
Dangerous Liaisons: Caspase-11 and Reactive Oxygen Species Crosstalk in Pathogen Elimination

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

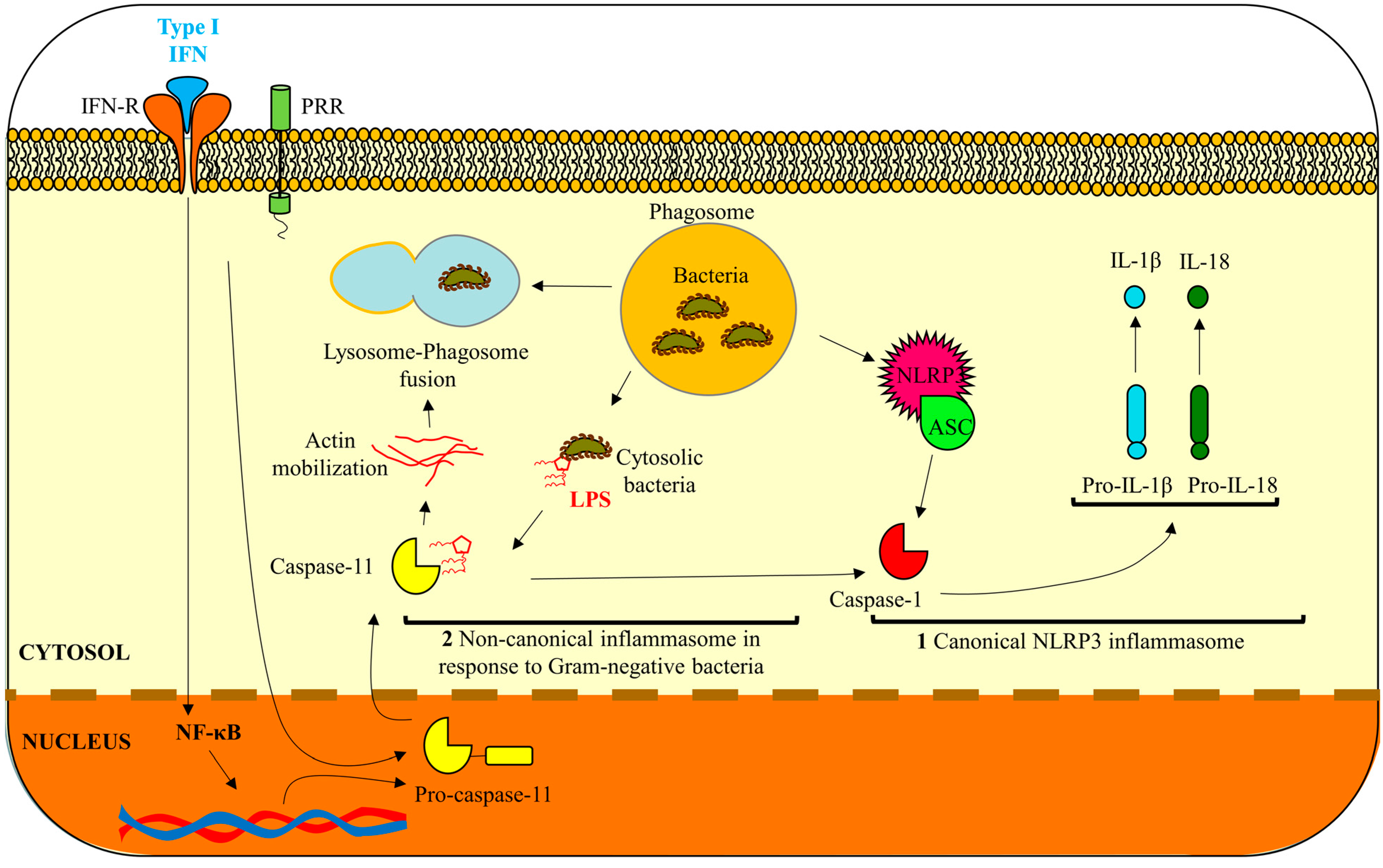
2. Inflammatory Murine Caspase-11
3. Are Human Caspase-4 and -5 Functional Orthologs of Murine Caspase-11?
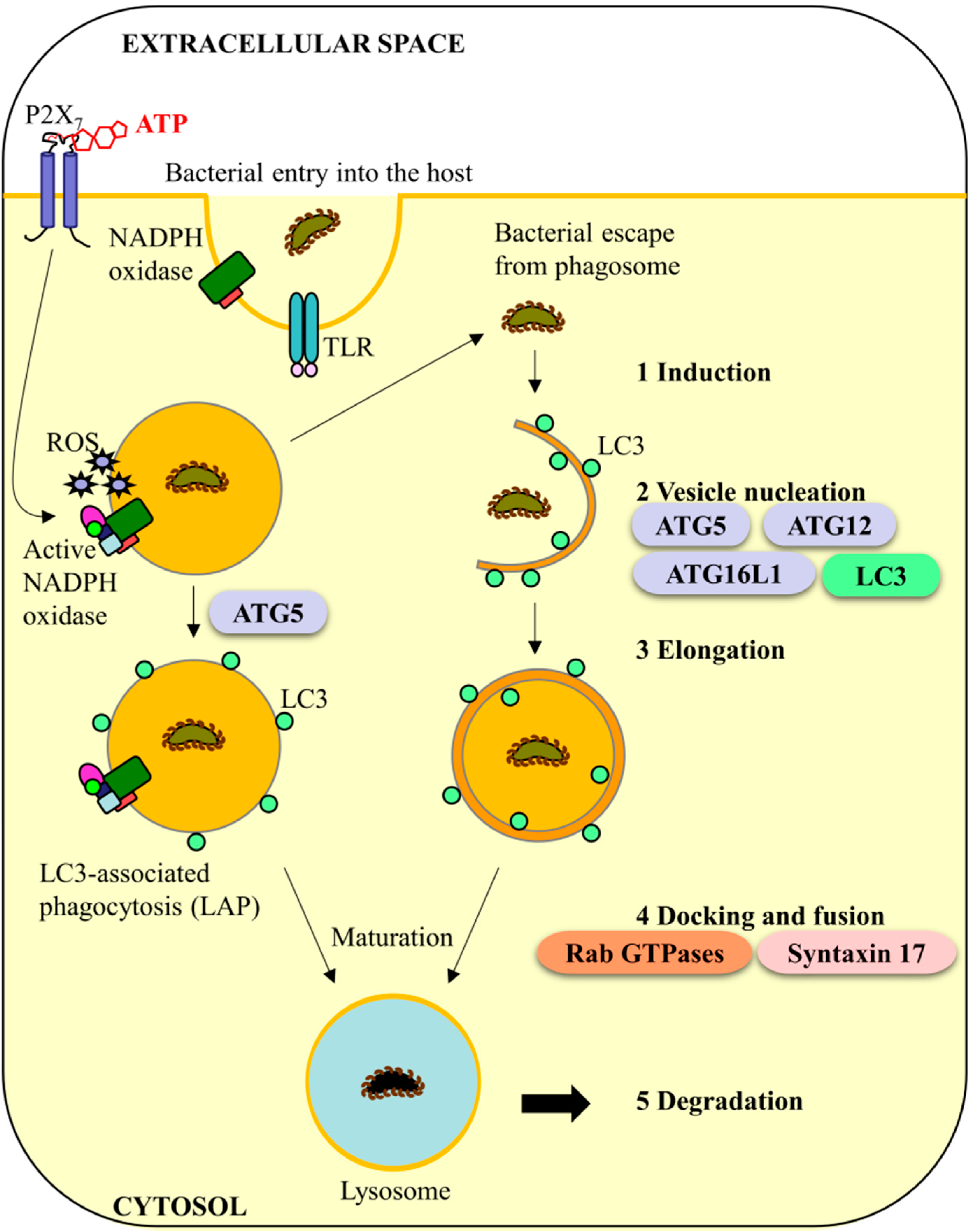
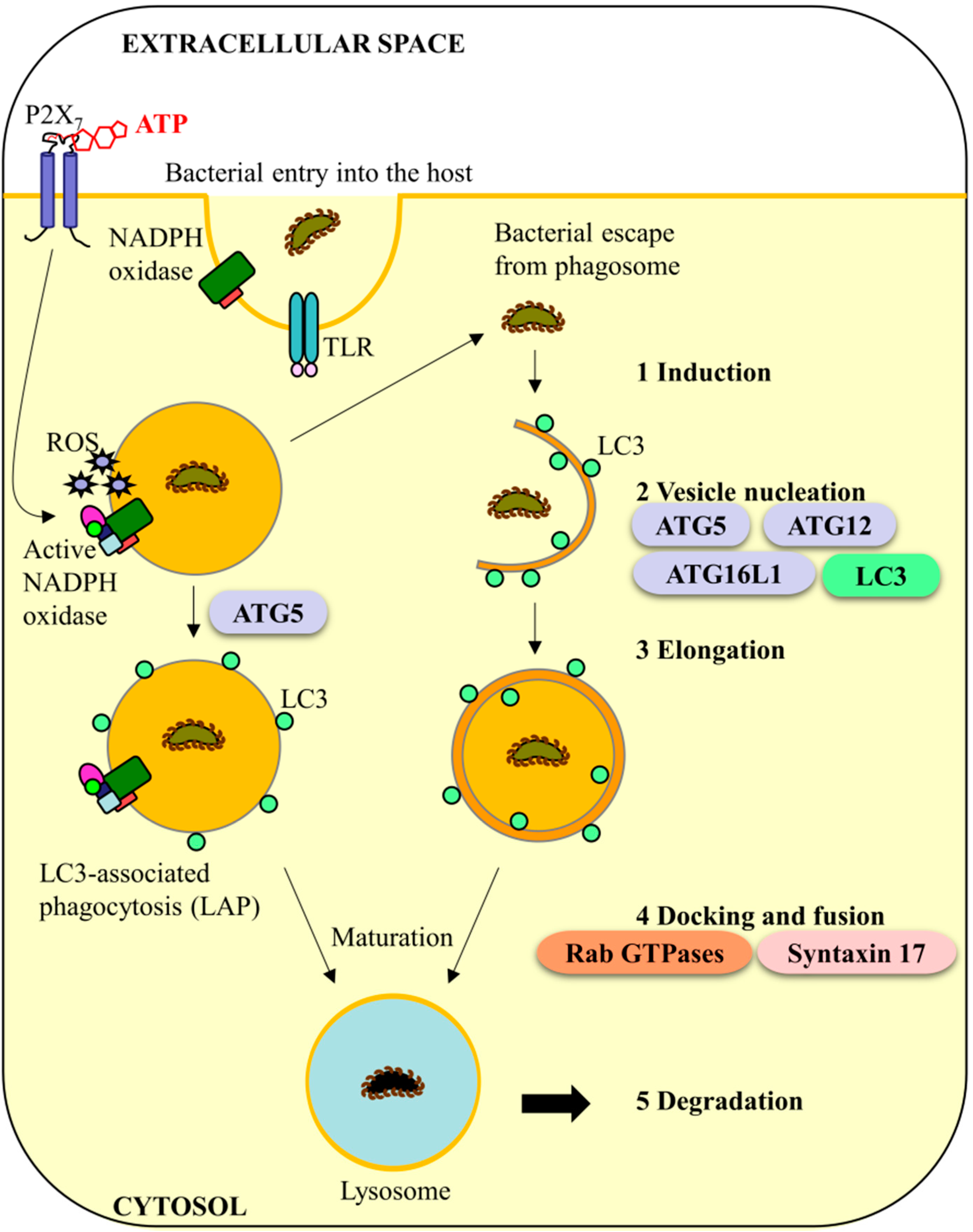
4. Murine Caspase-11 and Anti-Bacterial Autophagy Systems: A Conserved Role in Humans?
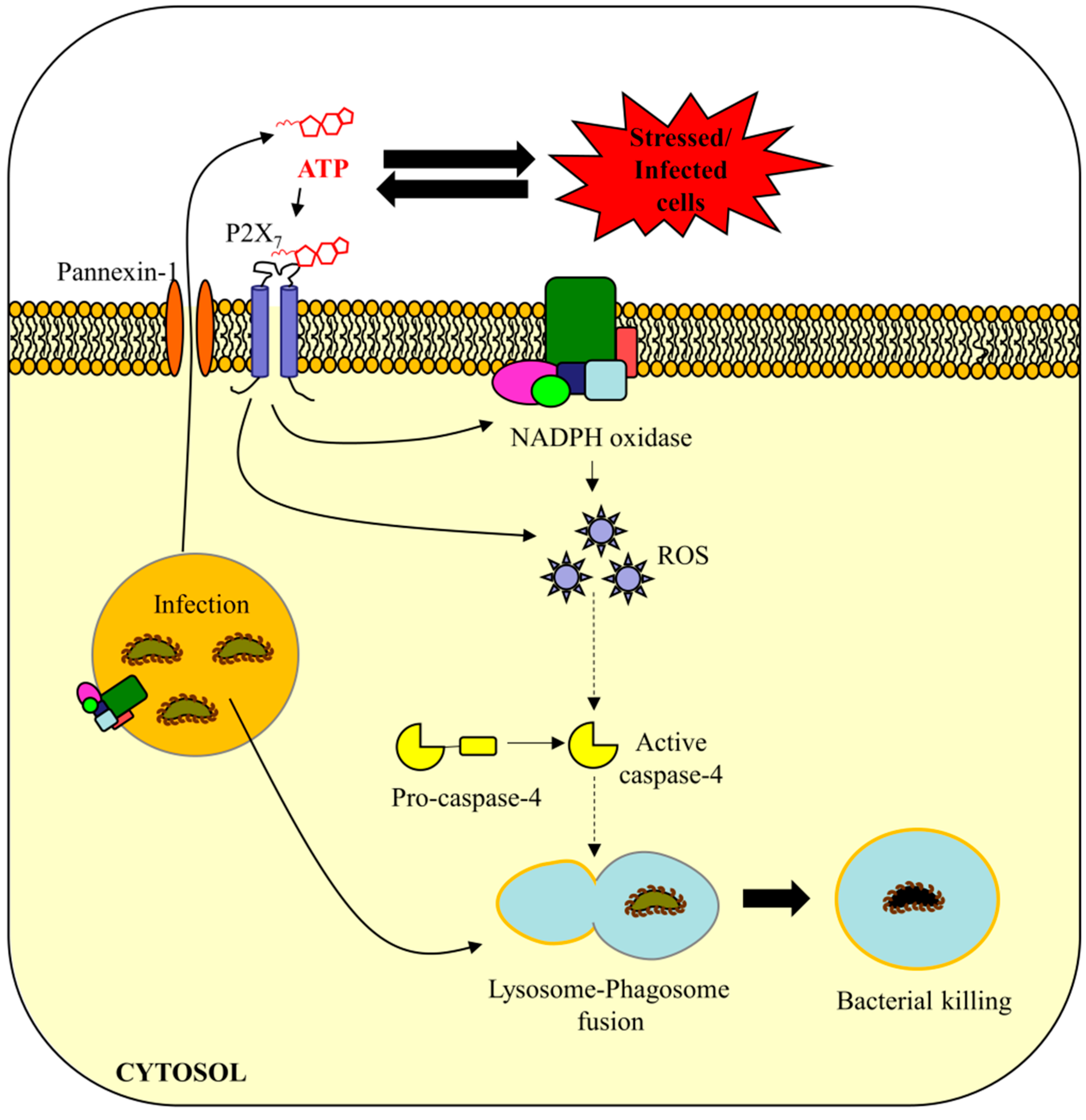
5. Are eATP and ROS Important in Caspase-4/11-Mediated Bacterial Defense?
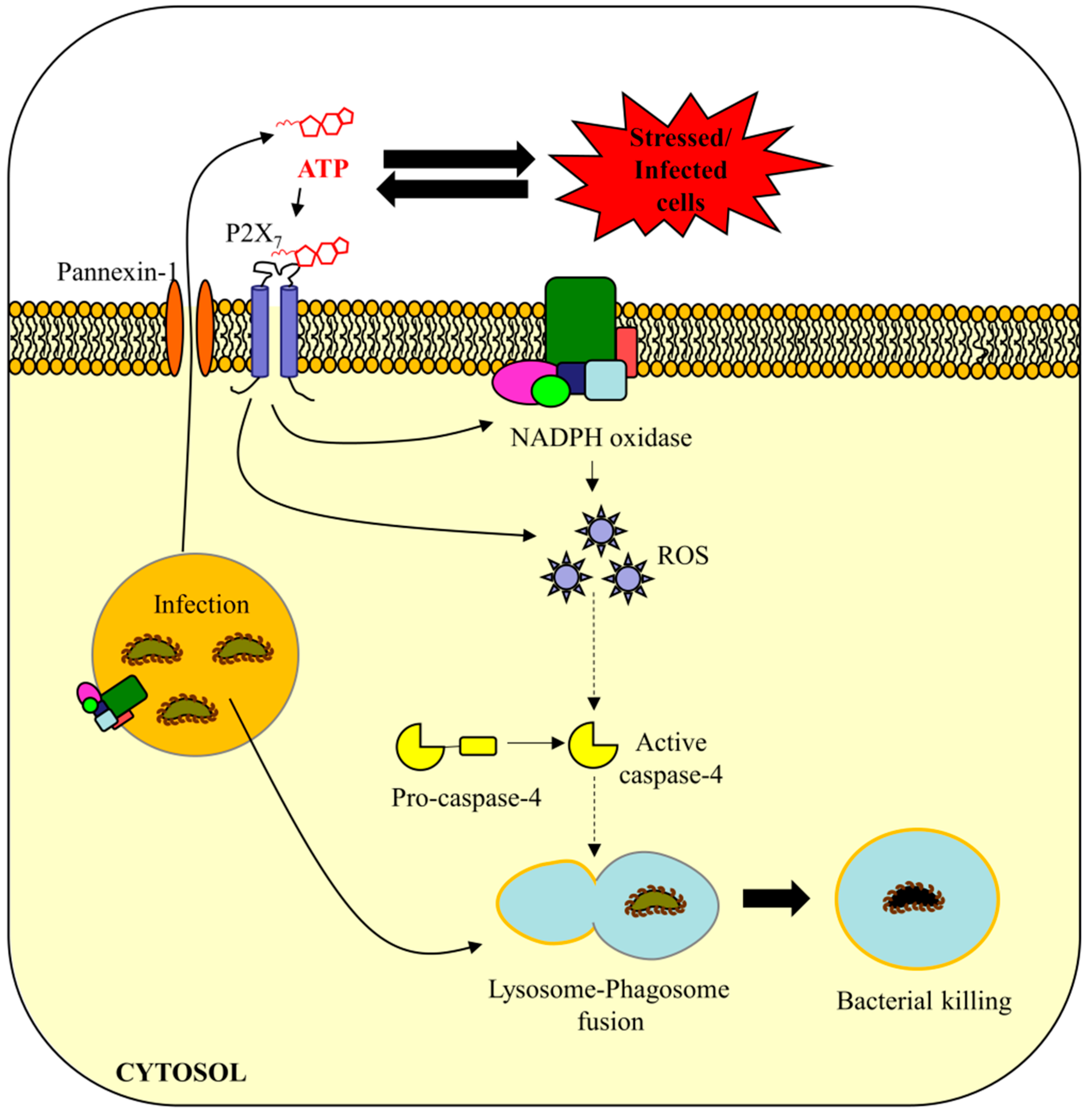
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Lee, K.L. The inflammasome and danger molecule signaling: At the crossroads of inflammation and pathogen persistence in the oral cavity. Periodontology 2000 2015, 69, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Said-Sadier, N.; Ojcius, D.M. Alarmins, inflammasomes and immunity. Biomed. J. 2012, 35, 437–449. [Google Scholar] [PubMed]
- Akhter, A.; Caution, K.; Abu Khweek, A.; Tazi, M.; Abdulrahman, B.A.; Abdelaziz, D.H.A.; Voss, O.H.; Doseff, A.I.; Hassan, H.; Azad, A.K.; et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 2012, 37, 35–47. [Google Scholar] [CrossRef] [PubMed]
- De Veerdonk, F.L.V.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Inflammasome activation and il-1 beta and il-18 processing during infection. Trends Immunol. 2011, 32, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.J.G.; Shenoy, A.R. Antimicrobial inflammasomes: Unified signalling against diverse bacterial pathogens. Curr. Opin. Microbiol. 2015, 23, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Creagh, E.M. Caspase crosstallk: Integration of apoptotic and innate immune signalliing pathways. Trends Immunol. 2014, 35, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Casson, C.N.; Yu, J.; Reyes, V.M.; Taschuk, F.O.; Yadav, A.; Copenhaver, A.M.; Nguyen, H.T.; Collman, R.G.; Shin, S. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 2015, 112, 6688–6693. [Google Scholar] [CrossRef] [PubMed]
- Knodler, L.A.; Crowley, S.M.; Sham, H.P.; Yang, H.J.; Wrande, M.; Ma, C.X.; Ernst, R.K.; Steele-Mortimer, O.; Celli, J.; Vallance, B.A. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Stowe, I.; Lee, B.; Kayagaki, N. Caspase-11: Arming the guards against bacterial infection. Immunol. Rev. 2015, 265, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Vigano, E.; Mortellaro, A. Caspase-11: The driving factor for noncanonical inflammasomes. Eur. J. Immunol. 2013, 43, 2240–2245. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Case, C.L.; Kohler, L.J.; Lima, J.B.; Strowig, T.; de Zoete, M.R.; Flavell, R.A.; Zamboni, D.S.; Roy, C.R. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to legionella pneumophila. Proc. Natl. Acad. Sci. USA 2013, 110, 1851–1856. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.F.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, U117–U146. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.J.; Zhao, Y.; Wang, Y.P.; Gao, W.Q.; Ding, J.J.; Li, P.; Hu, L.Y.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular lps. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Miura, M.; Jung, Y.K.; Zhu, H.; Gagliardini, V.; Shi, L.F.; Greenberg, A.H.; Yuan, J.Y. Identification and characterization of ich-3, a member of the interleukin-1 beta converting enzyme (ice)/ced-3 family and an upstream regulator of ice. J. Biol. Chem. 1996, 271, 20580–20587. [Google Scholar] [CrossRef]
- Wang, S.Y.; Miura, M.; Jung, Y.K.; Zhu, H.; Li, E.; Yuan, J.Y. Murine caspase-11, an ice-interacting protease, is essential for the activation of ice. Cell 1998, 92, 501–509. [Google Scholar] [CrossRef]
- Demon, D.; Kuchmiy, A.; Fossoul, A.; Zhu, Q.; Kanneganti, T.D.; Lamkanfi, M. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal. Immunol. 2014, 7, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Popat, R.; Bragdon, C.; O’Donnell, K.; Phelan, S.; Yuan, J.Y.; Sonis, S.T. Caspase-11 is not necessary for chemotherapy-induced intestinal mucositis. DNA Cell Biol. 2004, 23, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Leaf, I.A.; Hagar, J.A.; Fontana, M.F.; Campos, C.G.; Zak, D.E.; Tan, M.H.; Cotter, P.A.; Vance, R.E.; Aderem, A.; et al. Caspase-11 protects against bacteria that escape the vacuole. Science 2013, 339, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Noncanonical inflammasomes: Caspase-11 activation and effector mechanisms. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic lps activates caspase-11: Implications in tlr4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Gabrielli, E.; Pericolini, E.; Luciano, E.; Sabbatini, S.; Roselletti, E.; Perito, S.; Kasper, L.; Hube, B.; Vecchiarelli, A. Induction of caspase-11 by aspartyl proteinases of candida albicans and implication in promoting inflammatory response. Infect. Immun. 2015, 83, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Ogawa, M.; Sanada, T.; Mimuro, H.; Kim, M.; Ashida, H.; Akakura, R.; Yoshida, M.; Kawalec, M.; Reichhart, J.M.; et al. The shigella ospc3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 2013, 13, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.R.; Anand, P.K.; Liu, Z.P.; Stokes, K.L.; Vogel, P.; Lamkanfi, M.; Kanneganti, T.D. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical nlrp3 inflammasome during enteric pathogen infection. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Schauvliege, R.; Vanrobaeys, J.; Schotte, P.; Beyaert, R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa b and signal transducer and activator of transcription (stat) 1. J. Biol. Chem. 2002, 277, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O′Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signaling. Nature 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Shao, F. Non-canonical activation of inflammatory caspases by cytosolic lps in innate immunity. Curr. Opin. Immunol. 2015, 32, 78–83. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Schiff, T.; Voloudakis, G.; Sosa, M.A.G.; Elder, G.; Bozdagi, O.; Buxbaum, J.D. A critical role for human caspase-4 in endotoxin sensitivity. J. Immunol. 2014, 193, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Kistowska, M.; French, L.E.; Beer, H.D. Caspase-4 is required for activation of inflammasomes. J. Immunol. 2012, 188, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Wang, M.; Wang, L.; Cheng, B.F.; Lin, X.Y.; Feng, Z.W. Nf-kappa b regulates caspase-4 expression and sensitizes neuroblastoma cells to fas-induced apoptosis. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.Y.; Choi, M.S.; Porter, A.G. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the casp5 gene by lipopolysaccharide and interferon-gamma. J. Biol. Chem. 2000, 275, 39920–39926. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Cemma, M.; Brumell, J.H. Interactions of pathogenic bacteria with autophagy systems. Curr. Biol. 2012, 22, R540–R545. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Henault, J.; Kolbeck, R.; Sanjuan, M.A. Noncanonical autophagy: One small step for lc3, one giant leap for immunity. Curr. Opin. Immunol. 2014, 26, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Fairn, G.D.; Grinstein, S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 2012, 33, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. Nadph oxidases contribute to autophagy regulation. Autophagy 2009, 5, 887–889. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.B.; Randow, F. Rubicon swaps autophagy for lap. Nat. Cell Biol. 2015, 17, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Wu, J.M.; Wang, F.T.; Liu, B.; Huang, C.H.; Wei, Y.Q. Deconvoluting the role of reactive oxygen species and autophagy in human diseases. Free Radic. Biol. Med. 2013, 65, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored snare syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Nordenfelt, P.; Tapper, H. Phagosome dynamics during phagocytosis by neutrophils. J. Leukoc. Biol. 2011, 90, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Campoy, E.; Colombo, M.I. Autophagy in intracellular bacterial infection. BBA-Mol. Cell Res. 2009, 1793, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Dick, M.S.; Dreier, R.F.; Schurmann, N.; Broz, D.K.; Warming, S.; Roose-Girma, M.; Bumann, D.; Kayagaki, N.; Takeda, K.; et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by ifn-induced gtpases. Nature 2014, 509, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Pilla, D.M.; Hagar, J.A.; Haldar, A.K.; Mason, A.K.; Degrandi, D.; Pfeffer, K.; Ernst, R.K.; Yamamoto, M.; Miao, E.A.; Coers, J. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic lps. Proc. Natl. Acad. Sci. USA 2014, 111, 6046–6051. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Miao, E.A. Detection of cytosolic bacteria by inflammatory caspases. Curr. Opin. Microbiol. 2014, 17, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brumell, J.H. Bacteria-autophagy interplay: A battle for survival. Nat. Rev. Microbiol. 2014, 12, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Alix, E.; Mukherjee, S.; Roy, C.R. Host-pathogen interactions subversion of membrane transport pathways by vacuolar pathogens. J. Cell Biol. 2011, 195, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Cambier, C.J.; Falkow, S.; Ramakrishnan, L. Host evasion and exploitation schemes of mycobacterium tuberculosis. Cell 2014, 159, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Lee, K.; Atanasova, K.R.; Spooner, R.; Yilmaz, O. P. Gingivalis traffics into er-rich-autophagosomes for successful survival in gingival-epithelial-cells. In Proceedings of the International Association for Dental Research, Seattle, WA, USA, 20–23 March 2013.
- Yilmaz, O. The chronicles of porphyromonas gingivalis: The microbium, the human oral epithelium and their interplay. Microbiology 2008, 154, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Sater, A.A.; Yao, L.Y.; Koutouzis, T.; Pettengill, M.; Ojcius, D.M. Atp-dependent activation of an inflammasome in primary gingival epithelial cells infected by porphyromonas gingivalis. Cell. Microbiol. 2010, 12, 188–198. [Google Scholar] [CrossRef]
- Yilmaz, O.; Verbeke, P.; Lamont, R.J.; Ojcius, D.M. Intercellular spreading of porphyromonas gingivalis infection in primary gingival epithelial cells. Infect. Immun. 2006, 74, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Yao, L.; Maeda, K.; Rose, T.M.; Lewis, E.L.; Duman, M.; Lamont, R.J.; Ojcius, D.M. Atp scavenging by the intracellular pathogen porphyromonas gingivalis inhibits p2x7-mediated host-cell apoptosis. Cell. Microbiol. 2008, 10, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits atp-induced reactive-oxygen-species via p2x7 receptor/nadph-oxidase signalling and contributes to persistence. Cell Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, K.R.; Yilmaz, O. Prelude to oral microbes and chronic diseases: Past, present and future. Microbes Infect. 2015, 17, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Atanasova, K.R.; Bui, P.Q.; Lee, J.; Hung, S.C.; Yilmaz, O.; Ojcius, D.M. Porphyromonas gingivalis attenuates atp-mediated inflammasome activation and hmgb1 release through expression of a nucleoside-diphosphate kinase. Microbes Infect. 2015, 17, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; DeGuzman, J.V.; Lamont, R.J.; Yilmaz, O. Genetic transformation of an obligate anaerobe, p. Gingivalis for fmn-green fluorescent protein expression in studying host-microbe interaction. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.H.; Belanger, M.; Dunn, W., Jr.; Progulske-Fox, A. Porphyromonas gingivalis and the autophagic pathway: An innate immune interaction? Front Biosci. 2008, 13, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Mishra, B.B.; Jordao, L.; Elliott, E.; Anes, E.; Griffiths, G. Nf-kappa b activation controls phagolysosome fusion-mediated killing of mycobacteria by macrophages. J. Immunol. 2008, 181, 2651–2663. [Google Scholar] [CrossRef] [PubMed]
- Coats, S.R.; Jones, J.W.; Do, C.T.; Braham, P.H.; Bainbridge, B.W.; To, T.T.; Goodlett, D.R.; Ernst, R.K.; Darveau, R.P. Human toll-like receptor 4 responses to p. Gingivalis are regulated by lipid a 1- and 4′-phosphatase activities. Cell Microbiol. 2009, 11, 1587–1599. [Google Scholar] [CrossRef] [PubMed]
- Slocum, C.; Coats, S.R.; Hua, N.; Kramer, C.; Papadopoulos, G.; Weinberg, E.O.; Gudino, C.V.; Hamilton, J.A.; Darveau, R.P.; Genco, C.A. Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence 2015, 6, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hasturk, H.; Nguyen, D.; van Dyke, T.E.; Kantarci, A.; Darveau, R.P. Porphyromonas gingivalis lipid a phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect. Immun. 2014, 82, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Signaling by ros drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, A. Extracellular atp in the immune system: More than just a “danger signal”. Sci. Signal. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Liaisons dangereuses: P2x7 and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Schwiebert, E.M.; Zsembery, A. Extracellular atp as a signaling molecule for epithelial cells. BBA-Biomembranes 2003, 1615, 7–32. [Google Scholar] [CrossRef]
- Boots, A.W.; Hristova, M.; Kasahara, D.I.; Haenen, G.R.M.M.; Bast, A.; van der Vliet, A. Atp-mediated activation of the nadph oxidase duox1 mediates airway epithelial responses to bacterial stimuli. J. Biol. Chem. 2009, 284, 17858–17867. [Google Scholar] [CrossRef] [PubMed]
- Bucheimer, R.E.; Linden, J. Purinergic regulation of epithelial transport. J. Physiol. 2004, 555, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.F.; MacKenzie, A.B. Nadph oxidase nox2 mediates rapid cellular oxidation following atp stimulation of endotoxin-primed macrophages. J. Immunol. 2009, 183, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Coutinho-Silva, R.; Ojcius, D.M. Role of extracellular nucleotides in the immune response against intracellular bacteria and protozoan parasites. Microbes Infect. 2012, 14, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic p2z receptor for extracellular atp identified as a p2x receptor (p2x7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Chiozzi, P.; Falzoni, S.; Hanau, S.; di Virgilio, F. Purinergic modulation of interleukin-1 beta release from microglial cells stimulated with bacterial endotoxin. J. Exp. Med. 1997, 185, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Morandini, A.C.; Savio, L.E.; Coutinho-Silva, R. The role of p2x7 receptor in infectious inflammatory diseases and the influence of ectonucleotidases. Biomed. J. 2014, 37, 169–177. [Google Scholar] [PubMed]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.; Qureshi, O.S.; Lee, W.Y.; Croudace, J.E.; Mura, M.; Lammas, D.A. Atp-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The activation of p2x7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Selvarajan, K.; Moldovan, L.; Chandrakala, A.N.; Litvinov, D.; Parthasarathy, S. Peritoneal macrophages are distinct from monocytes and adherent macrophages. Atherosclerosis 2011, 219, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 2008, 9, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.O.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by nadph oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [PubMed]
- Sokolovska, A.; Becker, C.E.; Ip, W.K.E.; Rathinam, V.A.K.; Brudner, M.; Paquette, N.; Tanne, A.; Vanaja, S.K.; Moore, K.J.; Fitzgerald, K.A.; et al. Activation of caspase-1 by the nlrp3 inflammasome regulates the nadph oxidase nox2 to control phagosome function. Nat. Immunol. 2013, 14, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Lee, J.S.; Rodgers, M.; Min, C.K.; Lee, J.Y.; Kim, H.J.; Lee, K.H.; Kim, C.J.; Oh, B.; Zandi, E.; et al. Autophagy protein rubicon mediates phagocytic nadph oxidase activation in response to microbial infection or tlr stimulation. Cell Host Microbe 2012, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, J.S.; Yilmaz, Ӧ. Dangerous Liaisons: Caspase-11 and Reactive Oxygen Species Crosstalk in Pathogen Elimination. Int. J. Mol. Sci. 2015, 16, 23337-23354. https://doi.org/10.3390/ijms161023337
Roberts JS, Yilmaz Ӧ. Dangerous Liaisons: Caspase-11 and Reactive Oxygen Species Crosstalk in Pathogen Elimination. International Journal of Molecular Sciences. 2015; 16(10):23337-23354. https://doi.org/10.3390/ijms161023337
Chicago/Turabian StyleRoberts, JoAnn Simone, and Ӧzlem Yilmaz. 2015. "Dangerous Liaisons: Caspase-11 and Reactive Oxygen Species Crosstalk in Pathogen Elimination" International Journal of Molecular Sciences 16, no. 10: 23337-23354. https://doi.org/10.3390/ijms161023337
APA StyleRoberts, J. S., & Yilmaz, Ӧ. (2015). Dangerous Liaisons: Caspase-11 and Reactive Oxygen Species Crosstalk in Pathogen Elimination. International Journal of Molecular Sciences, 16(10), 23337-23354. https://doi.org/10.3390/ijms161023337