
Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”





Abstract
:1. Introduction
2. Results
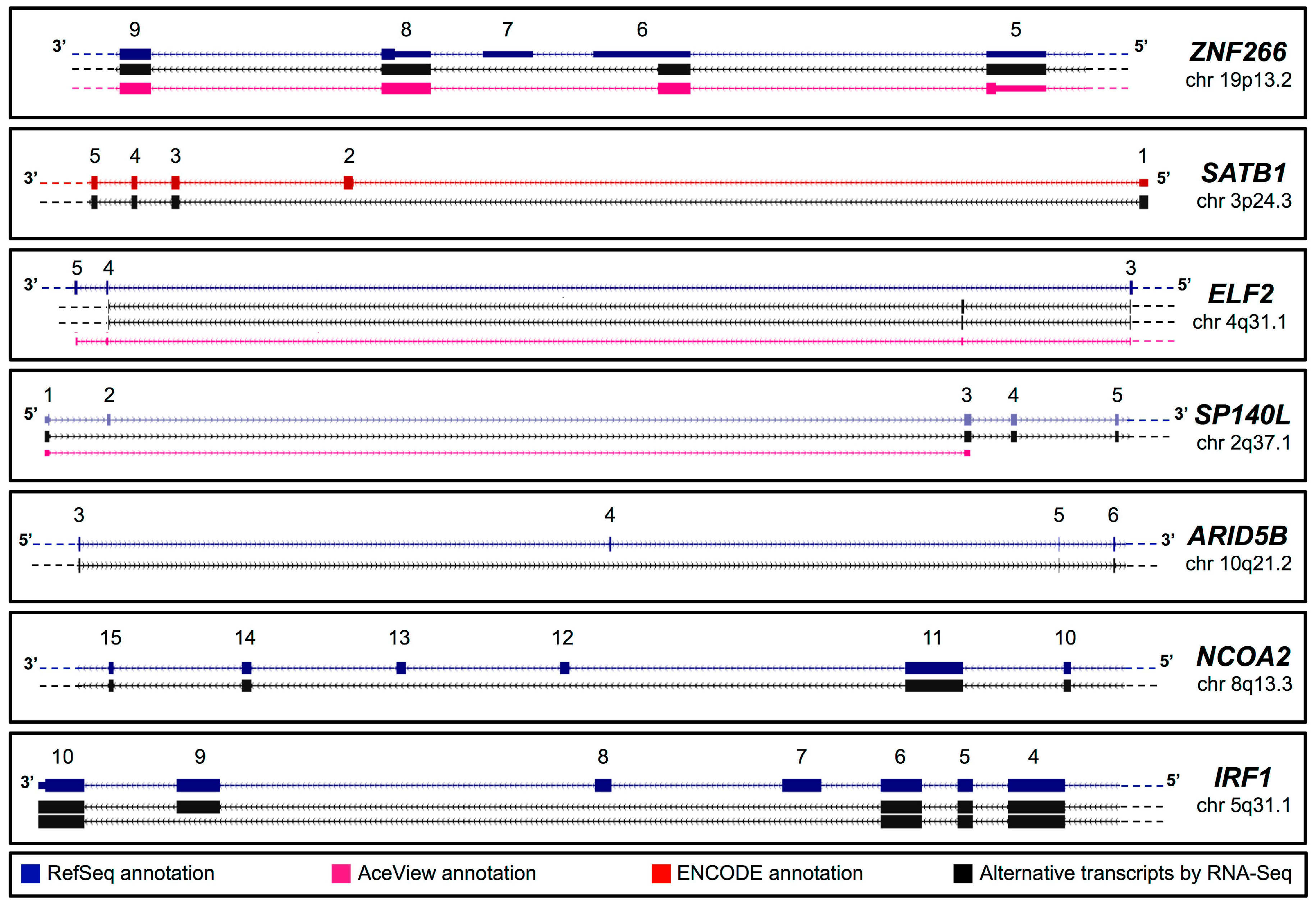
2.1. In Silico Identification of New Splice Isoforms of Genes Encoding Transcription Factors (TFs)
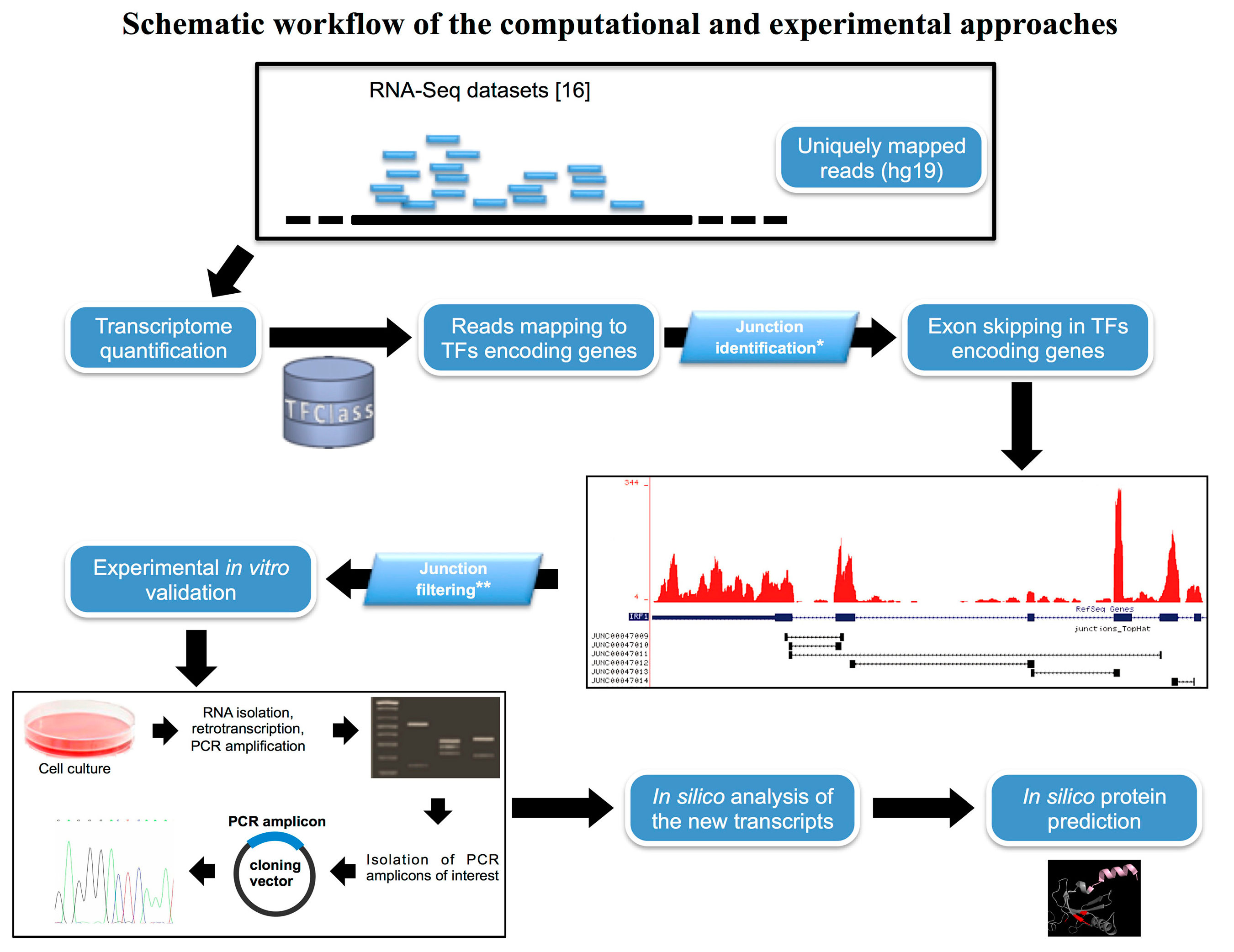
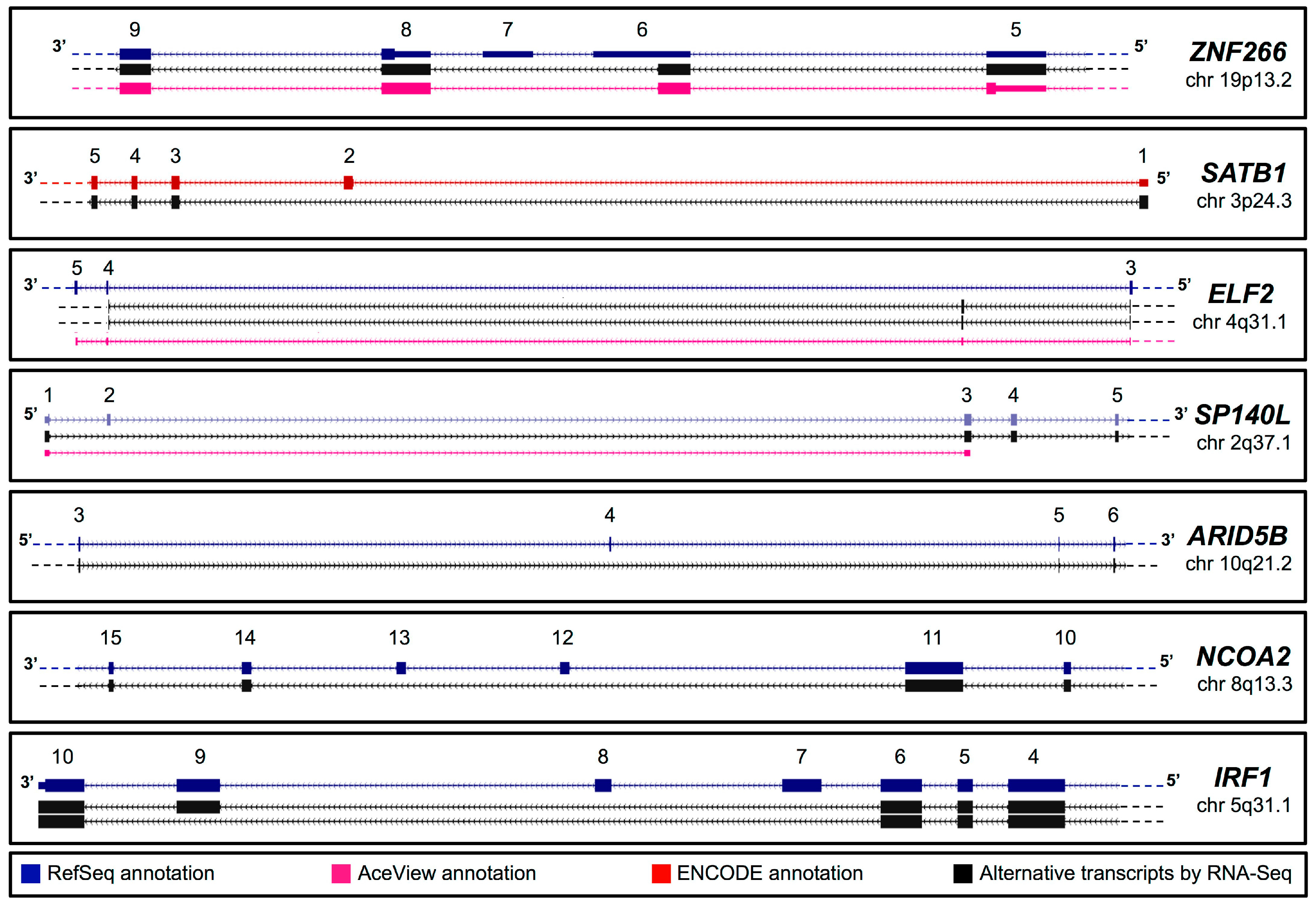

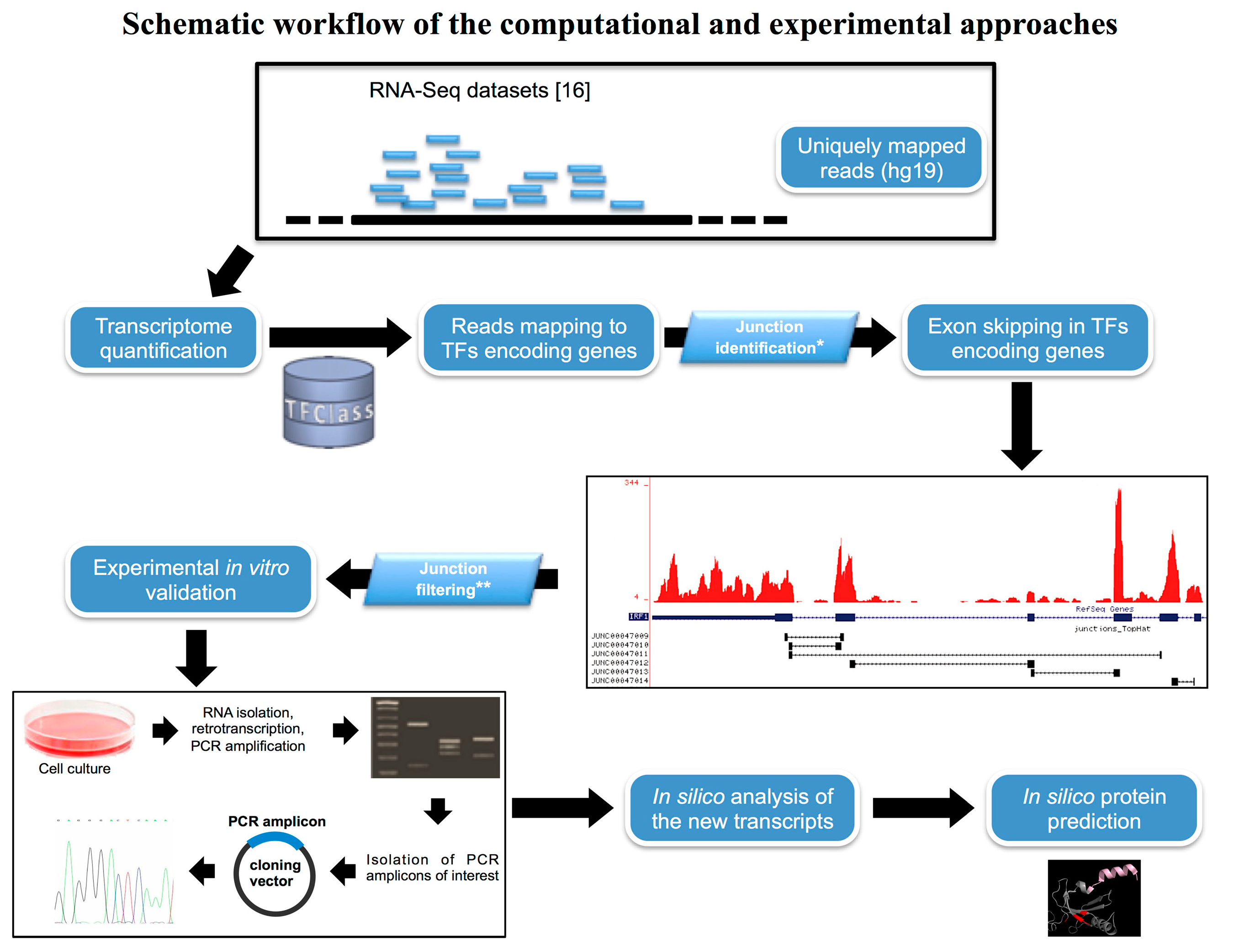{kind=link}
{kind=link}
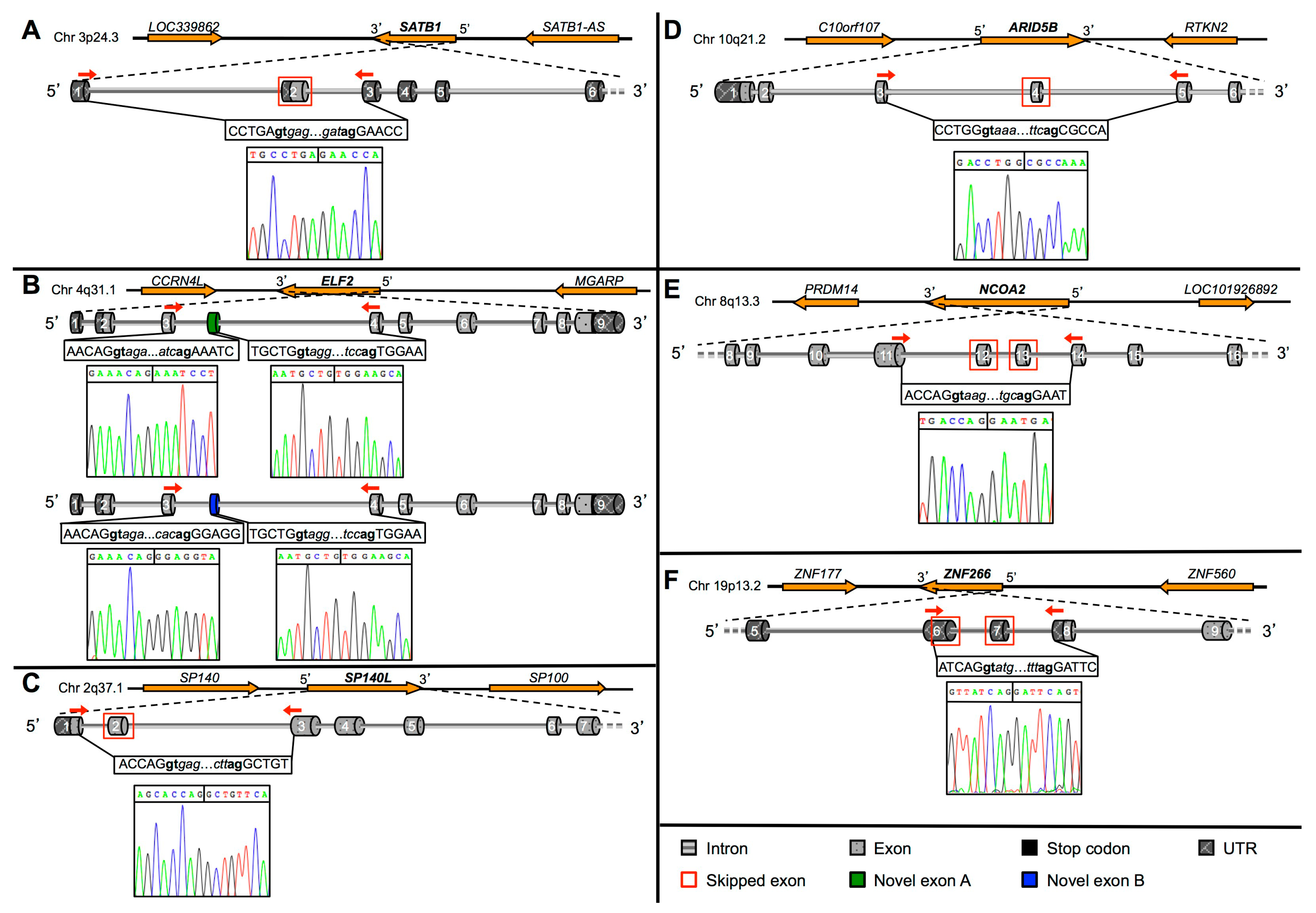{kind=link}
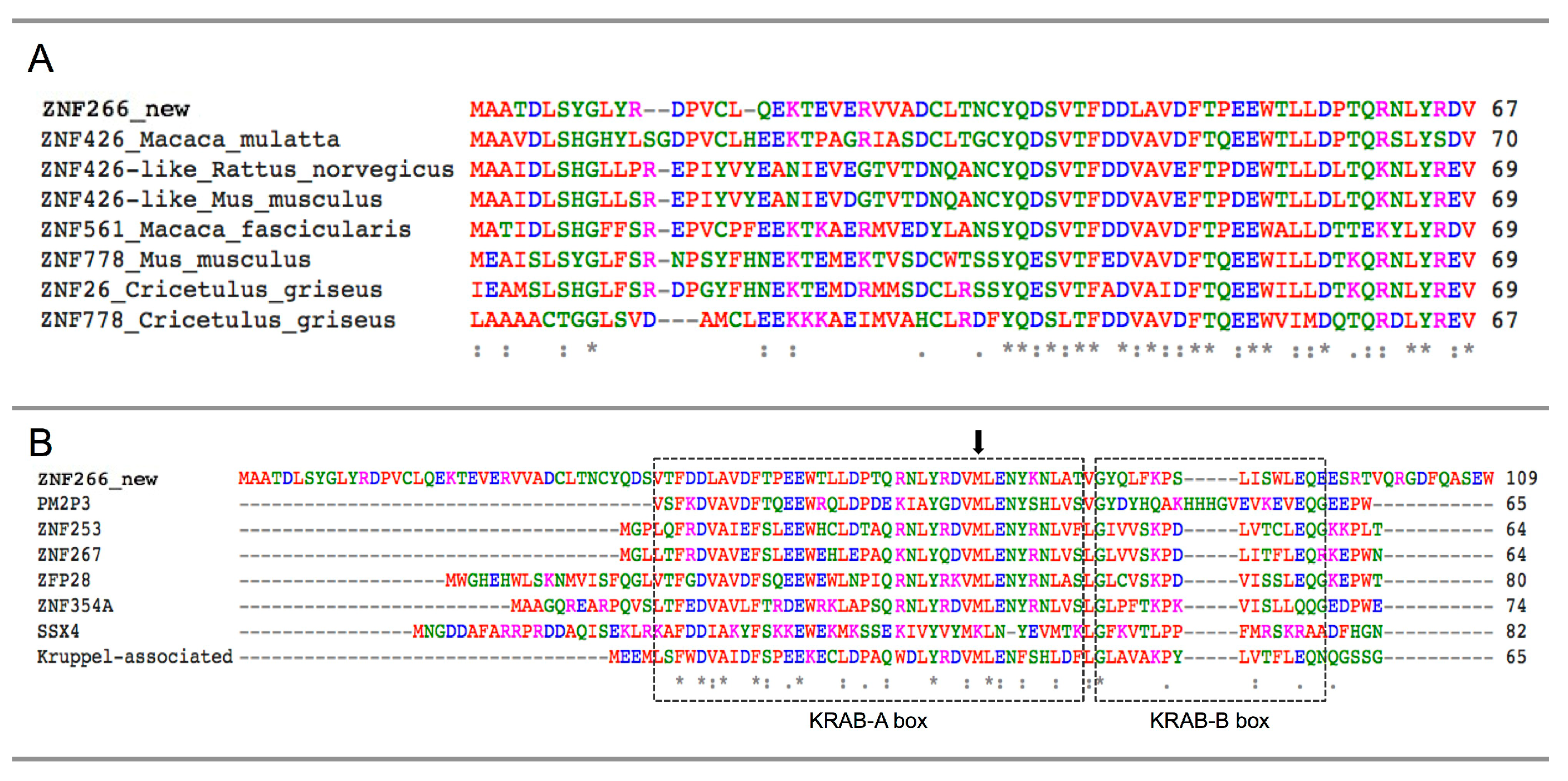
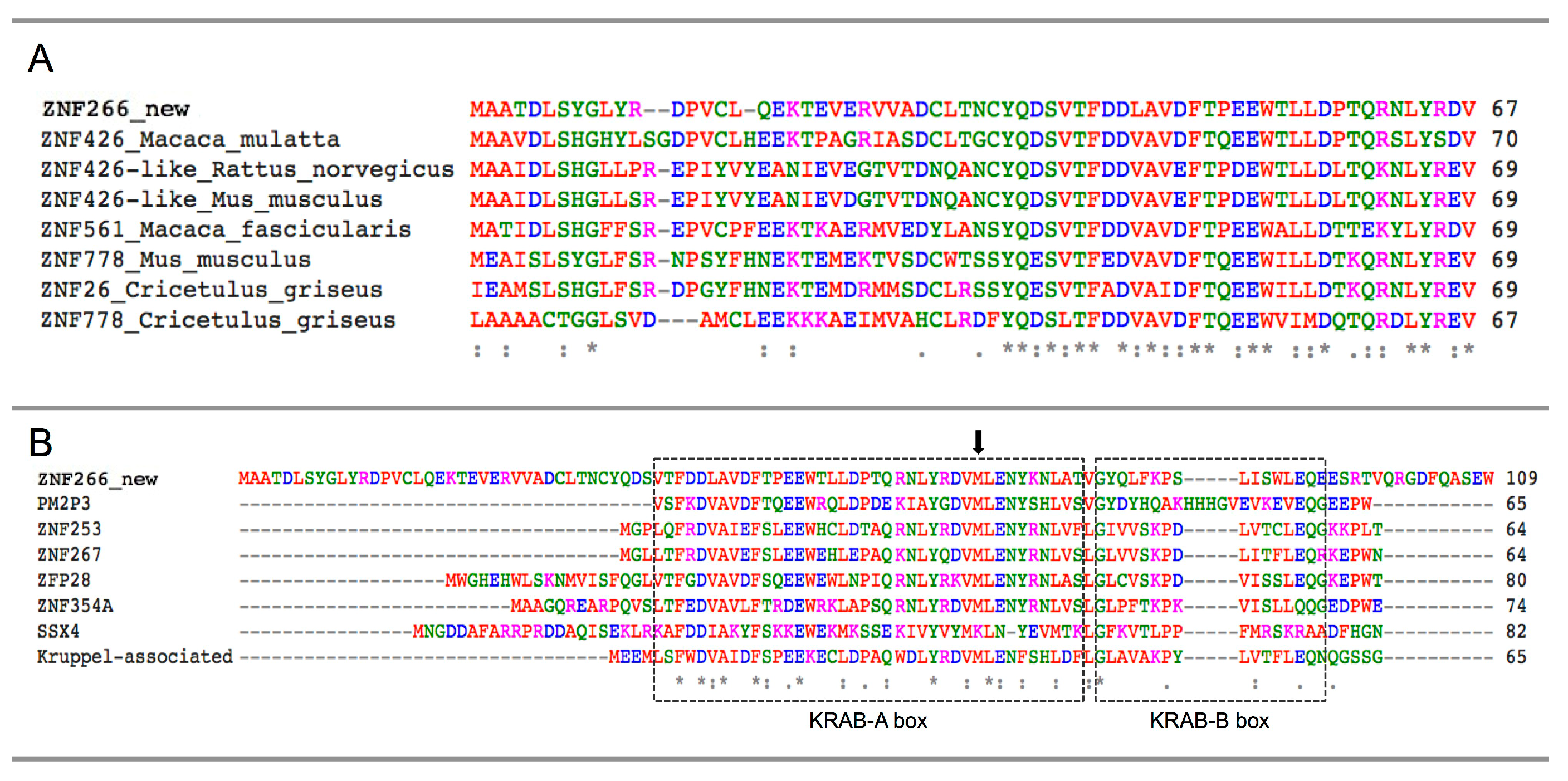{kind=link}
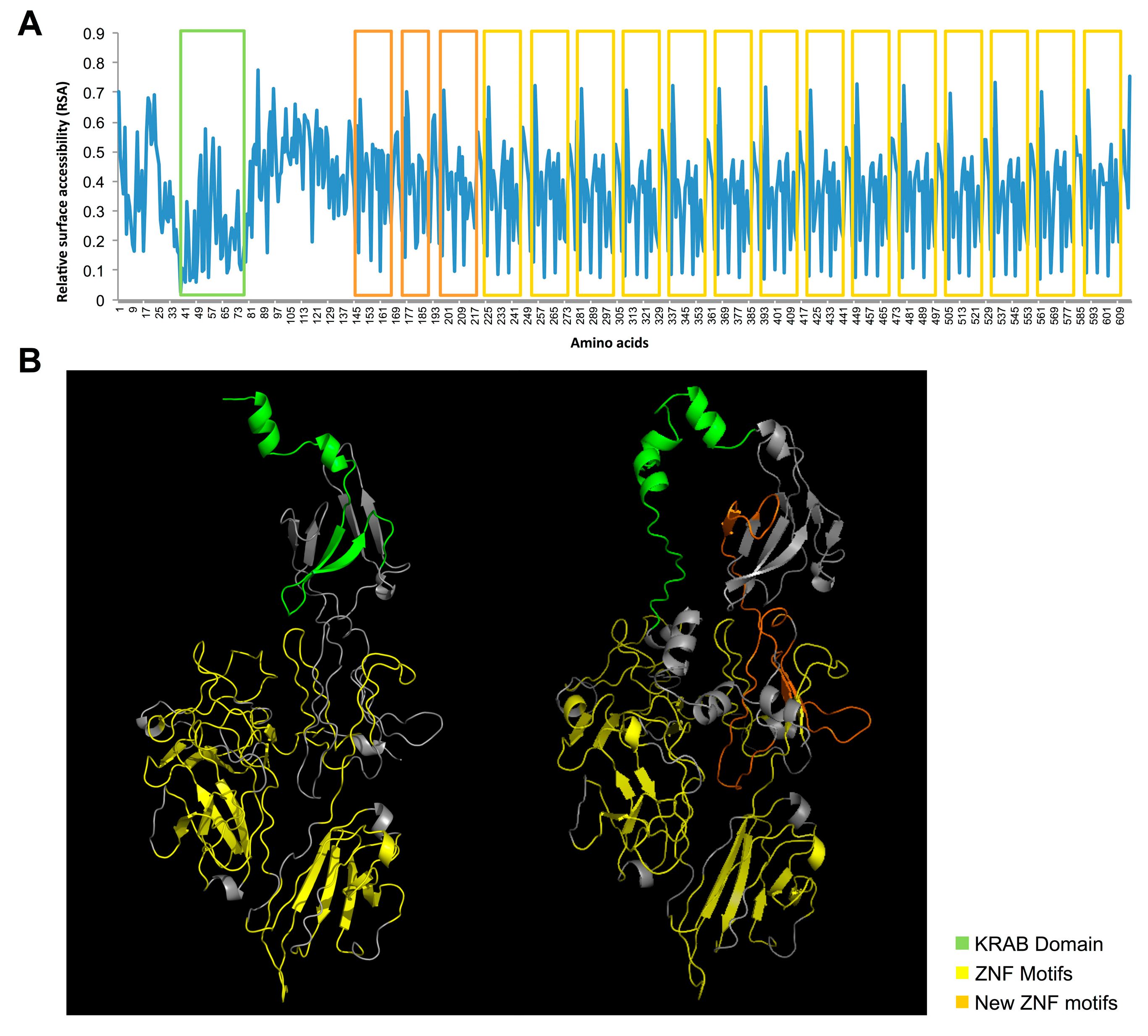{kind=link}
| Gene Symbol | Chromosome Position | Accession Number | Primer Sequence (5'–3') | |
|---|---|---|---|---|
| Forward Primer | Reverse Primer | |||
| ZNF266 | 19p13.2 | LN607832 | GAAGTAGAAAGGGTGGTGGC | TTCTTGTAGTTCTCCAGCATC |
| SATB1 | 3p24.3 | LN626687 | CGTATGGGGAAAGAGGACAA | GCGTTTTCATAATGTTCCACC |
| ELF2 | 4q31.1 | LN626691 | GAGACCGAGAATGTGGAAAC | TACTGCTGTGAACTGATGCT |
| ELF2 | 4q31.1 | LN626692 | GAGACCGAGAATGTGGAAAC | TACTGCTGTGAACTGATGCT |
| SP140L | 2q37.1 | KF419365/6/7 * | GGTGGGACGATGGCAGGT | CAAGTCCCTCATCTACATCC |
| ARID5B | 10q21.2 | LN607831 | AGGAATGGACAGAAGGAAGC | ATGGTTTCTTTTTGCGTGGTC |
| NCOA2 | 8q13.3 | LN607830 | GTGAGCCCCAAGAAGAAAGA | GACTCTCACAGCCGAACTC |
| IRF1 | 5q31.1 | LN607829 | CTCCACTCTGCCTGATGAC | GATGGAGGGCAACCGGACT |
| IRF1 | 5q31.1 | LN626686 | CTCCACTCTGCCTGATGAC | GATGGAGGGCAACCGGACT |
2.2. Validation of Novel TFs Transcripts

2.3. Protein Prediction of the Novel TFs Isoforms
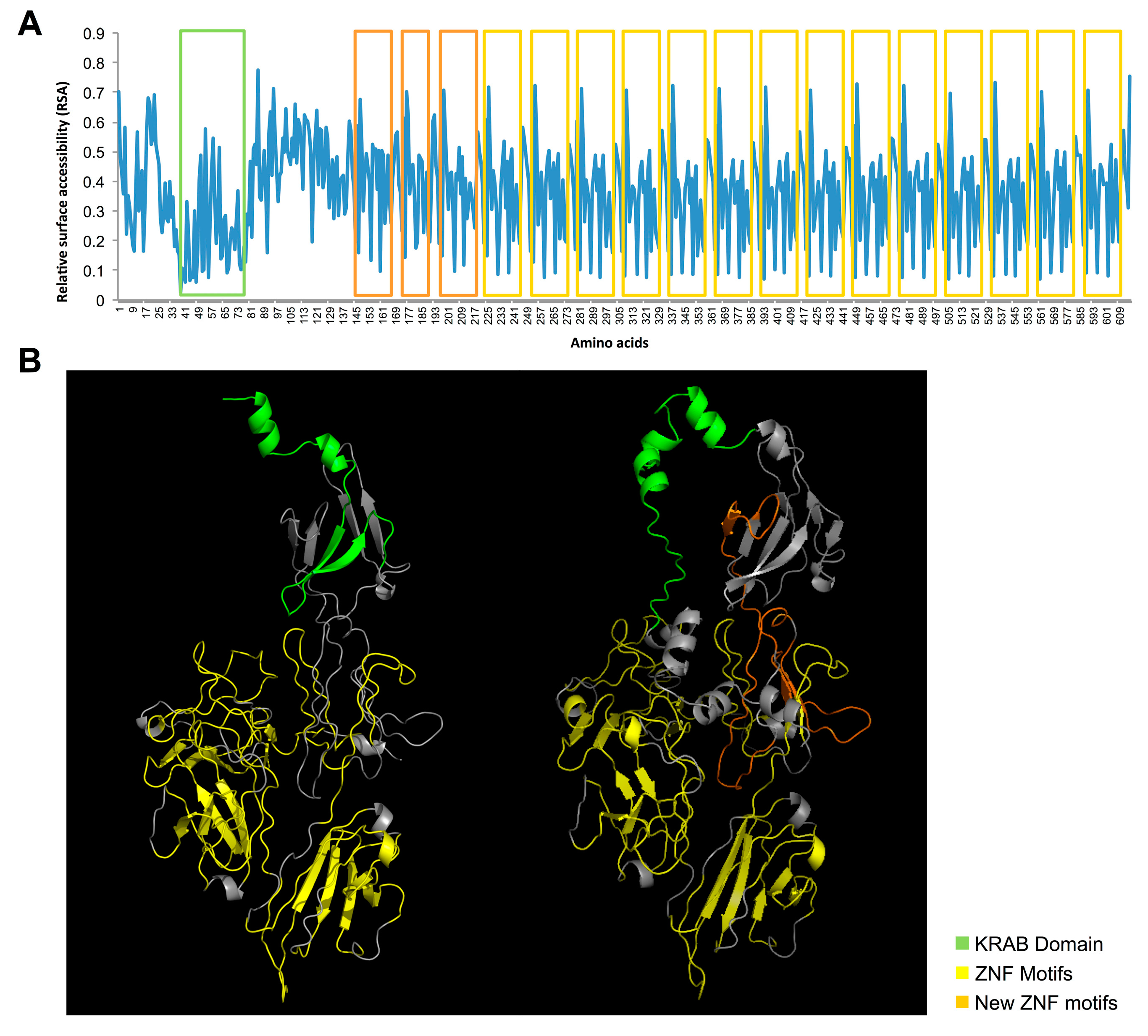
2.4. Identification and Characterization of a Novel ZNF266 Variant
3. Discussion
4. Experimental Section
4.1. Computational Analysis of RNA-Seq Datasets
4.2. Cell Cultures
4.3. RNA Extraction and RT-PCR Assays
4.4. DNA Gel Extraction, Cloning and Sequencing
4.5. In Silico Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Lassmann, T.; Itoh, M.; Summers, K.M.; Suzuki, H.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Haupt, L.M.; Griffiths, L.R. Alternative splicing (AS) of genes as an approach for generating protein complexity. Curr. Genomics 2013, 14, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007, 35, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Stranger, B.E. Genomics of alternative splicing: Evolution, development and pathophysiology. Hum. Genet. 2014, 133, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Talavera, D.; Orozco, M.; de la Cruz, X. Alternative splicing of transcription factors’ genes: Beyond the increase of proteome diversity. Comp. Funct. Genomics 2009, 2009, 905894. [Google Scholar] [CrossRef]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Aprile, M.; Ambrosio, M.R.; D’Esposito, V.; Beguinot, F.; Formisano, P.; Costa, V.; Ciccodicola, A. PPARG in human adipogenesis differential contribution of canonical transcripts and dominant negative isoforms. PPAR Res. 2014, 2014, 537865. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Casamassimi, A.; Peluso, G.; Barone, M.V.; Capaccio, D.; Migliore, C.; Bonelli, P.; Pedicini, A.; Febbraro, A.; Ciccodicola, A.; et al. A novel peroxisome proliferator-activated receptor gamma isoform with dominant negative activity generated by alternative splicing. J. Biol. Chem. 2005, 280, 26517–26525. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Aprile, M.; Esposito, R.; Ciccodicola, A. RNA-Seq and human complex diseases: Recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; D’Apice, L.; Mutarelli, M.; Casamassimi, A.; Sommese, L.; Gallo, M.A.; Aprile, M.; Esposito, R.; Leone, L.; et al. Massive-scale RNA-Seq analysis of non ribosomal transcriptome in human trisomy 21. PLoS One 2011, 6, e18493. [Google Scholar] [CrossRef] [PubMed]
- Scarpato, M.; Esposito, R.; Evangelista, D.; Aprile, M.; Ambrosio, M.R.; Angelini, C.; Ciccodicola, A.; Costa, V. AnaLysis of Expression on human chromosome 21, ALE-HSA21: A pilot integrated web resource. Database 2014, 2014, bau009. [Google Scholar] [CrossRef] [PubMed]
- Wingender, E.; Schoeps, T.; Dönitz, J. TFClass: An expandable hierarchical classification of human transcription factors. Nucleic Acids Res. 2013, 41, 165–170. [Google Scholar] [CrossRef]
- Thierry-Mieg, D.; Thierry-Mieg, J. AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006, 7, S12.1–S14. [Google Scholar] [CrossRef] [PubMed]
- Boguski, M.S.; Lowe, T.M.; Tolstoshev, C.M. dbEST—Database for “expressed sequence tags”. Nat. Genet. 1993, 4, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ortín, J.E.; Alepuz, P.; Chávez, S.; Choder, M. Eukaryotic mRNA decay: Methodologies, pathways, and links to other stages of gene expression. J. Mol. Biol. 2013, 425, 3750–3775. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Jo, M.; Park, J.; Zhang, W.; Lee, J.H. Alternative splicing variants of IRF-1 lacking exons 7, 8, and 9 in cervical cancer. Biochem. Biophys. Res. Commun. 2006, 347, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Sommese, L.; Casamassimi, A.; Colicchio, R.; Angelini, C.; Marchesano, V.; Milone, L.; Farzati, B.; Giovane, A.; Fiorito, C.; et al. Impairment of circulating endothelial progenitors in Down syndrome. BMC Med. Genomics 2010, 3, 40. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 1984, 12, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Du, Z.W.; Zhang, J.W. Identification and characterization of a novel zinc finger protein (HZF1) gene and its function in erythroid and megakaryocytic differentiation of K562 cells. Leukemia 2006, 20, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.D.; Horng, J.T.; Lee, C.C.; Liu, B.J. ProSplicer: A database of putative alternative splicing information derived from protein, mRNA and expressed sequence tag sequence data. Genome Biol. 2003, 4, R29. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.; Suzuki, Y.; Sakate, R.; Sato, Y.; Gojobori, T.; Imanishi, T.; Sugano, S. H-DBAS: human-transcriptome database for alternative splicing: Update 2010. Nucleic Acids Res. 2010, 38, D86–D90. [Google Scholar] [CrossRef] [PubMed]
- Kosti, I.; Radivojac, P.; Mandel-Gutfreund, Y. An integrated regulatory network reveals pervasive cross-regulation among transcription and splicing factors. PLoS Comput. Biol. 2012, 8, e1002603. [Google Scholar] [CrossRef] [PubMed]
- Rienzo, M.; Costa, V.; Scarpato, M.; Schiano, C.; Casamassimi, A.; Grimaldi, V.; Ciccodicola, A.; Napoli, C. RNA-Seq for the identification of novel Mediator transcripts in endothelial progenitor cells. Gene 2014, 547, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Chepelev, I.; Chen, X. Alternative splicing switching in stem cell lineages. Front. Biol. 2013, 8, 50–59. [Google Scholar] [CrossRef]
- Das, S.; Jena, S.; Levasseur, D.N. Alternative splicing produces Nanog protein variants with different capacities for self-renewal and pluripotency in embryonic stem cells. J. Biol. Chem. 2011, 286, 42690–42703. [Google Scholar] [CrossRef] [PubMed]
- Gabut, M.; Samavarchi-Tehrani, P.; Wang, X.; Slobodeniuc, V.; O’Hanlon, D.; Sung, H.K.; Alvarez, M.; Talukder, S.; Pan, Q.; Mazzoni, E.O.; et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011, 147, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Mowla, S.J.; Ziaee, S.A.; Gokhale, P.J.; Andrews, P.W. OCT4 spliced variants are differentially expressed in human pluripotent and nonpluripotent cells. Stem Cells 2008, 26, 3068–3074. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Pradhan, S.J.; Galande, S. Chromatin organizer SATB1 as a novel molecular target for cancer therapy. Curr. Drug Targets 2012, 13, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Oettgen, P.; Akbarali, Y.; Boltax, J.; Best, J.; Kunsch, C.; Libermann, T.A. Characterization of NERF, a novel transcription factor related to the Ets factor ELF1. Mol. Cell Biol. 1996, 16, 5091–5106. [Google Scholar] [PubMed]
- Dube, A.; Akbarali, Y.; Sato, T.N.; Libermann, T.A.; Oettgen, P. Role of the Ets transcription factors in the regulation of the vascular-specific Tie2 gene. Circ. Res. 1999, 84, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Akbarali, Y.; Zerbini, L.F.; Gu, X.; Boltax, J.; Wang, Y.; Oettgen, P.; Zhang, D.E.; Libermann, T.A. Isoforms of the Ets transcription factor NERF/ELF2 physically interact with AML1 and mediate opposing effects on AML1-mediated transcription of the B cell-specific blk gene. J. Biol. Chem. 2004, 279, 19512–19522. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Miyachi, M.; Sakamoto, K.; Ouchi, K.; Yagyu, S.; Kikuchi, K.; Kuwahara, Y.; Tsuchiya, K.; Imamura, T.; Iehara, T.; et al. PAX3-NCOA2 fusion gene has a dual role in promoting the proliferation and inhibiting the myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2013, 33, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Boye, K.; Heim, S. Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncol. Rep. 2014, 32, 40–44. [Google Scholar] [PubMed]
- Carapeti, M.; Aguiar, R.C.; Goldman, J.M.; Cross, N.C. A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 1998, 91, 3127–3133. [Google Scholar] [PubMed]
- Grondin, B.; Bazinet, M.; Aubry, M. The KRAB zinc finger gene ZNF74 encodes an RNA-binding protein tightly associated with the nuclear matrix. J. Biol. Chem. 1996, 271, 15458–15467. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Angelini, C.; de Feis, I.; Ciccodicola, A. Uncovering the complexity of transcriptomes with RNA-Seq. J. Biomed. Biotechnol. 2010, 2010, 853916. [Google Scholar] [CrossRef]
- Kameyama, T.; Suzuki, H.; Mayeda, A. Re-splicing of mature mRNA in cancer cells promotes activation of distant weak alternative splice sites. Nucleic Acids Res. 2012, 40, 7896–7906. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Shamir, R.; Ast, G. How prevalent is functional alternative splicing in the human genome? Trends Genet. 2004, 20, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Soreq, L.; Guffanti, A.; Salomonis, N.; Simchovitz, A.; Israel, Z.; Bergman, H.; Soreq, H. Long non-coding RNA and alternative splicing modulations in Parkinson’s leukocytes identified by RNA sequencing. PLoS Comput. Biol. 2014, 10, e1003517. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Conte, I.; Ziviello, C.; Casamassimi, A.; Alfano, G.; Banfi, S.; Ciccodicola, A. Identification and expression analysis of novel Jakmip1 transcripts. Gene 2007, 402, 1–8. [Google Scholar] [CrossRef] [PubMed]
- A Plasmid Editor (ApE). Available online: http://biologylabs.utah.edu/jorgensen/wayned/ape/ (accessed on 3 October 2013).
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Database. Available online: http://www.uniprot.org (accessed on 29 September 2014).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpato, M.; Federico, A.; Ciccodicola, A.; Costa, V. Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”. Int. J. Mol. Sci. 2015, 16, 1755-1771. https://doi.org/10.3390/ijms16011755
Scarpato M, Federico A, Ciccodicola A, Costa V. Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”. International Journal of Molecular Sciences. 2015; 16(1):1755-1771. https://doi.org/10.3390/ijms16011755
Chicago/Turabian StyleScarpato, Margherita, Antonio Federico, Alfredo Ciccodicola, and Valerio Costa. 2015. "Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”" International Journal of Molecular Sciences 16, no. 1: 1755-1771. https://doi.org/10.3390/ijms16011755
APA StyleScarpato, M., Federico, A., Ciccodicola, A., & Costa, V. (2015). Novel Transcription Factor Variants through RNA-Sequencing: The Importance of Being “Alternative”. International Journal of Molecular Sciences, 16(1), 1755-1771. https://doi.org/10.3390/ijms16011755