
Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer

 ,
, 
Abstract
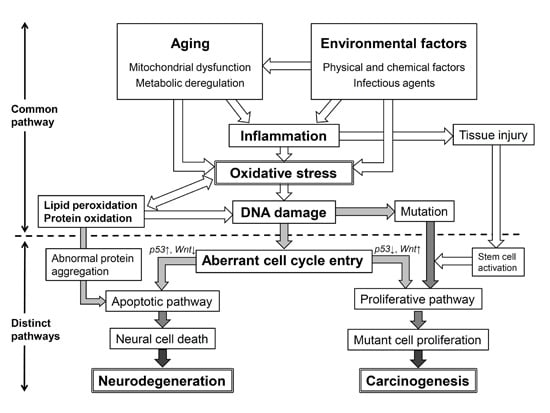
:
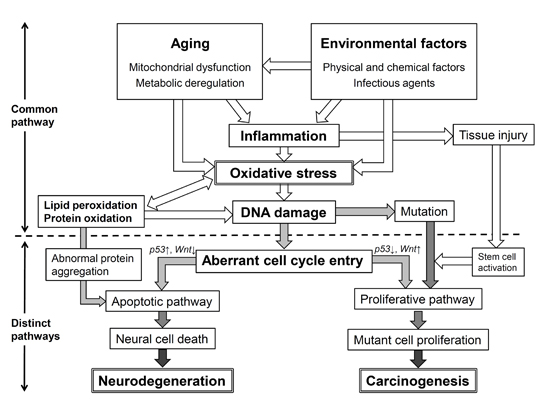{kind=link}
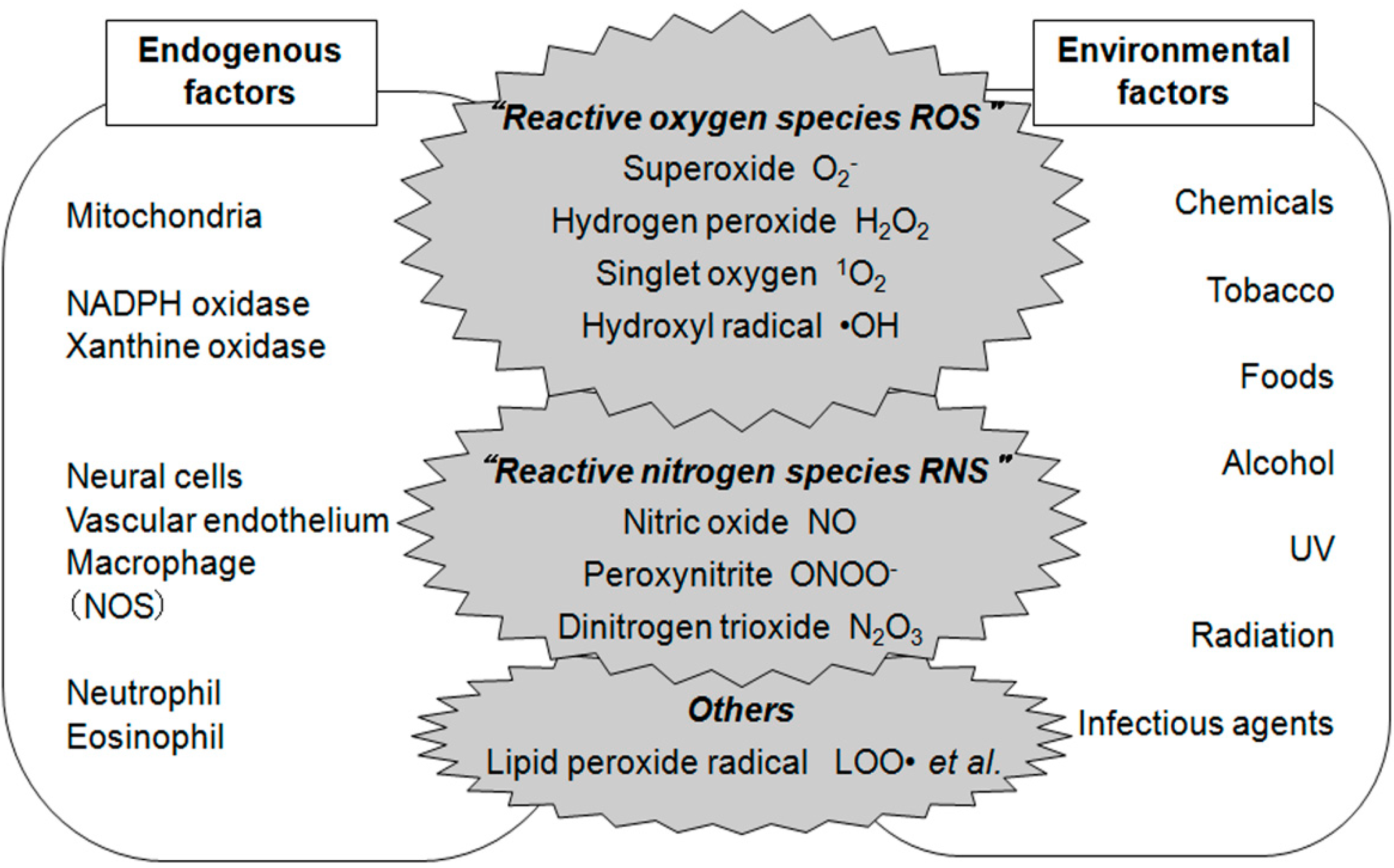{kind=link}
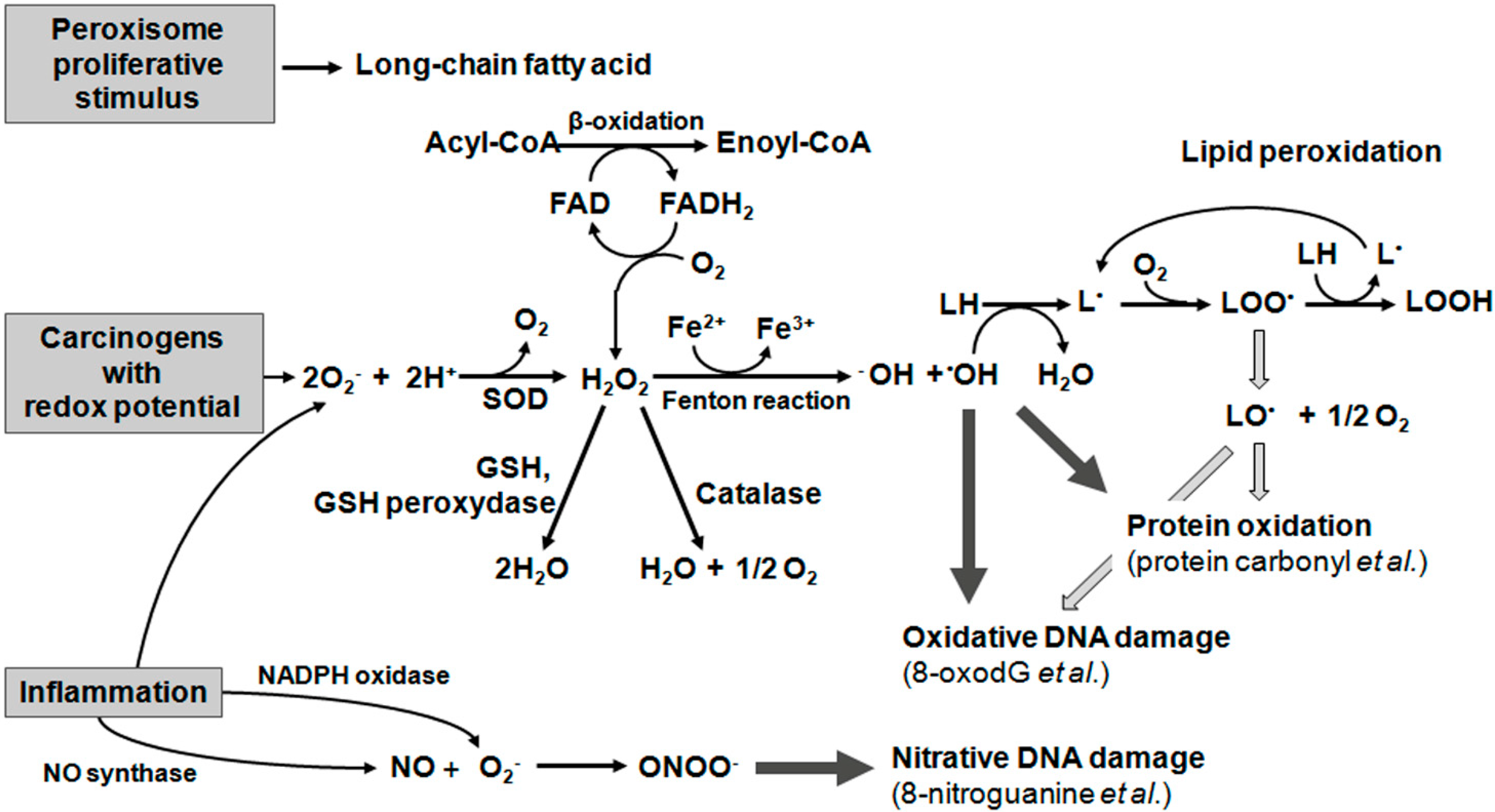{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Oxidative Damage to Lipids
2.1. Lipid Peroxidation
2.2. Oxysterols
3. Oxidative Damage to Protein
Carbonyl Protein as a Potential Oxidative Damage Biomarker
4. Oxidative Damage to DNA
4.1. DNA Damage Formation from Oxidative Modification
4.2. DNA Damage Formation from Nitrative Modification
4.3. 8-OxodG and 8-Nitroguanine Are Potential Mutagenic DNA Lesions
5. Oxidative Stress and Neurodegenerative Diseases
5.1. Lipid Oxidation in Neurodegenerative Diseases
5.2. Carbonyl Protein in Neurodegenerative Diseases
5.3. DNA Damage in Neurodegenerative Diseases
6. Oxidative Stress and Cancers
6.1. Lipid Oxidation in Cancers
6.2. Carbonyl Protein in Cancers
6.3. DNA Damage in Inflammation-Related Cancers
6.3.1. DNA Damage in Parasite-Associated Cancers
6.3.2. DNA Damage in Bacterial and Viral Infection-Related Cancers
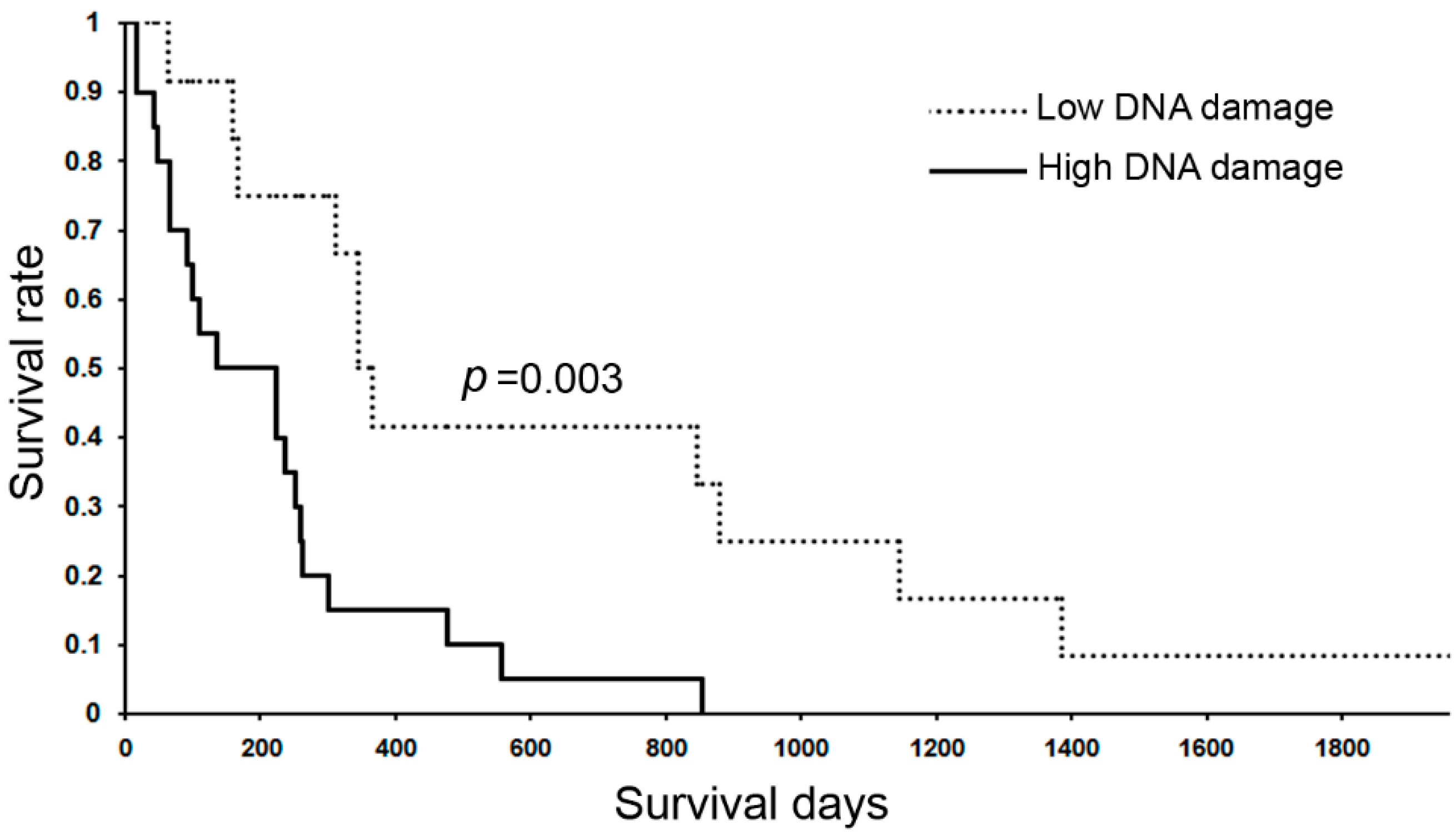
6.3.3. DNA Damage in Inflammation-Related Diseases and Cancers
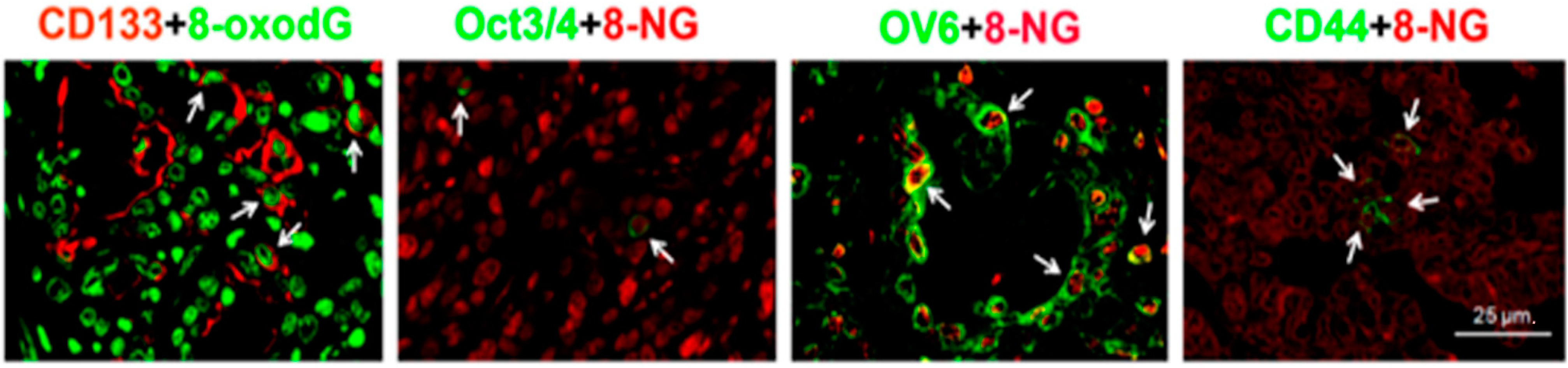
6.4. DNA Damage in Stemness Cells of Inflammation-Related Cancers
6.5. Oxidative Stress and DNA Methylation
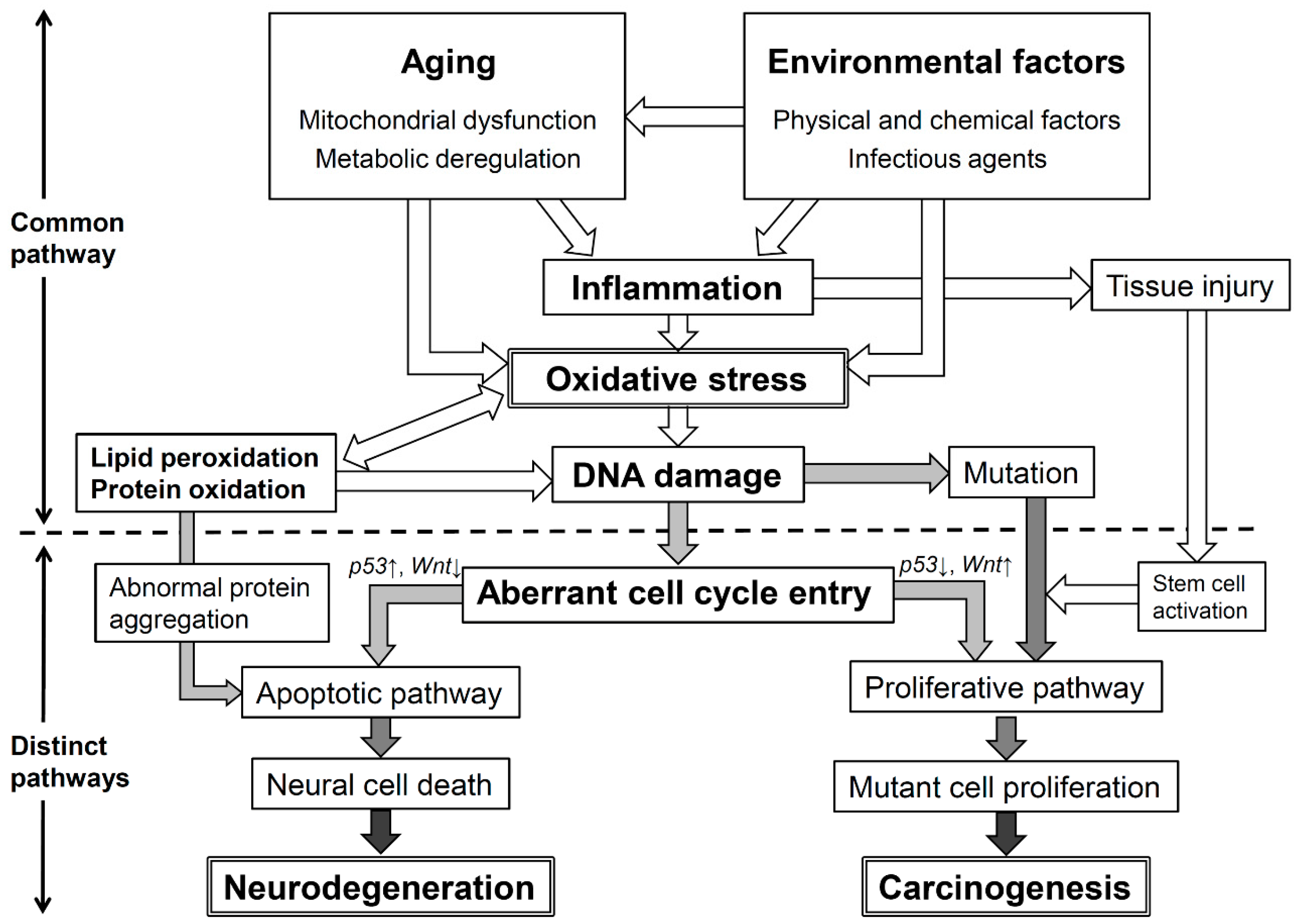
7. Conclusions: Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of nox and duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Hiraku, Y.; Kawanishi, S.; Ichinose, T.; Murata, M. The role of inos-mediated DNA damage in infection- and asbestos-induced carcinogenesis. Ann. N. Y. Acad. Sci. 2010, 1203, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012. [Google Scholar] [CrossRef]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Stewart, A.J.; Pellegrini, N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Yongvanit, P.; Loilome, W.; Namwat, N.; Kuver, R. Mechanisms of oxysterol-induced carcinogenesis. Lipids Health Dis. 2011, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Caccia, C. Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 2011, 164, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Natoli, S.; Micheletta, F.; Riggi, S.; di Angelantonio, E.; Montali, A.; Antonini, T.M.; Antonini, R.; Diczfalusy, U.; Iuliano, L. Increased plasma levels of oxysterols, in vivo markers of oxidative stress, in patients with familial combined hyperlipidemia: Reduction during atorvastatin and fenofibrate therapy. Free Radic. Biol. Med. 2007, 42, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Lemaire-Ewing, S.; Prunet, C.; Montange, T.; Vejux, A.; Berthier, A.; Bessede, G.; Corcos, L.; Gambert, P.; Neel, D.; Lizard, G. Comparison of the cytotoxic, pro-oxidant and pro-inflammatory characteristics of different oxysterols. Cell Biol. Toxicol. 2005, 21, 97–114. [Google Scholar]
- Lizard, G.; Gueldry, S.; Sordet, O.; Monier, S.; Athias, A.; Miguet, C.; Bessede, G.; Lemaire, S.; Solary, E.; Gambert, P. Glutathione is implied in the control of 7-ketocholesterol-induced apoptosis, which is associated with radical oxygen species production. FASEB J. 1998, 12, 1651–1663. [Google Scholar] [PubMed]
- Yoon, J.H.; Canbay, A.E.; Werneburg, N.W.; Lee, S.P.; Gores, G.J. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: Implications for biliary tract carcinogenesis. Hepatology 2004, 39, 732–738. [Google Scholar] [PubMed]
- Bjorkhem, I.; Diczfalusy, U. Oxysterols: Friends, foes, or just fellow passengers? Arterioscler. Thromb. Vasc. Biol. 2002, 22, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.M.; Mallozzi, C.; Macchia, G.; Petrucci, T.C.; Minetti, M. Peroxynitrite induces tryosine nitration and modulates tyrosine phosphorylation of synaptic proteins. J. Neurochem. 1999, 73, 727–735. [Google Scholar]
- He, K.; Nukada, H.; McMorran, P.D.; Murphy, M.P. Protein carbonyl formation and tyrosine nitration as markers of oxidative damage during ischaemia-reperfusion injury to rat sciatic nerve. Neuroscience 1999, 94, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Telci, A.; Cakatay, U.; Karaca, C.; Akcay, T.; Sivas, A.; Altug, T. Oxidative protein damage parameters in plasma in chronic experimental diabetes in rats. Eur. J. Med. Res. 2003, 8, 307–312. [Google Scholar]
- Lamb, N.J.; Gutteridge, J.M.; Baker, C.; Evans, T.W.; Quinlan, G.J. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: Evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit. Care Med. 1999, 27, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Lyall, F.; Gibson, J.L.; Greer, I.A.; Brockman, D.E.; Eis, A.L.; Myatt, L. Increased nitrotyrosine in the diabetic placenta: Evidence for oxidative stress. Diabetes Care 1998, 21, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; de Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (ages and ales): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl. 1), 3–27. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990, 186, 464–478. [Google Scholar]
- Nakamura, A.; Goto, S. Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. J. Biochem. 1996, 119, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kobayashi, H.; Kitamura, Y.; Zhu, H.; Obata, K.; Minabe, Y.; Dazortsava, M.; Ohashi, K.; Tada-Oikawa, S.; Takahashi, H.; et al. Proteomic analysis of carbonylated proteins in the monkey substantia nigra after ischemia-reperfusion. Free Radic. Res. 2014, 48, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Yamada, T.; Minohata, T.; Kobayashi, H.; Furukawa, A.; Tada-Oikawa, S.; Hiraku, Y.; Murata, M.; Kikuchi, M.; Yamashima, T. Proteomic identification of carbonylated proteins in the monkey hippocampus after ischemia-reperfusion. Free Radic. Biol. Med. 2009, 46, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Yongvanit, P.; Hiraku, Y.; Ma, N.; Pinlaor, S.; Pairojkul, C.; Wongkham, C.; Sripa, B.; Khuntikeo, N.; et al. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic. Biol. Med. 2012, 52, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Regazzoni, L.; Orioli, M.; Rimoldi, I.; Facino, R.M.; Carini, M. A tandem ms precursor-ion scan approach to identify variable covalent modification of albumin cys34: A new tool for studying vascular carbonylation. J. Mass Spectrom. 2008, 43, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Prokai, L. To tag or not to tag: A comparative evaluation of immunoaffinity-labeling and tandem mass spectrometry for the identification and localization of posttranslational protein carbonylation by 4-hydroxy-2-nonenal, an end-product of lipid peroxidation. J. Proteomics 2011, 74, 2360–2369. [Google Scholar] [CrossRef] [PubMed]
- Meany, D.L.; Xie, H.; Thompson, L.V.; Arriaga, E.A.; Griffin, T.J. Identification of carbonylated proteins from enriched rat skeletal muscle mitochondria using affinity chromatography-stable isotope labeling and tandem mass spectrometry. Proteomics 2007, 7, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Regnier, F. Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal. Chem. 2005, 77, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Prokai-Tatrai, K.; Prokai, L. Identification of carbonylation sites in apomyoglobin after exposure to 4-hydroxy-2-nonenal by solid-phase enrichment and liquid chromatography-electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2010, 45, 398–410. [Google Scholar] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Ravanat, J.L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Langley, B. Chromatin modifications associated with DNA double-strand breaks repair as potential targets for neurological diseases. Neurotherapeutics 2013, 10, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Hiraku, Y.; Oikawa, S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat. Res. 2001, 488, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Rozalski, R.; Gackowski, D.; Foksinski, M.; Siomek, A.; Cooke, M.S. Urinary measurement of 8-oxodg, 8-oxogua, and 5HMUra: A noninvasive assessment of oxidative damage to DNA. Antioxid. Redox Signal. 2006, 8, 1011–1019. [Google Scholar]
- Cooke, M.S.; Olinski, R.; Loft, S. Measurement and meaning of oxidatively modified DNA lesions in urine. Cancer Epidemiol. Biomark. Prev. 2008, 17, 3–14. [Google Scholar] [CrossRef]
- Akaike, T.; Fujii, S.; Kato, A.; Yoshitake, J.; Miyamoto, Y.; Sawa, T.; Okamoto, S.; Suga, M.; Asakawa, M.; Nagai, Y.; et al. Viral mutation accelerated by nitric oxide production during infection in vivo. FASEB J. 2000, 14, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxygen and nitrogen are pro-carcinogens: Damage to DNA by reactive oxygen, chlorine and nitrogen species: Measurement, mechanism and the effects of nutrition. Mutat. Res. 1999, 443, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Yermilov, V.; Rubio, J.; Becchi, M.; Friesen, M.D.; Pignatelli, B.; Ohshima, H. Formation of 8-nitroguanine by the reaction of guanine with peroxynitrite in vitro. Carcinogenesis 1995, 16, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Nishino, H.; Ohshima, H. Formation of 8-nitroguanosine in cellular RNA as a biomarker of exposure to reactive nitrogen species. Chem. Biol. Interact. 2002, 139, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Adachi, Y.; Hiraku, Y.; Horiki, N.; Horiike, S.; Imoto, I.; Pinlaor, S.; Murata, M.; Semba, R.; Kawanishi, S. Accumulation of 8-nitroguanine in human gastric epithelium induced by helicobacter pylori infection. Biochem. Biophys. Res. Commun. 2004, 319, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Tatemichi, M.; Akaike, T.; Barbin, A.; Ohshima, H. Analysis of urinary 8-nitroguanine, a marker of nitrative nucleic acid damage, by high-performance liquid chromatography-electrochemical detection coupled with immunoaffinity purification: Association with cigarette smoking. Free Radic. Biol. Med. 2006, 40, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Nash, H.M.; Bruner, S.D.; Scharer, O.D.; Kawate, T.; Addona, T.A.; Spooner, E.; Lane, W.S.; Verdine, G.L. Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a base-excision DNA-repair protein superfamily. Curr. Biol. 1996, 6, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Earley, M.C.; Crouse, G.F. The role of mismatch repair in the prevention of base pair mutations in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1998, 95, 15487–15491. [Google Scholar] [CrossRef] [PubMed]
- Ni, T.T.; Marsischky, G.T.; Kolodner, R.D. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-Guanine in S. cerevisiae. Mol. Cell 1999, 4, 439–444. [Google Scholar] [CrossRef]
- Reardon, J.T.; Bessho, T.; Kung, H.C.; Bolton, P.H.; Sancar, A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA 1997, 94, 9463–9468. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.P.; Song, J.B.; Crouse, G.F. In vivo bypass of 8-oxodG. PLoS Genet. 2013, 9, e1003682. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Progress Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Bruner, S.D.; Norman, D.P.; Verdine, G.L. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 2000, 403, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Yermilov, V.; Rubio, J.; Ohshima, H. Formation of 8-nitroguanine in DNA treated with peroxynitrite in vitro and its rapid removal from DNA by depurination. FEBS Lett. 1995, 376, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Preston, B.D. Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet. 1986, 20, 201–230. [Google Scholar] [CrossRef]
- Suzuki, N.; Yasui, M.; Geacintov, N.E.; Shafirovich, V.; Shibutani, S. Miscoding events during DNA synthesis past the nitration-damaged base 8-nitroguanine. Biochemistry 2005, 44, 9238–9245. [Google Scholar] [CrossRef] [PubMed]
- Haracska, L.; Unk, I.; Johnson, R.E.; Johansson, E.; Burgers, P.M.; Prakash, S.; Prakash, L. Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 2001, 15, 945–954. [Google Scholar] [CrossRef]
- Wu, X.; Takenaka, K.; Sonoda, E.; Hochegger, H.; Kawanishi, S.; Kawamoto, T.; Takeda, S.; Yamazoe, M. Critical roles for polymerase ζ in cellular tolerance to nitric oxide-induced DNA damage. Cancer Res. 2006, 66, 748–754. [Google Scholar] [CrossRef]
- Chan-On, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Urosevic, N.; Krtolica, K.; Skaro-Milic, A.; Knezevic-Usaj, S.; Dujic, A. Prevalence of G-to-T transversions among K-ras oncogene mutations in human colorectal tumors in Yugoslavia. Int. J. Cancer 1993, 54, 249–254. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Patterns of p53 G→T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 2001, 22, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Hyde, J.S. Concentration of oxygen in lipid bilayers using a spin-label method. Biophys. J. 1983, 41, 283–286. [Google Scholar] [CrossRef]
- Bjorkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Asp. Med. 2009, 30, 171–179. [Google Scholar] [CrossRef]
- Lund, E.G.; Guileyardo, J.M.; Russell, D.W. Cdna cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA 1999, 96, 7238–7243. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.G.; Li, L.; Muller, W.E.; Eckert, G.P. Cholesterol as a causative factor in Alzheimer’s disease: A debatable hypothesis. J. Neurochem. 2014, 129, 559–572. [Google Scholar] [CrossRef]
- Huang, Z.; Ichihara, S.; Oikawa, S.; Chang, J.; Zhang, L.; Takahashi, M.; Subramanian, K.; Mohideen, S.S.; Wang, Y.; Ichihara, G. Proteomic analysis of hippocampal proteins of F344 rats exposed to 1-bromopropane. Toxicol. Appl. Pharmacol. 2011, 257, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Yim, M.B.; Chock, P.B. Free radicals and disease. Arch. Biochem. Biophys. 2002, 397, 354–359. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein oxidation in aging: Does it play a role in aging progression? Antioxid. Redox Signal. 2014. [Google Scholar] [CrossRef]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New α- and γ-synuclein immunopathological lesions in human brain. Acta Neuropathol. Commun. 2014, 2, 132. [Google Scholar] [PubMed]
- Picklo, M.J.; Montine, T.J.; Amarnath, V.; Neely, M.D. Carbonyl toxicology and Alzheimer’s disease. Toxicol. Appl. Pharmacol. 2002, 184, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010, 20, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Kawamoto, Y.; Chiba, Y.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Yoshikawa, K.; Hosokawa, M.; Oikawa, S.; Kato, M.; et al. Proteomic identification of hippocampal proteins vulnerable to oxidative stress in excitotoxin-induced acute neuronal injury. Neurobiol. Dis. 2011, 43, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Yamashima, T.; Oikawa, S. The role of lysosomal rupture in neuronal death. Progress Neurobiol. 2009, 89, 343–358. [Google Scholar] [CrossRef]
- Brasnjevic, I.; Hof, P.R.; Steinbusch, H.W.; Schmitz, C. Accumulation of nuclear DNA damage or neuron loss: Molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair 2008, 7, 1087–1097. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Wang, H.; Boldogh, I.; Rao, K.S.; Mitra, S.; Hegde, M.L. New perspectives on oxidized genome damage and repair inhibition by pro-oxidant metals in neurological diseases. Biomolecules 2014, 4, 678–703. [Google Scholar] [CrossRef]
- Halliwell, B. The wanderings of a free radical. Free Radic. Biol. Med. 2009, 46, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Hirosawa, I.; Tada-Oikawa, S.; Furukawa, A.; Nishiura, K.; Kawanishi, S. Mechanism for manganese enhancement of dopamine-induced oxidative DNA damage and neuronal cell death. Free Radic. Biol. Med. 2006, 41, 748–756. [Google Scholar] [CrossRef]
- Kobayashi, H.; Oikawa, S.; Umemura, S.; Hirosawa, I.; Kawanishi, S. Mechanism of metal-mediated DNA damage and apoptosis induced by 6-hydroxydopamine in neuroblastoma SH-SY5Y cells. Free Radic. Res. 2008, 42, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Piao, F.; Ma, N.; Hiraku, Y.; Murata, M.; Oikawa, S.; Cheng, F.; Zhong, L.; Yamauchi, T.; Kawanishi, S.; Yokoyama, K. Oxidative DNA damage in relation to neurotoxicity in the brain of mice exposed to arsenic at environmentally relevant levels. J. Occup. Health 2005, 47, 445–449. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Kanski, J. Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 2001, 122, 945–962. [Google Scholar] [CrossRef]
- Hands, S.; Sajjad, M.U.; Newton, M.J.; Wyttenbach, A. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J. Biol. Chem. 2011, 286, 44512–44520. [Google Scholar] [PubMed]
- Dechakhamphu, S.; Pinlaor, S.; Sitthithaworn, P.; Nair, J.; Bartsch, H.; Yongvanit, P. Lipid peroxidation and etheno DNA adducts in white blood cells of liver fluke-infected patients: Protection by plasma α-tocopherol and praziquantel. Cancer Epidemiol. Biomark. Prev. 2010, 19, 310–318. [Google Scholar] [CrossRef]
- Dechakhamphu, S.; Yongvanit, P.; Nair, J.; Pinlaor, S.; Sitthithaworn, P.; Bartsch, H. High excretion of etheno adducts in liver fluke-infected patients: Protection by praziquantel against DNA damage. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1658–1664. [Google Scholar] [CrossRef]
- Dechakhamphu, S.; Pinlaor, S.; Sitthithaworn, P.; Bartsch, H.; Yongvanit, P. Accumulation of miscoding etheno-DNA adducts and highly expressed DNA repair during liver fluke-induced cholangiocarcinogenesis in hamsters. Mutat. Res. 2010, 691, 9–16. [Google Scholar] [CrossRef]
- Loilome, W.; Wechagama, P.; Namwat, N.; Jusakul, A.; Sripa, B.; Miwa, M.; Kuver, R.; Yongvanit, P. Expression of oxysterol binding protein isoforms in opisthorchiasis-associated cholangiocarcinoma: A potential molecular marker for tumor metastasis. Parasitol. Int. 2012, 61, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Loilome, W.; Namwat, N.; Haigh, W.G.; Kuver, R.; Dechakhamphu, S.; Sukontawarin, P.; Pinlaor, S.; Lee, S.P.; Yongvanit, P. Liver fluke-induced hepatic oxysterols stimulate DNA damage and apoptosis in cultured human cholangiocytes. Mutat. Res. 2012, 731, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Loilome, W.; Namwat, N.; Techasen, A.; Kuver, R.; Ioannou, G.N.; Savard, C.; Haigh, W.G.; Yongvanit, P. Anti-apoptotic phenotypes of cholestan-3β,5α,6β-triol-resistant human cholangiocytes: Characteristics contributing to the genesis of cholangiocarcinoma. J. Steroid Biochem. Mol. Biol. 2013, 138, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Manucha, W.; Valles, P. Hsp70/nitric oxide relationship in apoptotic modulation during obstructive nephropathy. Cell Stress Chaperones 2008, 13, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gettins, P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Masson, V. Roles of serine proteases and matrix metalloproteinases in tumor invasion and angiogenesis. Bull. Mem. Acad. R. Med. Belg. 2006, 161, 320–326. [Google Scholar] [PubMed]
- Jamnongkan, W.; Techasen, A.; Thanan, R.; Duenngai, K.; Sithithaworn, P.; Mairiang, E.; Loilome, W.; Namwat, N.; Pairojkul, C.; Yongvanit, P. Oxidized α-1 antitrypsin as a predictive risk marker of opisthorchiasis-associated cholangiocarcinoma. Tumor Biol. 2013, 34, 695–704. [Google Scholar] [CrossRef]
- Mannello, F.; Tonti, G.A.; Medda, V. Protein oxidation in breast microenvironment: Nipple aspirate fluid collected from breast cancer women contains increased protein carbonyl concentration. Cell. Oncol. 2009, 31, 383–392. [Google Scholar] [PubMed]
- Rossner, P., Jr.; Terry, M.B.; Gammon, M.D.; Agrawal, M.; Zhang, F.F.; Ferris, J.S.; Teitelbaum, S.L.; Eng, S.M.; Gaudet, M.M.; Neugut, A.I.; et al. Plasma protein carbonyl levels and breast cancer risk. J. Cell. Mol. Med. 2007, 11, 1138–1148. [Google Scholar] [CrossRef]
- Zipprich, J.; Terry, M.B.; Liao, Y.; Agrawal, M.; Gurvich, I.; Senie, R.; Santella, R.M. Plasma protein carbonyls and breast cancer risk in sisters discordant for breast cancer from the new york site of the breast cancer family registry. Cancer Res. 2009, 69, 2966–2972. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Thanan, R.; Ma, N.; Kawanishi, S. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Ma, N.; Thanan, R.; Kobayashi, H.; Hammam, O.; Wishahi, M.; el Leithy, T.; Hiraku, Y.; Amro el, K.; Oikawa, S.; Ohnishi, S.; et al. Nitrative DNA damage and Oct3/4 expression in urinary bladder cancer with Schistosoma haematobium infection. Biochem. Biophys. Res. Commun. 2011, 414, 344–349. [Google Scholar] [CrossRef]
- Yongvanit, P.; Pinlaor, S.; Bartsch, H. Oxidative and nitrative DNA damage: Key events in opisthorchiasis-induced carcinogenesis. Parasitol. Int. 2012, 61, 130–135. [Google Scholar] [CrossRef]
- Thanan, R.; Murata, M.; Pinlaor, S.; Sithithaworn, P.; Khuntikeo, N.; Tangkanakul, W.; Hiraku, Y.; Oikawa, S.; Yongvanit, P.; Kawanishi, S. Urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine in patients with parasite infection and effect of antiparasitic drug in relation to cholangiocarcinogenesis. Cancer Epidemiol. Biomark. Prev. 2008, 17, 518–524. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef]
- Hiraku, Y.; Tabata, T.; Ma, N.; Murata, M.; Ding, X.; Kawanishi, S. Nitrative and oxidative DNA damage in cervical intraepithelial neoplasia associated with human papilloma virus infection. Cancer Sci. 2007, 98, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Zhang, B.B.; Ma, N.; Murata, M.; Tang, A.Z.; Huang, G.W. Nitrative and oxidative DNA damage as potential survival biomarkers for nasopharyngeal carcinoma. Med. Oncol. 2011, 28, 377–384. [Google Scholar] [CrossRef]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.W.; Huang, Y.; Luo, D.Z.; Mo, W.G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in ebv-associated nasopharyngeal carcinoma: The relation to stat3 activation and egfr expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef]
- Fujita, N.; Horiike, S.; Sugimoto, R.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Hasegawa, K.; Ma, N.; Kawanishi, S.; Adachi, Y.; et al. Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis c patients. Free Radic. Biol. Med. 2007, 42, 353–362. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujita, N.; Sugimoto, R.; Urawa, N.; Horiike, S.; Kobayashi, Y.; Iwasa, M.; Ma, N.; Kawanishi, S.; Watanabe, S.; et al. Hepatic oxidative DNA damage is associated with increased risk for hepatocellular carcinoma in chronic hepatitis c. Br. J. Cancer 2008, 98, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Pairojkul, C.; Pinlaor, S.; Khuntikeo, N.; Wongkham, C.; Sripa, B.; Ma, N.; Vaeteewoottacharn, K.; Furukawa, A.; Kobayashi, H.; et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free Radic. Biol. Med. 2013, 65, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- IARC. IARC Mongraphs on the Evalution of Carcinogenic Risk to Humans; World Health Organization: Geneva, Switzerland, 2012; Volume 100, pp. 219–309. [Google Scholar]
- Hiraku, Y.; Sakai, K.; Shibata, E.; Kamijima, M.; Hisanaga, N.; Ma, N.; Kawanishi, S.; Murata, M. Formation of the nitrative DNA lesion 8-nitroguanine is associated with asbestos contents in human lung tissues: A pilot study. J. Occup. Health 2014, 56, 186–196. [Google Scholar] [CrossRef]
- Scully, C.; Beyli, M.; Ferreiro, M.C.; Ficarra, G.; Gill, Y.; Griffiths, M.; Holmstrup, P.; Mutlu, S.; Porter, S.; Wray, D. Update on oral lichen planus: Etiopathogenesis and management. Crit. Rev. Oral. Biol. Med. 1998, 9, 86–122. [Google Scholar]
- Mignogna, M.D.; Fedele, S.; Lo Russo, L.; Lo Muzio, L.; Bucci, E. Immune activation and chronic inflammation as the cause of malignancy in oral lichen planus: Is there any evidence ? Oral. Oncol. 2004, 40, 120–130. [Google Scholar] [CrossRef]
- Chaiyarit, P.; Ma, N.; Hiraku, Y.; Pinlaor, S.; Yongvanit, P.; Jintakanon, D.; Murata, M.; Oikawa, S.; Kawanishi, S. Nitrative and oxidative DNA damage in oral lichen planus in relation to human oral carcinogenesis. Cancer Sci. 2005, 96, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Tagawa, T.; Hiraku, Y.; Murata, M.; Ding, X.; Kawanishi, S. 8-Nitroguanine formation in oral leukoplakia, a premalignant lesion. Nitric Oxide 2006, 14, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ekbom, A.; Helmick, C.; Zack, M.; Adami, H.O. Increased risk of large-bowel cancer in Crohn’s disease with colonic involvement. Lancet 1990, 336, 357–359. [Google Scholar] [CrossRef]
- Ding, X.; Hiraku, Y.; Ma, N.; Kato, T.; Saito, K.; Nagahama, M.; Semba, R.; Kuribayashi, K.; Kawanishi, S. Inducible nitric oxide synthase-dependent DNA damage in mouse model of inflammatory bowel disease. Cancer Sci. 2005, 96, 157–163. [Google Scholar] [PubMed]
- Hoki, Y.; Hiraku, Y.; Ma, N.; Murata, M.; Matsumine, A.; Nagahama, M.; Shintani, K.; Uchida, A.; Kawanishi, S. Inos-dependent DNA damage in patients with malignant fibrous histiocytoma in relation to prognosis. Cancer Sci. 2007, 98, 163–168. [Google Scholar] [CrossRef]
- Alison, M.R. Liver stem cells: Implications for hepatocarcinogenesis. Stem Cell Rev. 2005, 1, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Collins, A.T. Inflammation as the primary aetiological agent of human prostate cancer: A stem cell connection? J. Cell Biochem. 2008, 105, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Polyak, K. The cancer stem cell hypothesis: In search of definitions, markers, and relevance. Lab. Investig. 2008, 88, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M.; Kawanishi, S. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxid. Med. Cell. Longev. 2014, 2013, 387014. [Google Scholar]
- Thanan, R.; Murata, M.; Ma, N.; Hammam, O.; Wishahi, M.; El Leithy, T.; Hiraku, Y.; Oikawa, S.; Kawanishi, S. Nuclear localization of COX-2 in relation to the expression of stemness markers in urinary bladder cancer. Mediat. Inflamm. 2012. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+ human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef]
- Guo, Y.; Einhorn, L.; Kelley, M.; Hirota, K.; Yodoi, J.; Reinbold, R.; Scholer, H.; Ramsey, H.; Hromas, R. Redox regulation of the embryonic stem cell transcription factor Oct-4 by thioredoxin. Stem Cells 2004, 22, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc− and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive oxygen species in cancer stem cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Domann, F.E. An epigenetic perspective on the free radical theory of development. Free Radic. Biol. Med. 2007, 43, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Laget, S.; Miotto, B.; Chin, H.G.; Esteve, P.O.; Roberts, R.J.; Pradhan, S.; Defossez, P.A. MBD4 cooperates with DNMT1 to mediate methyl-DNA repression and protects mammalian cells from oxidative stress. Epigenetics 2014, 9, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Soberanes, S.; Gonzalez, A.; Urich, D.; Chiarella, S.E.; Radigan, K.A.; Osornio-Vargas, A.; Joseph, J.; Kalyanaraman, B.; Ridge, K.M.; Chandel, N.S.; et al. Particulate matter air pollution induces hypermethylation of the p16 promoter via a mitochondrial ROS-JNK-DNMT1 pathway. Sci. Rep. 2012. [Google Scholar] [CrossRef]
- Hodge, D.R.; Cho, E.; Copeland, T.D.; Guszczynski, T.; Yang, E.; Seth, A.K.; Farrar, W.L. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genomics Proteomics 2007, 4, 387–398. [Google Scholar]
- Li, Y.; Deuring, J.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C.J. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis 2012, 33, 1889–1896. [Google Scholar] [CrossRef] [PubMed]
- Thaler, R.; Agsten, M.; Spitzer, S.; Paschalis, E.P.; Karlic, H.; Klaushofer, K.; Varga, F. Homocysteine suppresses the expression of the collagen cross-linker lysyl oxidase involving IL-6, Fli1, and epigenetic DNA methylation. J. Biol. Chem. 2007, 286, 5578–5588. [Google Scholar]
- Niller, H.H.; Wolf, H.; Minarovits, J. Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin. Cancer Biol. 2009, 19, 158–164. [Google Scholar] [CrossRef]
- Chen, F.; Mo, Y.; Ding, H.; Xiao, X.; Wang, S.Y.; Huang, G.; Zhang, Z.; Wang, S.Z. Frequent epigenetic inactivation of Myocardin in human nasopharyngeal carcinoma. Head Neck 2011, 33, 54–59. [Google Scholar] [PubMed]
- Du, C.; Huang, T.; Sun, D.; Mo, Y.; Feng, H.; Zhou, X.; Xiao, X.; Yu, N.; Hou, B.; Huang, G.; et al. CDH4 as a novel putative tumor suppressor gene epigenetically silenced by promoter hypermethylation in nasopharyngeal carcinoma. Cancer Lett. 2011, 309, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Midorikawa, K.; Zhang, Z.; Zhou, X.; Ma, N.; Huang, G.; Hiraku, Y.; Oikawa, S.; Murata, M. Promoter hypermethylation of Ras-related GTPase gene RRAD inactivates a tumor suppressor function in nasopharyngeal carcinoma. Cancer Lett. 2012, 323, 147–154. [Google Scholar] [PubMed]
- Wang, S.; Xiao, X.; Zhou, X.; Huang, T.; Du, C.; Yu, N.; Mo, Y.; Lin, L.; Zhang, J.; Ma, N.; et al. TFPI-2 is a putative tumor suppressor gene frequently inactivated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer 2010, 10, 617. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, D.; van, D.N.; Tang, A.; Hu, L.; Huang, G. Inactivation of RASSF2A by promoter methylation correlates with lymph node metastasis in nasopharyngeal carcinoma. Int. J. Cancer 2007, 120, 32–38. [Google Scholar] [CrossRef]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflammation and ageing: Current theories and an overview of the data. Rev. Recent Clin. Trials 2011, 6, 189–203. [Google Scholar]
- Aseervatham, G.S.; Sivasudha, T.; Jeyadevi, R.; Arul Ananth, D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans—An overview. Environ. Sci. Pollut. Res. Int. 2013, 20, 4356–4369. [Google Scholar]
- Mylonas, C.; Kouretas, D. Lipid peroxidation and tissue damage. In Vivo (Athens, Greece) 1999, 13, 295–309. [Google Scholar]
- Giordano, S.; Darley-Usmar, V.; Zhang, J. Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol. 2014, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A. Inverse association between cancer and neurodegenerative disease: Review of the epidemiologic and biological evidence. Biogerontology 2014, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2015, 16, 193-217. https://doi.org/10.3390/ijms16010193
Thanan R, Oikawa S, Hiraku Y, Ohnishi S, Ma N, Pinlaor S, Yongvanit P, Kawanishi S, Murata M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. International Journal of Molecular Sciences. 2015; 16(1):193-217. https://doi.org/10.3390/ijms16010193
Chicago/Turabian StyleThanan, Raynoo, Shinji Oikawa, Yusuke Hiraku, Shiho Ohnishi, Ning Ma, Somchai Pinlaor, Puangrat Yongvanit, Shosuke Kawanishi, and Mariko Murata. 2015. "Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer" International Journal of Molecular Sciences 16, no. 1: 193-217. https://doi.org/10.3390/ijms16010193
APA StyleThanan, R., Oikawa, S., Hiraku, Y., Ohnishi, S., Ma, N., Pinlaor, S., Yongvanit, P., Kawanishi, S., & Murata, M. (2015). Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. International Journal of Molecular Sciences, 16(1), 193-217. https://doi.org/10.3390/ijms16010193