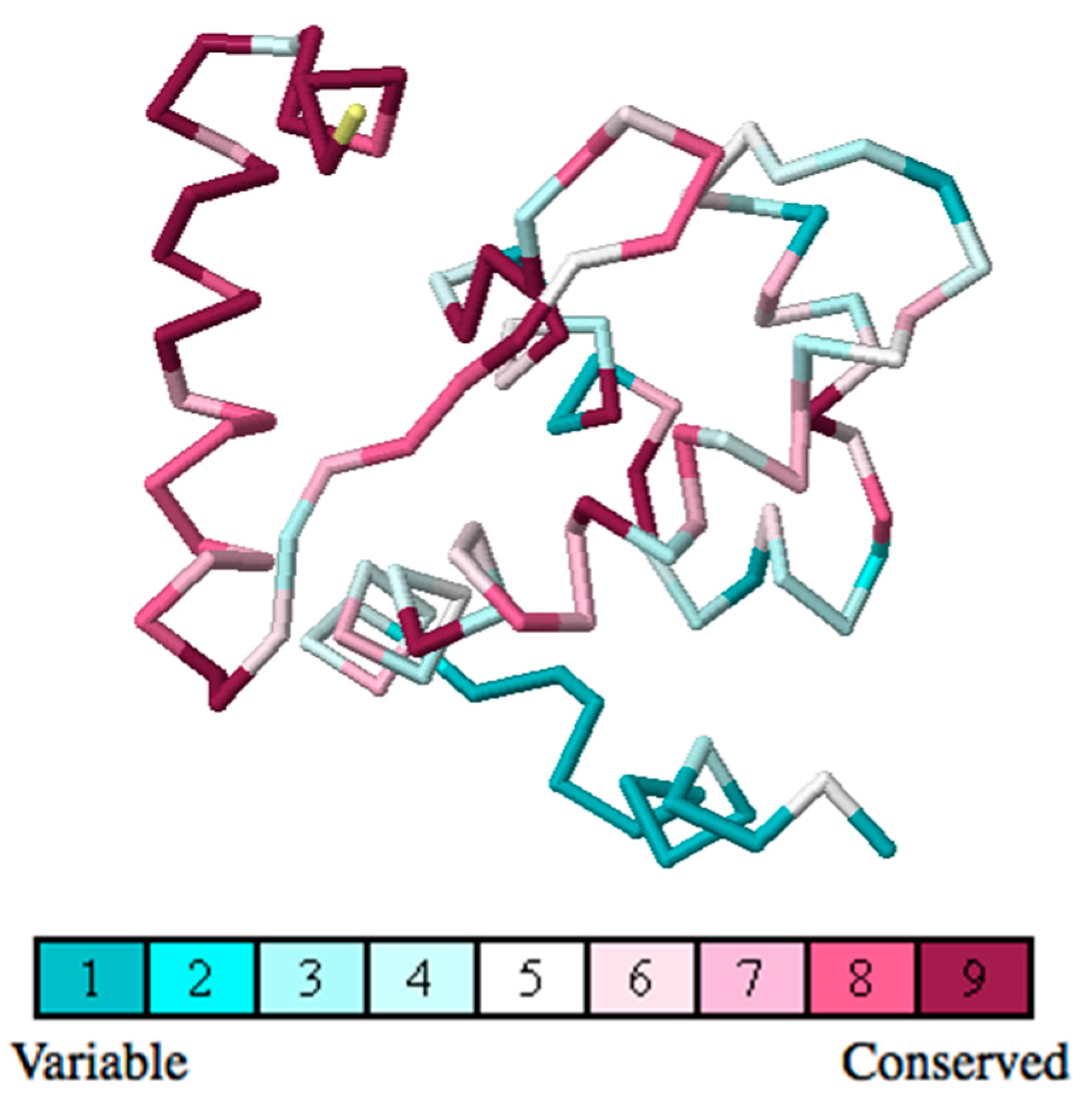
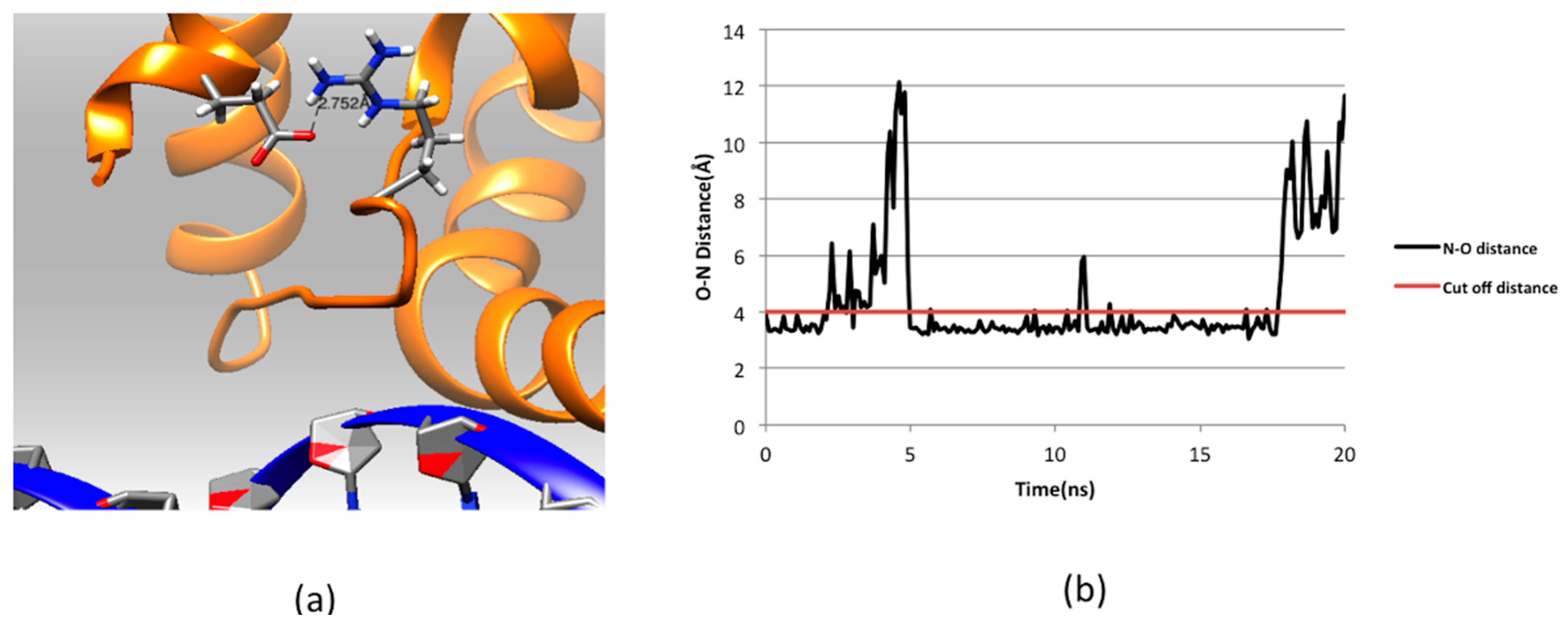
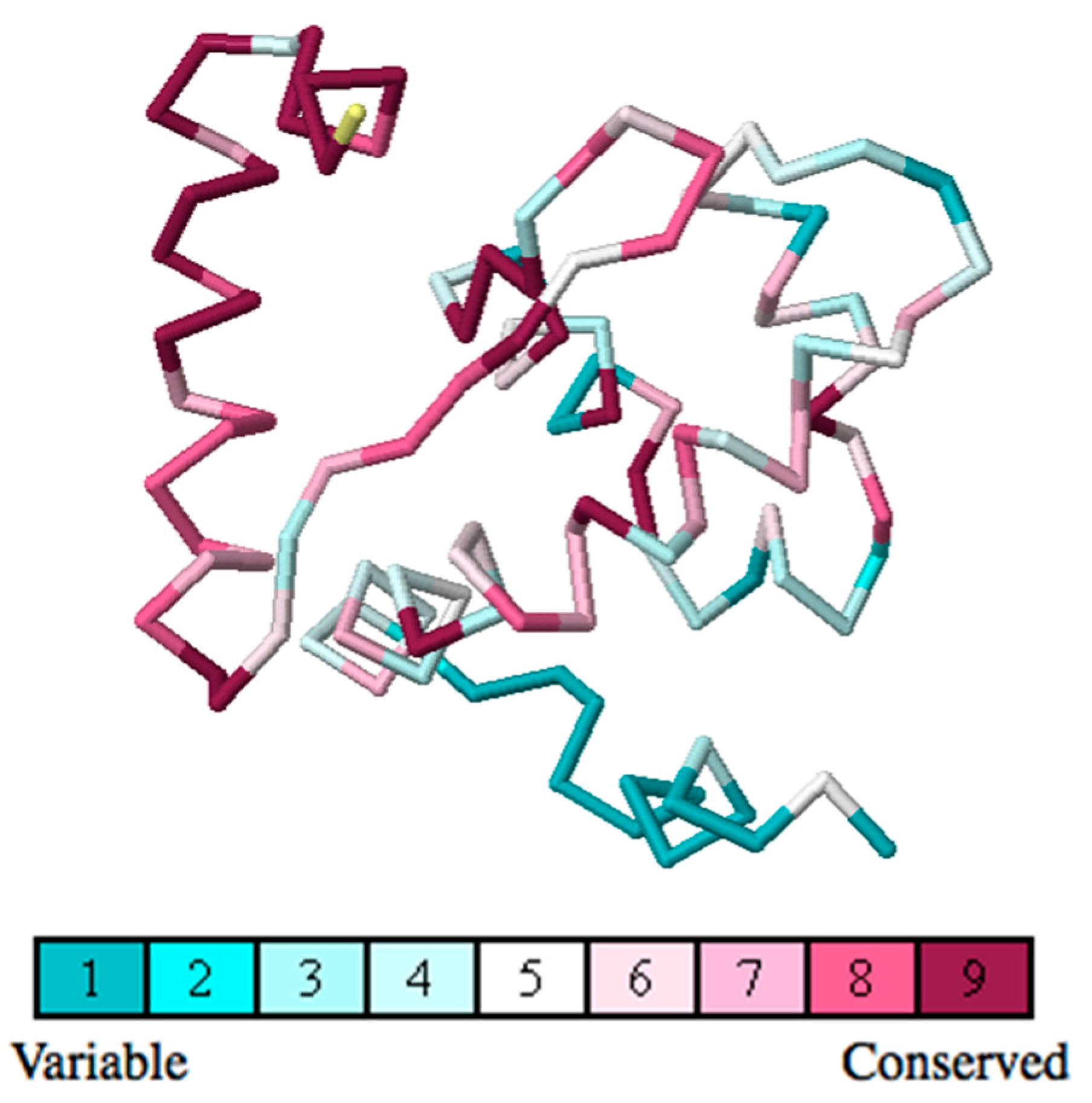
2.4. Residue Conservation via Multiple Sequence Alignment

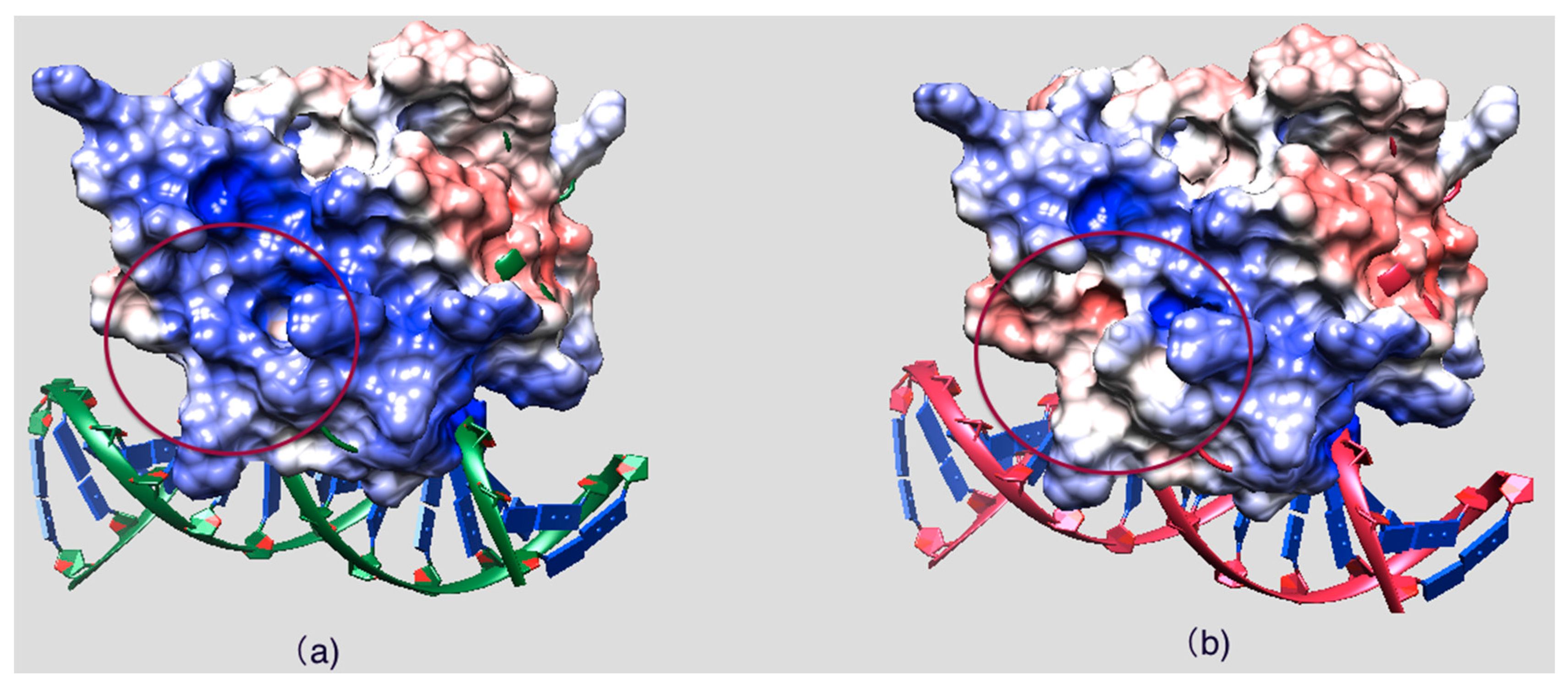
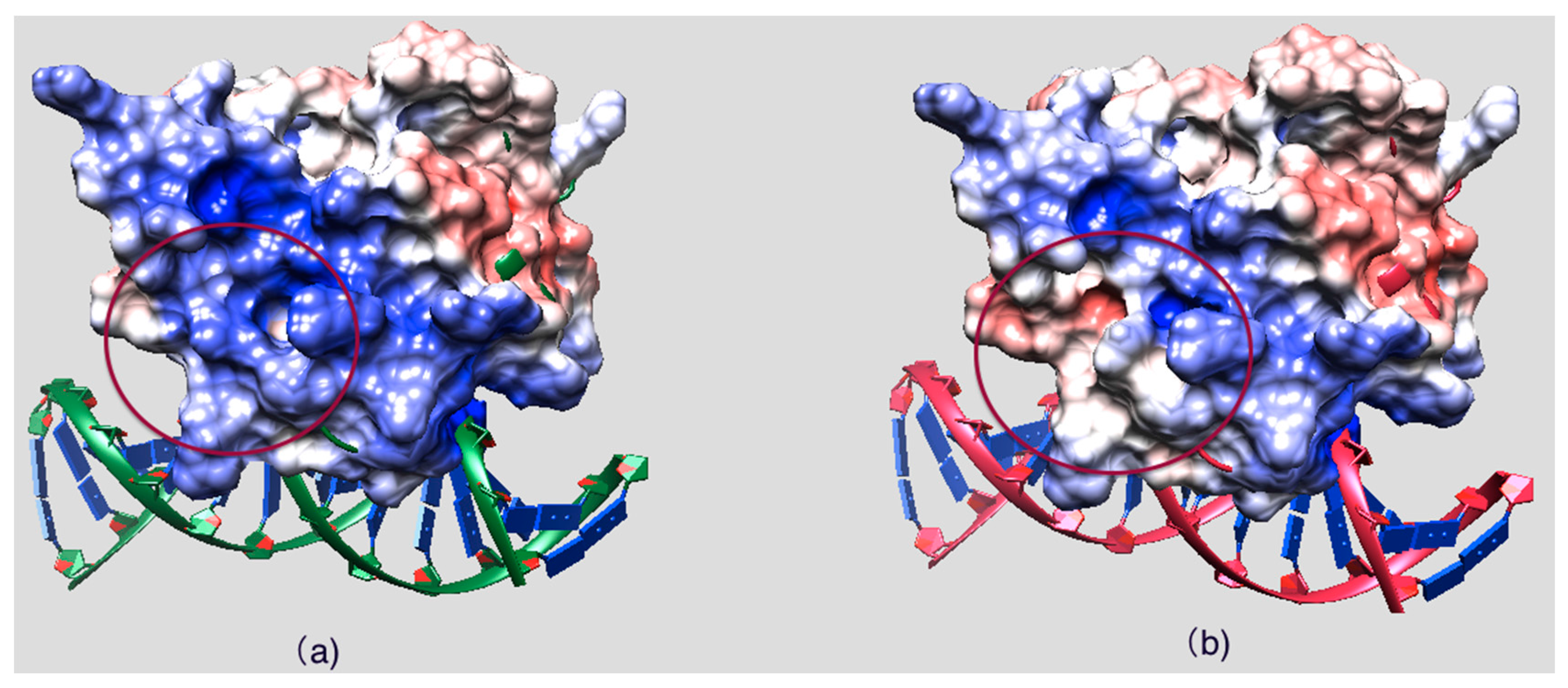
2.5. Evolutionary Conservation and Protein Interacting Investigation Using the ConSurf Server and IBIS Server
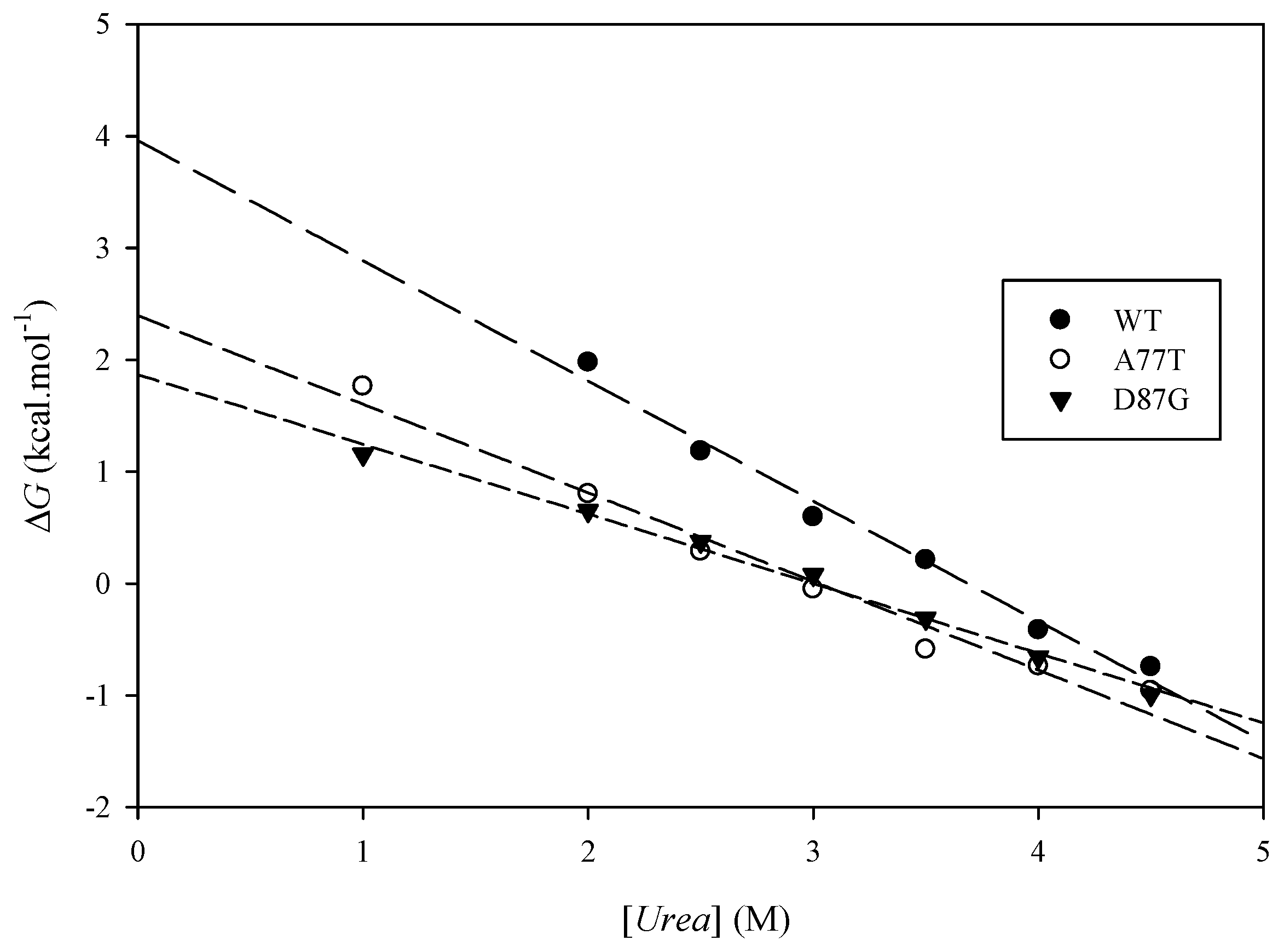
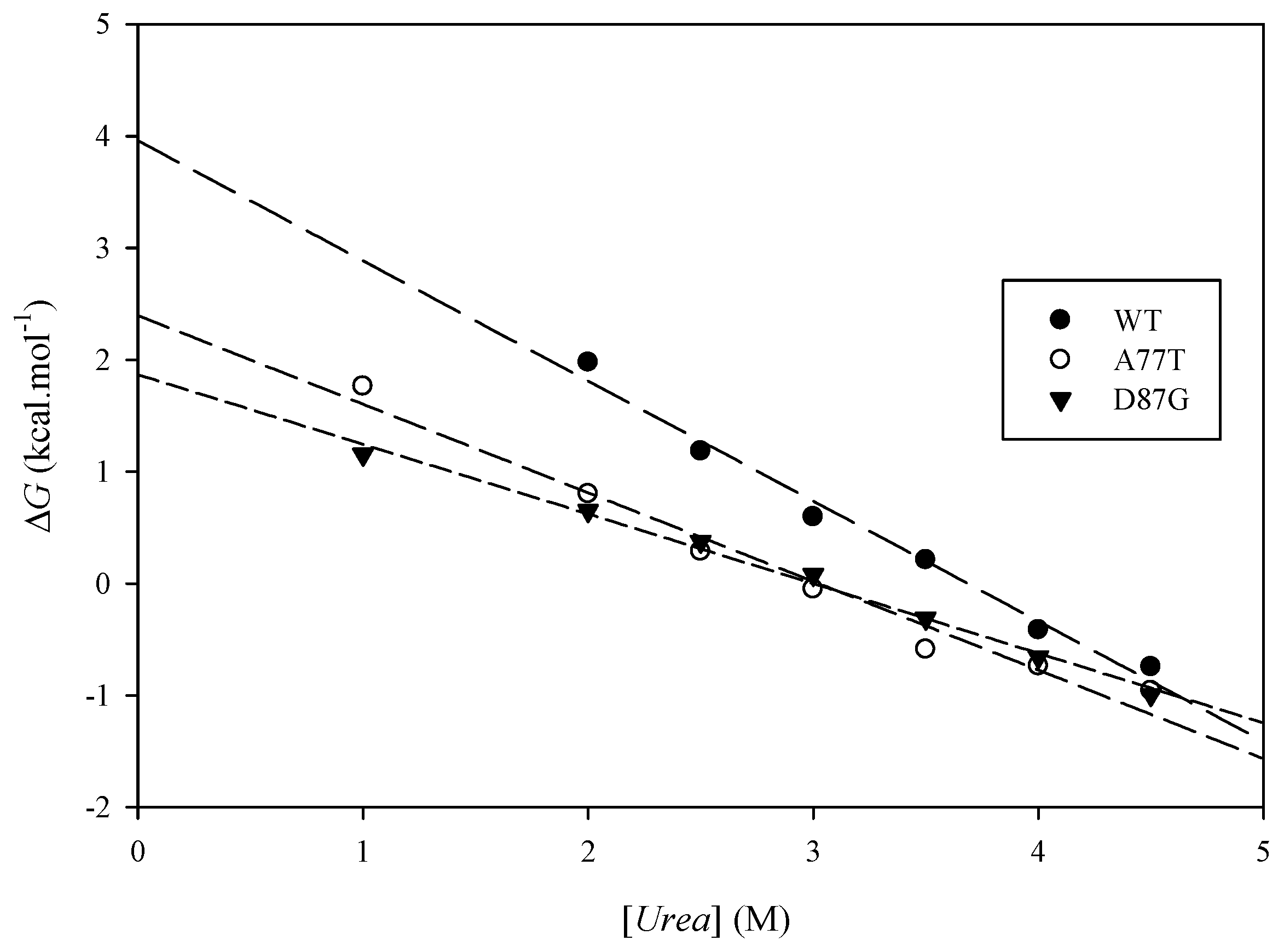
2.6. Experimental Results
| Protein | Helix | Strand | Turns | Unordered |
|---|---|---|---|---|
| WT | 14% | 31% | 20% | 35% |
| A77T | 13% | 31% | 19% | 38% |
| D87G | 13% | 30% | 20% | 38% |
| Protein | (kcal·mol−1) | (kcal·mol−1) | Urea Concentration (M) | (kcal·mol−1) |
|---|---|---|---|---|
| ARID WT | 3.51 ± 0.32 | 3.99 ± 0.02 | ||
| A77T | 2.41 ± 0.05 | 1.10 | 3.07 ± 0.02 | 0.70 |
| D87G | 1.82 ± 0.01 | 1.70 | 2.99 ± 0.03 | 0.76 |
3. Methods and Experimental Section
3.1. Structures
3.2. ARID Folding and Binding Free Energy Changes
3.3. Utilizing Webservers and Third Party Software
3.4. Molecular Dynamics Simulation
3.5. Electrostatic Potential Calculation
3.6. ARID Domain Protein Expression and Purification
- ARID A77T NdeI F1 (5’-CATATGAATGAGCTAGAGACCCAGACGAGAGTGAAAC-3’)
- ARID A77T NdeI R2 (5’-GTTTCACTCTCGTCTGGGTCTCTAGCTCATTCATATG-3’)
- ARID D87G F1 (5’-GAAACTGAACTACTTGGGCCAGATTGCCAAATTCTG-3’)
- ARID D87G R2 (5’-CAGAATTTGGCAATCTGGCCCAAGTAGTTCAGTTTC-3’)
- Initial denaturing at 95 °C for 1:30 min
- 95 °C for 30 s
- Annealing at 60 °C for 35 s
- Extension at 72 °C for 1 min
3.7. Circular Dichroism Spectrum and Urea-Induced Unfolding
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl.), 245–254. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.P.; Cao, J.; Zou, M.R.; Sayegh, J.; Yan, Q. Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers 2011, 3, 1383–1404. [Google Scholar] [CrossRef] [PubMed]
- Benevolenskaya, E.V. Histone h3k4 demethylases are essential in development and differentiation. Biochem. Cell Biol. Biochim. Biol. Cell. 2007, 85, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The x-linked mental retardation gene smcx/jarid1c defines a family of histone h3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Outchkourov, N.S.; Muino, J.M.; Kaufmann, K.; van Ijcken, W.F.; Groot Koerkamp, M.J.; van Leenen, D.; de Graaf, P.; Holstege, F.C.; Grosveld, F.G.; Timmers, H.T. Balancing of histone h3k4 methylation states by the kdm5c/smcx histone demethylase modulates promoter and enhancer function. Cell Rep. 2013, 3, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Grafodatskaya, D.; Chung, B.H.; Butcher, D.T.; Turinsky, A.L.; Goodman, S.J.; Choufani, S.; Chen, Y.A.; Lou, Y.; Zhao, C.; Rajendram, R.; et al. Multilocus loss of DNA methylation in individuals with mutations in the histone h3 lysine 4 demethylase kdm5c. BMC Med. Genom. 2013, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Patsialou, A.; Wilsker, D.; Moran, E. DNA-binding properties of arid family proteins. Nucleic Acids Res. 2005, 33, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Probst, L.; Wain, H.M.; Maltais, L.; Tucker, P.W.; Moran, E. Nomenclature of the arid family of DNA-binding proteins. Genomics 2005, 86, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chandrasekharan, M.B.; Chen, Y.C.; Bhaskara, S.; Hiebert, S.W.; Sun, Z.W. The jmjn domain of jhd2 is important for its protein stability, and the plant homeodomain (phd) finger mediates its chromatin association independent of h3k4 methylation. J. Biol. Chem. 2010, 285, 24548–24561. [Google Scholar] [CrossRef] [PubMed]
- Mellor, J. It takes a phd to read the histone code. Cell 2006, 126, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Tzschach, A.; Lenzner, S.; Moser, B.; Reinhardt, R.; Chelly, J.; Fryns, J.P.; Kleefstra, T.; Raynaud, M.; Turner, G.; Ropers, H.H.; et al. Novel jarid1c/smcx mutations in patients with x-linked mental retardation. Hum. Mutat. 2006, 27, 389. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, T.F.; Goncalves, A.P.; Fintelman Rodrigues, N.; dos Santos, J.M.; Pimentel, M.M.; Santos-Reboucas, C.B. Kdm5c mutational screening among males with intellectual disability suggestive of x-linked inheritance and review of the literature. Eur. J. Med. Genet. 2014, 57, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Schalock, R.L.; Borthwick-Duffy, S.A.; Bradley, V.J.; Buntinx, W.H.E.; Coulter, D.L.; Craig, E.M.; Gomez, S.C.; Lachapelle, Y.; Luckasson, R.; Reeve, A.; et al. Yeager Intellectual Disability: Definition, Classification, and Systems of Supports, 11th ed.; American Association on Intellectual and Developmental Disabilities: Washington, DC, USA, 2010. [Google Scholar]
- Kaufman, L.; Ayub, M.; Vincent, J.B. The genetic basis of non-syndromic intellectual disability: A review. J. Neurodev. Disord. 2010, 2, 182–209. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.R.; Amende, M.; Gurok, U.; Moser, B.; Gimmel, V.; Tzschach, A.; Janecke, A.R.; Tariverdian, G.; Chelly, J.; Fryns, J.P.; et al. Mutations in the jarid1c gene, which is involved in transcriptional regulation and chromatin remodeling, cause x-linked mental retardation. Am. J. Hum. Genet. 2005, 76, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone h3k4 demethylase smcx links rest target genes to x-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Abidi, F.E.; Holloway, L.; Moore, C.A.; Weaver, D.D.; Simensen, R.J.; Stevenson, R.E.; Rogers, R.C.; Schwartz, C.E. Mutations in jarid1c are associated with x-linked mental retardation, short stature and hyperreflexia. J. Med. Genet. 2008, 45, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. Dbsnp: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Exome Aggregation Consortium (ExAC), Cambridge, MA, USA. Available online: http://exac.Broadinstitute.Org (accessed on 20 October 2015).
- Schaefer, C.; Bromberg, Y.; Achten, D.; Rost, B. Disease-related mutations predicted to impact protein function. BMC Genom. 2012, 13 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nssnps on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Schuster-Bockler, B.; Bateman, A. Protein interactions in human genetic diseases. Genome Biol. 2008, 9, R9. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Schork, N.J. Distribution analysis of nonsynonymous polymorphisms within the human kinase gene family. Genomics 2007, 90, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kucukkal, T.G.; Yang, Y.; Uvarov, O.; Cao, W.; Alexov, E. Impact of rett syndrome mutations on MeCP2 MBD stability. Biochemistry 2015, 54, 6357–6368. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Petukh, M.; Li, L.; Alexov, E. Continuous development of schemes for parallel computing of the electrostatics in biological systems: Implementation in delphi. J. Comput. Chem. 2013, 34, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.; Sarkar, S.; Zhang, J.; Witham, S.; Zhang, Z.; Wang, L.; Smith, N.; Petukh, M.; Alexov, E. Delphi: A comprehensive suite for delphi software and associated resources. BMC Biophys. 2012, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Subhra Sarkar, S.W.; Zhang, J.; Zhenirovskyy, M.; Rocchia, W.; Alexov, E. Delphi web server: A comprehensive online suite for electrostatic calculations of biological macromolecules and their complexes. Commun. Comput. Phys. 2013, 13, 269–284. [Google Scholar] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Teng, Y.C.; Yuan, C.; Wu, Y.T.; Chan, M.Y.; Cheng, A.N.; Lin, P.H.; Juan, L.J.; Tsai, M.D. The arid domain of the h3k4 demethylase rbp2 binds to a DNA ccgccc motif. Nat. Struct. Mol. Biol. 2008, 15, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, B.A.; Zhang, D.; Thangudu, R.R.; Tyagi, M.; Fong, J.H.; Marchler-Bauer, A.; Bryant, S.H.; Madej, T.; Panchenko, A.R. Inferred biomolecular interaction server—A web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 2010, 38, D518–D524. [Google Scholar] [CrossRef] [PubMed]
- Van Stokkum, I.H.; Spoelder, H.J.; Bloemendal, M.; van Grondelle, R.; Groen, F.C. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 1990, 191, 110–118. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. Dichroweb, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [PubMed]
- Koehler, C.; Bishop, S.; Dowler, E.F.; Schmieder, P.; Diehl, A.; Oschkinat, H.; Ball, L.J. Backbone and sidechain 1h, 13c and 15n resonance assignments of the bright/arid domain from the human jarid1c (smcx) protein. Biomol. NMR Assign. 2008, 2, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, J.; Iwahara, M.; Daughdrill, G.W.; Ford, J.; Clubb, R.T. The structure of the dead ringer-DNA complex reveals how at-rich interaction domains (arids) recognize DNA. EMBO J. 2002, 21, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Kofke, D.A. Accuracy of free-energy perturbation calculations in molecular simulation. I. Modeling. J. Chem. Phys. 2001, 114, 7303. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Thomas, L.L. Perspective on free-energy perturbation calculations for chemical equilibria. J. Chem. Theory Comput. 2008, 4, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with namd. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A. A comparison of alternative approaches to free energy calculations. J. Phys. Chem. 1994, 98, 1487–1493. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Dehez, F.; Cai, W.; Chipot, C. A toolkit for the analysis of free-energy perturbation calculations. J. Chem. Theory Comput. 2012, 8, 2606–2616. [Google Scholar] [CrossRef]
- Beveridge, D.L.; Dicapua, F.M. Free-energy via molecular simulation—Applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 431–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012, 28, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Teng, S.; Wang, L.; Schwartz, C.E.; Alexov, E. Computational analysis of missense mutations causing snyder-robinson syndrome. Hum. Mutat. 2010, 31, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Ofiteru, A.; Bucurenci, N.; Alexov, E.; Bertrand, T.; Briozzo, P.; Munier-Lehmann, H.; Gilles, A.M. Structural and functional consequences of single amino acid substitutions in the pyrimidine base binding pocket of escherichia coli cmp kinase. FEBS J. 2007, 274, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Norris, J.; Schwartz, C.; Alexov, E. In silico and in vitro investigations of the mutability of disease-causing missense mutation sites in spermine synthase. PLoS ONE 2011, 6, e20373. [Google Scholar] [CrossRef] [PubMed]
- Merz, K.M.; Kollman, P.A. Free energy perturbation simulations of the inhibition of thermolysin: Prediction of the free energy of binding of a new inhibitor. J. Am. Chem. Soc. 1989, 111, 5649–5658. [Google Scholar] [CrossRef]
- Brandsdal, B.O.; Osterberg, F.; Almlöf, M.; Feierberg, I.; Luzhkov, V.B.; Aqvist, J. Free energy calculations and ligand binding. Adv. Protein Chem. 2003, 123–158. [Google Scholar]
- Li, M.; Petukh, M.; Alexov, E.; Panchenko, A.R. Predicting the impact of missense mutations on protein-protein binding affinity. J. Chem. Theory Comput. 2014, 10, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Tyagi, M.; Teng, S.; Shoemaker, B.A.; Hashimoto, K.; Alexov, E.; Wuchty, S.; Panchenko, A.R. Cancer missense mutations alter binding properties of proteins and their interaction networks. PLoS ONE 2013, 8, e66273. [Google Scholar] [CrossRef] [PubMed]
- Giollo, M.; Martin, A.J.; Walsh, I.; Ferrari, C.; Tosatto, S.C. Neemo: A method using residue interaction networks to improve prediction of protein stability upon mutation. BMC Genom. 2014, 15 (Suppl. S4), S7. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, Y.; Grosfils, A.; Folch, B.; Gilis, D.; Bogaerts, P.; Rooman, M. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: Popmusic-2.0. Bioinformatics 2009, 25, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W303–W305. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. Duet: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. Cupsat: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The foldx web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.E.; Beltrao, P.; Stricher, F.; Schymkowitzm, J.; Ferkinghoff-Borg, J.; Rousseau, F.; Serrano, L. Genome-wide prediction of SH2 domain targets using structural information and the FoldX algorithm. PLoS Comput. Biol. 2008, e1000052. [Google Scholar] [CrossRef]
- DNASU Plasmid Repository; PSI:Biology-Materials Repository: Arizona State University, Tempe, AZ, USA. Available online: https://dnasu.org/DNASU/ (accessed on 20 October 2015).
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the expasy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [PubMed]
- Wei, Y.; Thyparambil, A.A.; Latour, R.A. Protein helical structure determination using cd spectroscopy for solutions with strong background absorbance from 190 to 230 nm. Biochim. Biophys. Acta 2014, 1844, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.L.; Scholtz, J.M.; Pace, C.N.; Grimsley, G.R. Determining the conformational stability of a protein using urea denaturation curves. Methods Mol. Biol. 2009, 490, 41–55. [Google Scholar] [PubMed]
- Ahmad, F.; Yadav, S.; Taneja, S. Determining stability of proteins from guanidinium chloride transition curves. Biochem. J. 1992, 287 Pt 2, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Suryadi, J.; Tran, E.J.; Maxwell, E.S.; Brown, B.A., 2nd. The crystal structure of the methanocaldococcus jannaschii multifunctional l7ae RNA-binding protein reveals an induced-fit interaction with the box c/d rnas. Biochemistry 2005, 44, 9657–9672. [Google Scholar] [CrossRef] [PubMed]
- Rohman, M.S.; Tadokoro, T.; Angkawidjaja, C.; Abe, Y.; Matsumura, H.; Koga, Y.; Takano, K.; Kanaya, S. Destabilization of psychrotrophic rnase hi in a localized fashion as revealed by mutational and x-ray crystallographic analyses. FEBS J. 2009, 276, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; DeDecker, B.S.; Johnson, C.M.; Nikolova, P.V.; Proctor, M.R.; Lane, D.P.; Fersht, A.R. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 1997, 94, 14338–14342. [Google Scholar] [CrossRef] [PubMed]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Consurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. Consurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. Consurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. Consurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Suryadi, J.; Yang, Y.; Kucukkal, T.G.; Cao, W.; Alexov, E. Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. Int. J. Mol. Sci. 2015, 16, 27270-27287. https://doi.org/10.3390/ijms161126022
Peng Y, Suryadi J, Yang Y, Kucukkal TG, Cao W, Alexov E. Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. International Journal of Molecular Sciences. 2015; 16(11):27270-27287. https://doi.org/10.3390/ijms161126022
Chicago/Turabian StylePeng, Yunhui, Jimmy Suryadi, Ye Yang, Tugba G. Kucukkal, Weiguo Cao, and Emil Alexov. 2015. "Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease" International Journal of Molecular Sciences 16, no. 11: 27270-27287. https://doi.org/10.3390/ijms161126022
APA StylePeng, Y., Suryadi, J., Yang, Y., Kucukkal, T. G., Cao, W., & Alexov, E. (2015). Mutations in the KDM5C ARID Domain and Their Plausible Association with Syndromic Claes-Jensen-Type Disease. International Journal of Molecular Sciences, 16(11), 27270-27287. https://doi.org/10.3390/ijms161126022