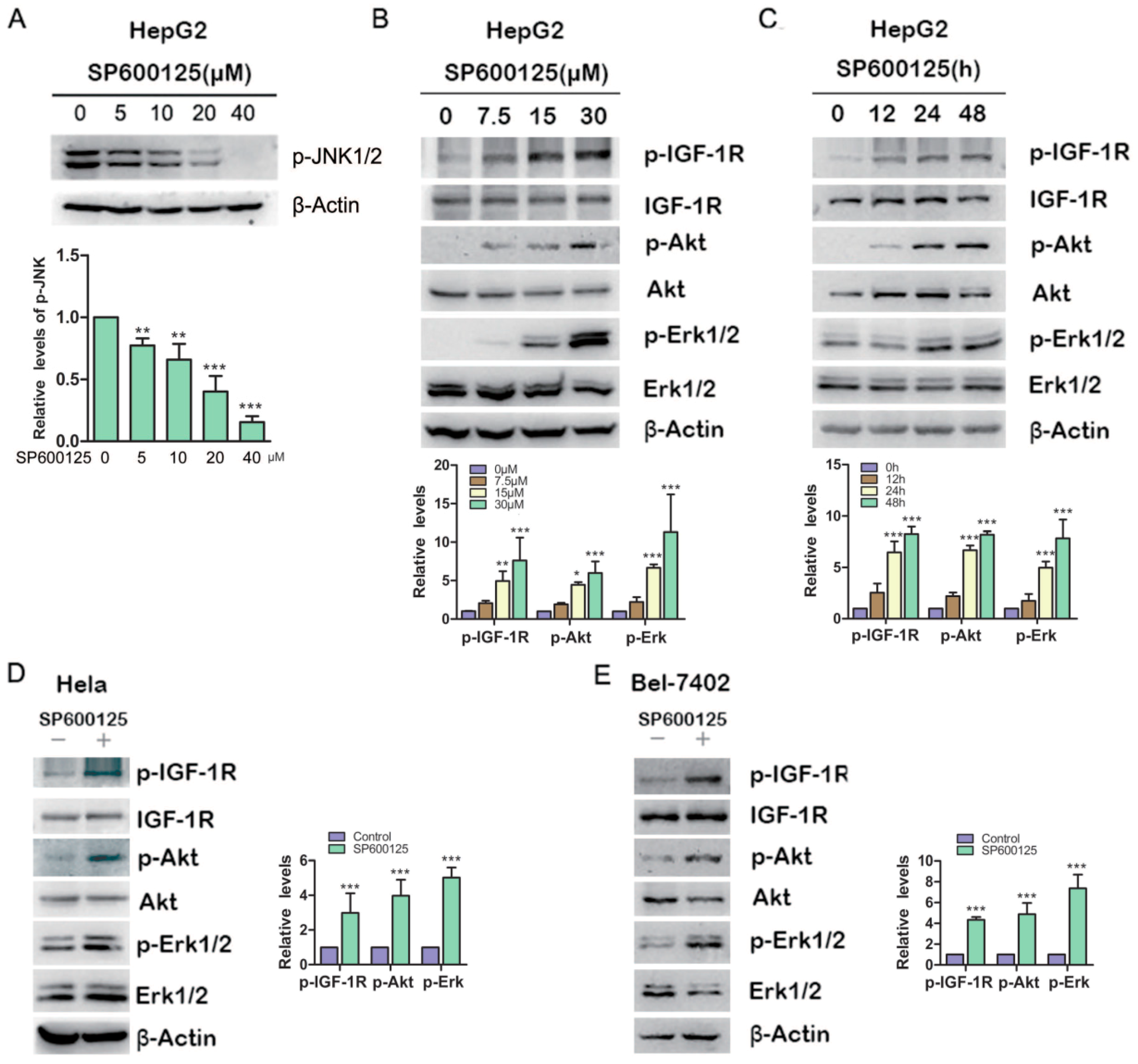
2.1. SP600125 Induces Type I Insulin-Like Growth Factor Receptor (IGF-IR), Akt and Erk1/2 Phosphorylation
To investigate the effects of SP600125 on Akt or Erk1/2 phosphorylation, HepG2 hepatoma cells were treated with different doses of SP600125 for 48 h, followed by western blot analysis of the levels of Akt and Erk1/2 phosphorylation. Treatment with SP600125 inhibited JNK phosphorylation and induced both Akt and Erk1/2 phosphorylaion in a dose-dependent manner (
Figure 1A,B). Moreover, Akt and Erk1/2 phosphorylation was stimulated by SP600125 in a time-dependent manner (
Figure 1C). Similar results were observed in BEL-7402 hepatoma cells and Hela cervical cancer cells (
Figure 1D,E). These data demonstrate that SP60125 can induce Akt and Erk1/2 phosphorylation. Akt, also known as protein Kinase B (PKB), is a serine/threonine protein kinase that plays a key role in multiple cellular processes such as glucose metabolism, apoptosis, cell proliferation and migration. Akt may be activated by PI3-kinase-dependent or PI3K-independent manner [
13]. In addition, Erk, the classical mitogen-activated protein kinase that is usually activated by Ras GTP-binding protein, is important intracellular signaling molecule regulating meiosis, mitosis, and postmitotic functions [
14]. Erk can activate many transcription factors and downstream protein kinases [
15]. Aberration in the Erk pathway is common in cancers, especially Ras, c-Raf and receptors such as EGFR and Her2. Thus, SP600125 may have important functions in signal transduction through regulating Akt and Erk1/2.
Figure 1.
SP600125 induces type I insulin-like growth factor receptor (IGF-IR), Akt, Erk1/2 phosphorylation. (A) HepG2 cells were treated with increasing doses of SP600125 for 48 h, followed by western blot analysis of c-Jun N-terminal kinases (JNK) phosphorylation; (B) HepG2 cells were treated with SP600125 for 48 h at the indicated doses, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; (C) HepG2 cells were treated with 15 µM SP600125 for indicated periods, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; and (D,E) Hela and BEL-7402 cells were treated with 15 µM SP600125 for 48 h, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. The blots were subjected to densitometric analysis and relative quantification. Levels in SP600125-untreated cells were set as 1. A representative of three experiments was shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control samples.
Figure 1.
SP600125 induces type I insulin-like growth factor receptor (IGF-IR), Akt, Erk1/2 phosphorylation. (A) HepG2 cells were treated with increasing doses of SP600125 for 48 h, followed by western blot analysis of c-Jun N-terminal kinases (JNK) phosphorylation; (B) HepG2 cells were treated with SP600125 for 48 h at the indicated doses, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; (C) HepG2 cells were treated with 15 µM SP600125 for indicated periods, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; and (D,E) Hela and BEL-7402 cells were treated with 15 µM SP600125 for 48 h, followed by western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. The blots were subjected to densitometric analysis and relative quantification. Levels in SP600125-untreated cells were set as 1. A representative of three experiments was shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control samples.
![]()
Both Akt and Erk1/2 are classical effectors downstream growth factor receptors including IGF-IR. We found that treatment with SP600125 also increased IGF-1R phosphorylation in dose-dependent and time-dependent manner in various cancer cell lines (
Figure 1). In addition, we found that SP600125 from another source, Sigma, had similar effects (data not shown). IGF-IR is well known as a positive regulator of a wide range of cellular processes, ranging from cellular proliferation, migration and survival. Upon IGF stimulation, IGF-IR is activated and then recruits insulin receptor substrates (IRS) such as IRS1 and IRS2, leading to the activation of multiple signaling cascades including PI3K/Akt and MAPK pathways [
16]. Although it has been reported that SP600125 can induce Akt phosphorylation [
7], it is unclear whether IGF-IR mediates this effect.
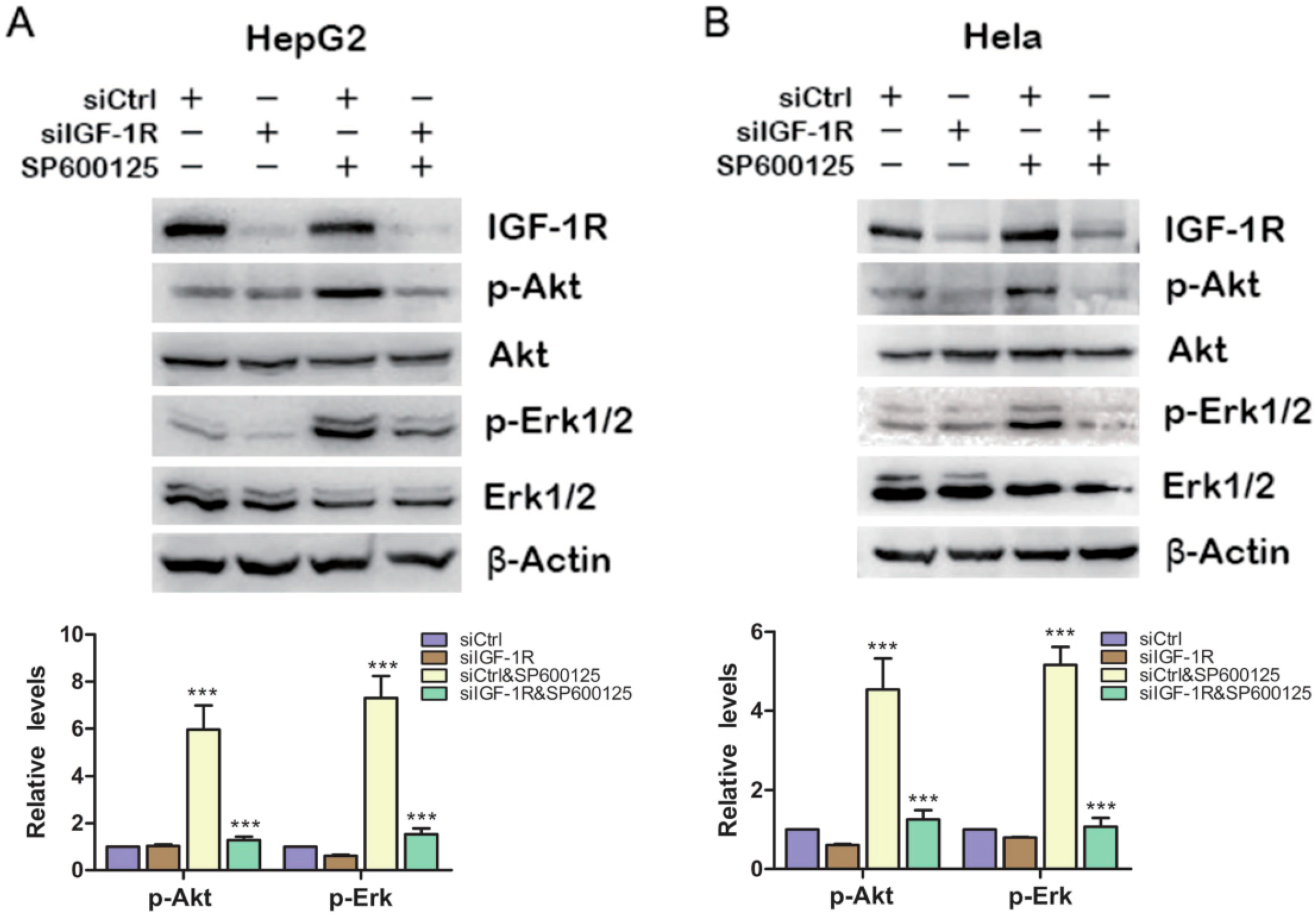
2.2. SP600125 Activates Akt and Erk via IGF-1R Activation
Because plenty of studies show that IGF-1R plays a pivotal role in regulating PI3K/Akt and Ras/MAPK signaling pathways [
16], we wonder whether the effect of SP600125 on Akt and Erk1/2 phosphorylation results from IGF-IR phosphorylation. To this end, HepG2 cells were transfected with siCtrl or siRNA against IGF-1R and treated with or without SP600125. SP600125 failed to induce Akt and Erk1/2 phosphorylation upon IGF-1R knockdown (
Figure 2A). Similar results were also observed in Hela cells (
Figure 2B). These results indicate that the effect of SP600125 on Akt and Erk1/2 phosphorylation is downstream of IGF-1R.
Figure 2.
IGF-1R is required for the induction of Akt and Erk1/2 phosphorylation by SP600125. HepG2 (A) or Hela (B) cells were transfected with negative siRNA (siCtrl) or siRNA against IGF-1R for 24 h, followed by treatment with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of total IGF-1R, Akt, Erk1/2 and phosphorylated Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. ***, p < 0.001, compared with control samples.
Figure 2.
IGF-1R is required for the induction of Akt and Erk1/2 phosphorylation by SP600125. HepG2 (A) or Hela (B) cells were transfected with negative siRNA (siCtrl) or siRNA against IGF-1R for 24 h, followed by treatment with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of total IGF-1R, Akt, Erk1/2 and phosphorylated Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. ***, p < 0.001, compared with control samples.
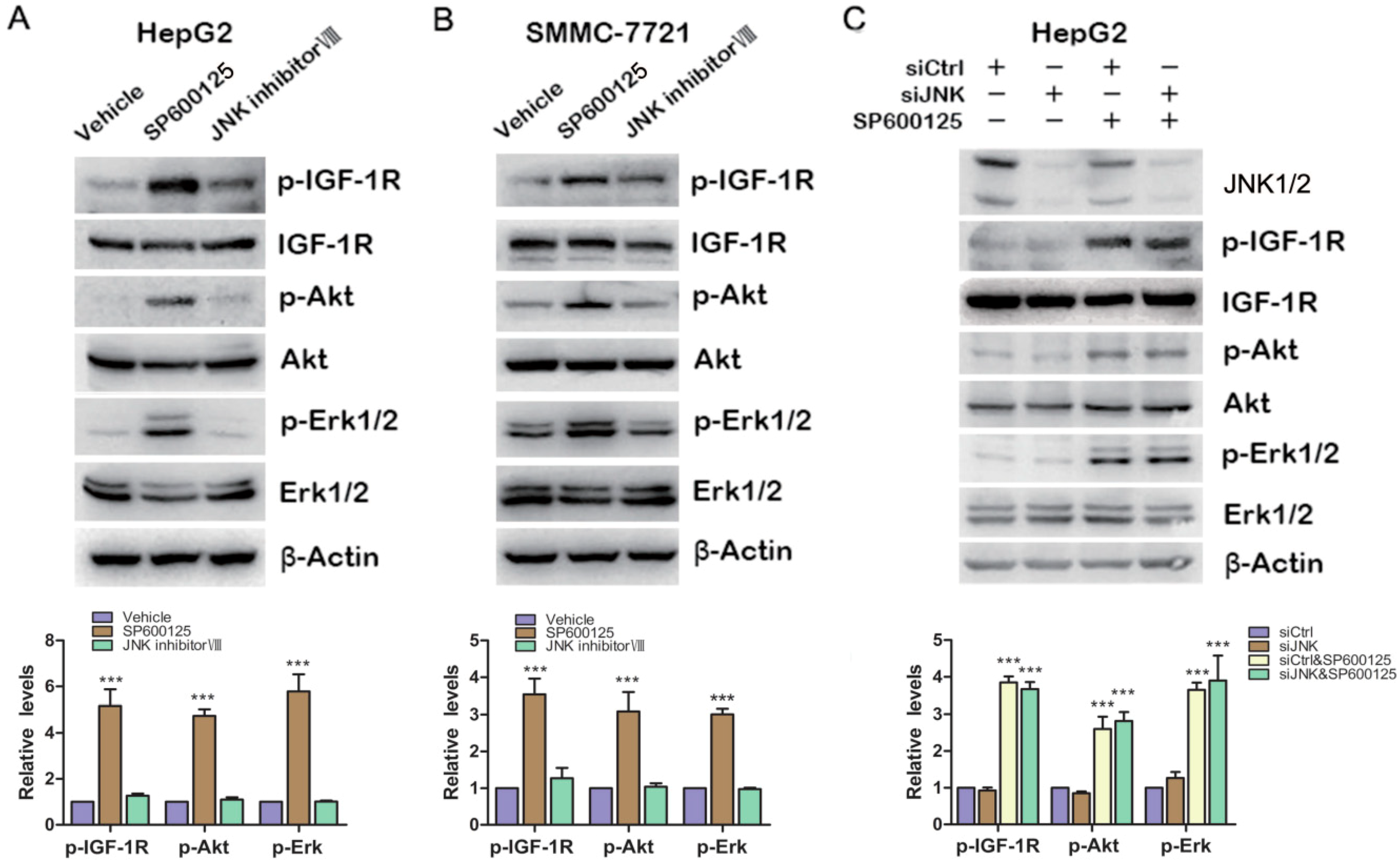
2.3. Effects of SP600125 on IGF-1R Signaling Pathway Is Independent of c-Jun N-Terminal Kinases (JNK)
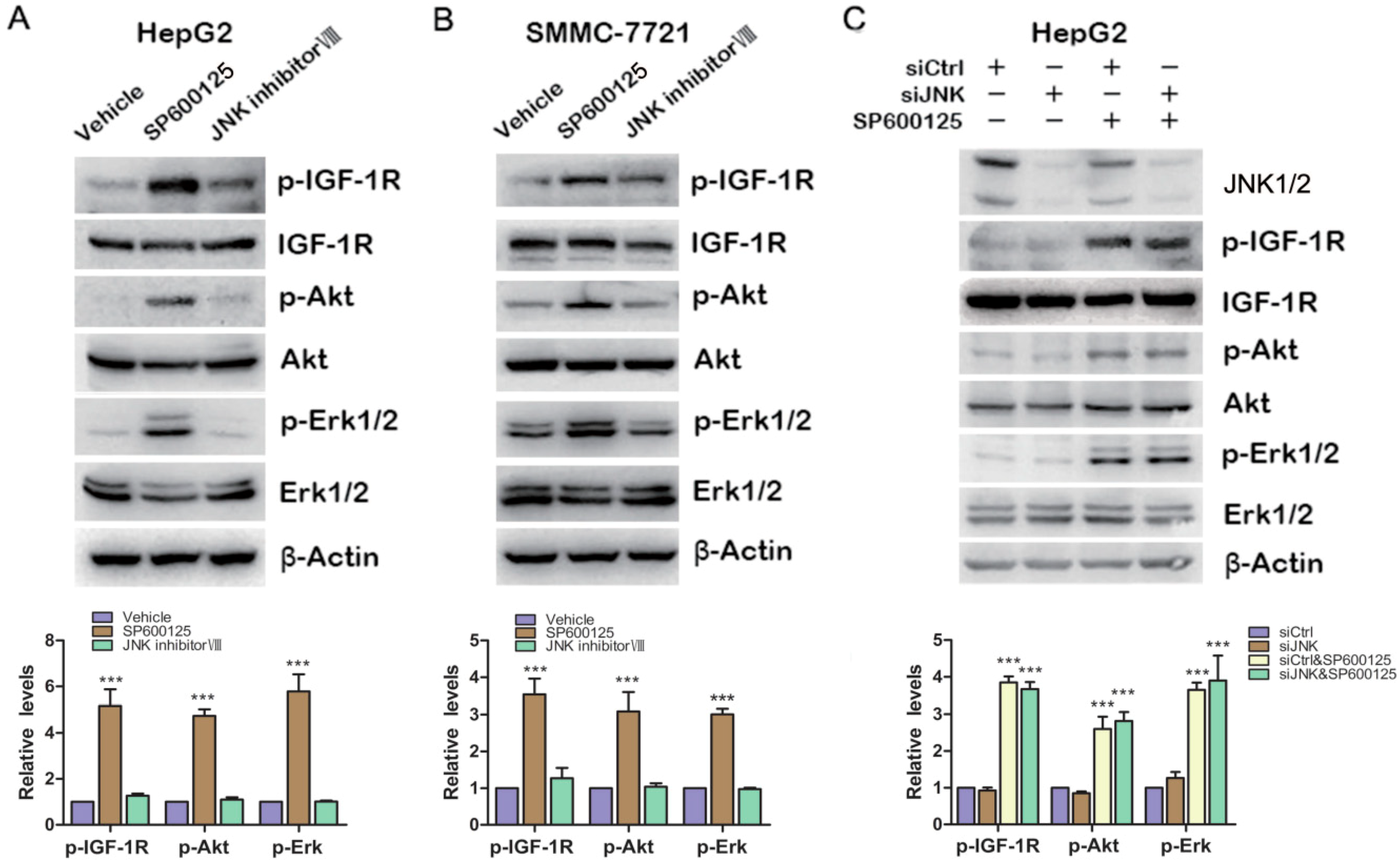
As we know, SP600125 is widely used as JNK inhibitor. To determine whether JNK is involved in SP600125-induced IGF-1R signaling, HepG2 cells and SMMC-7721 cells were treated with SP600125 or JNK inhibitor VIII, another specific JNK inhibitor. SP600125 induced IGF-1R, Akt and Erk1/2 phosphorylation in both HepG2 and SMMC-7721 cells (
Figure 3A,B). JNK inhibitor VIII slightly induced IGF-1R phosphorylation, while it did not induce Akt and Erk1/2 phosphorylation (
Figure 3A,B). In addition, HepG2 cells were transfected with sictrl or siRNA against JNK1/2 and treated with or without SP600125.Western blot analysis showed that JNK1/2 was efficiently knocked down. Knockdown of JNK1/2 alone had no effect on IGF-1R signaling cascades (
Figure 3C). In addition, SP600125 induced IGF-1R signaling both in the absence or presence of JNK1/2 (
Figure 3C). These data suggest that SP600125 may exert its effects on IGF-1R signaling pathway independent of JNK1/2.
Figure 3.
Effects of SP600125 on IGF-1R signaling pathway is independent of JNK. (A,B) HepG2 and SMMC-7721 cells were treated with 15 µM SP600125 or 20 µM JNK inhibitor VIII for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; and (C) HepG2 cells were transfected with siCtrl or siRNA against JNK for 24 h, followed by treatment with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. ***, p < 0.001, compared with other groups.
Figure 3.
Effects of SP600125 on IGF-1R signaling pathway is independent of JNK. (A,B) HepG2 and SMMC-7721 cells were treated with 15 µM SP600125 or 20 µM JNK inhibitor VIII for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2; and (C) HepG2 cells were transfected with siCtrl or siRNA against JNK for 24 h, followed by treatment with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. ***, p < 0.001, compared with other groups.
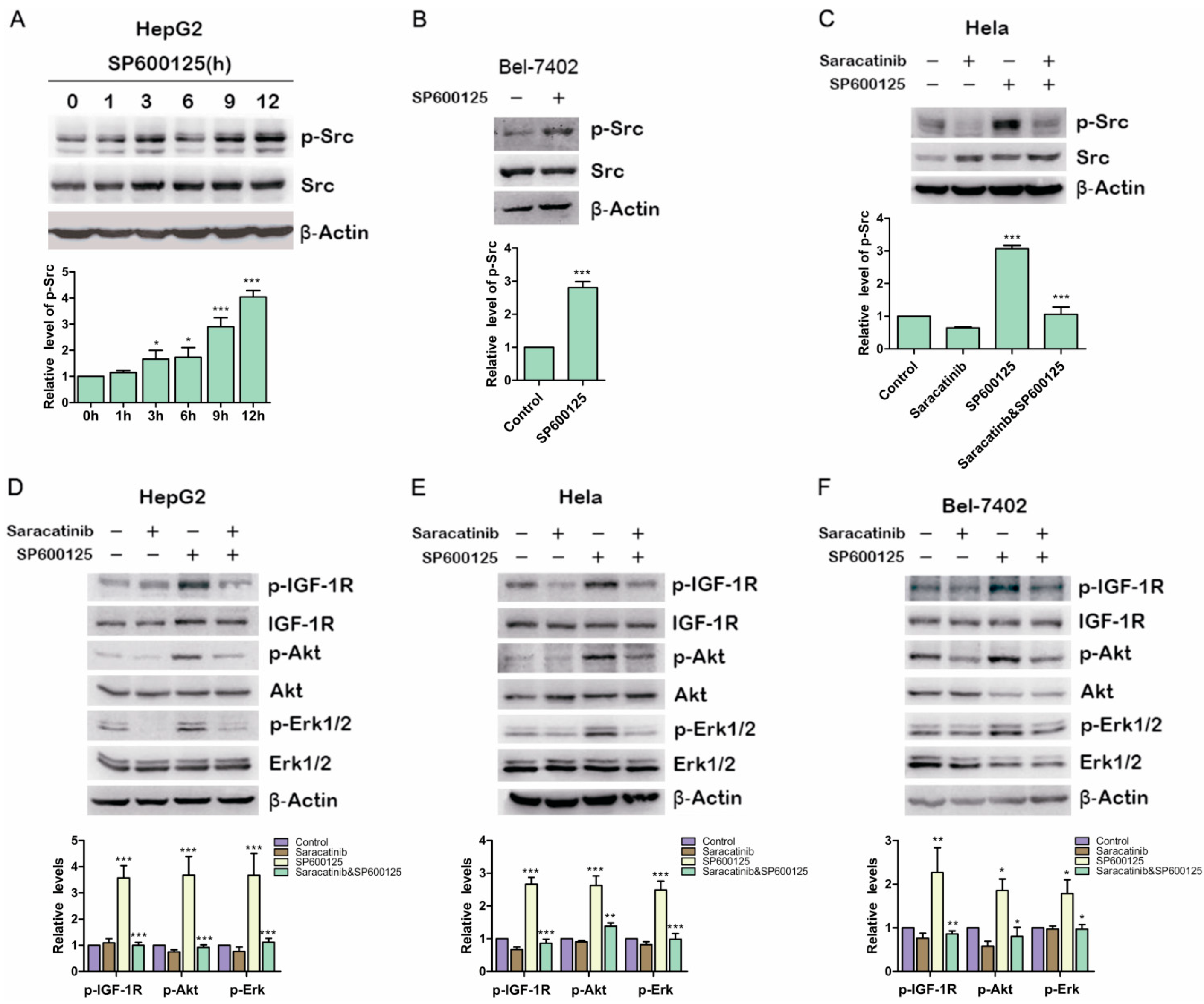
2.4. Src Is Involved in SP600125-Induced IGF-1R Phosphorylation
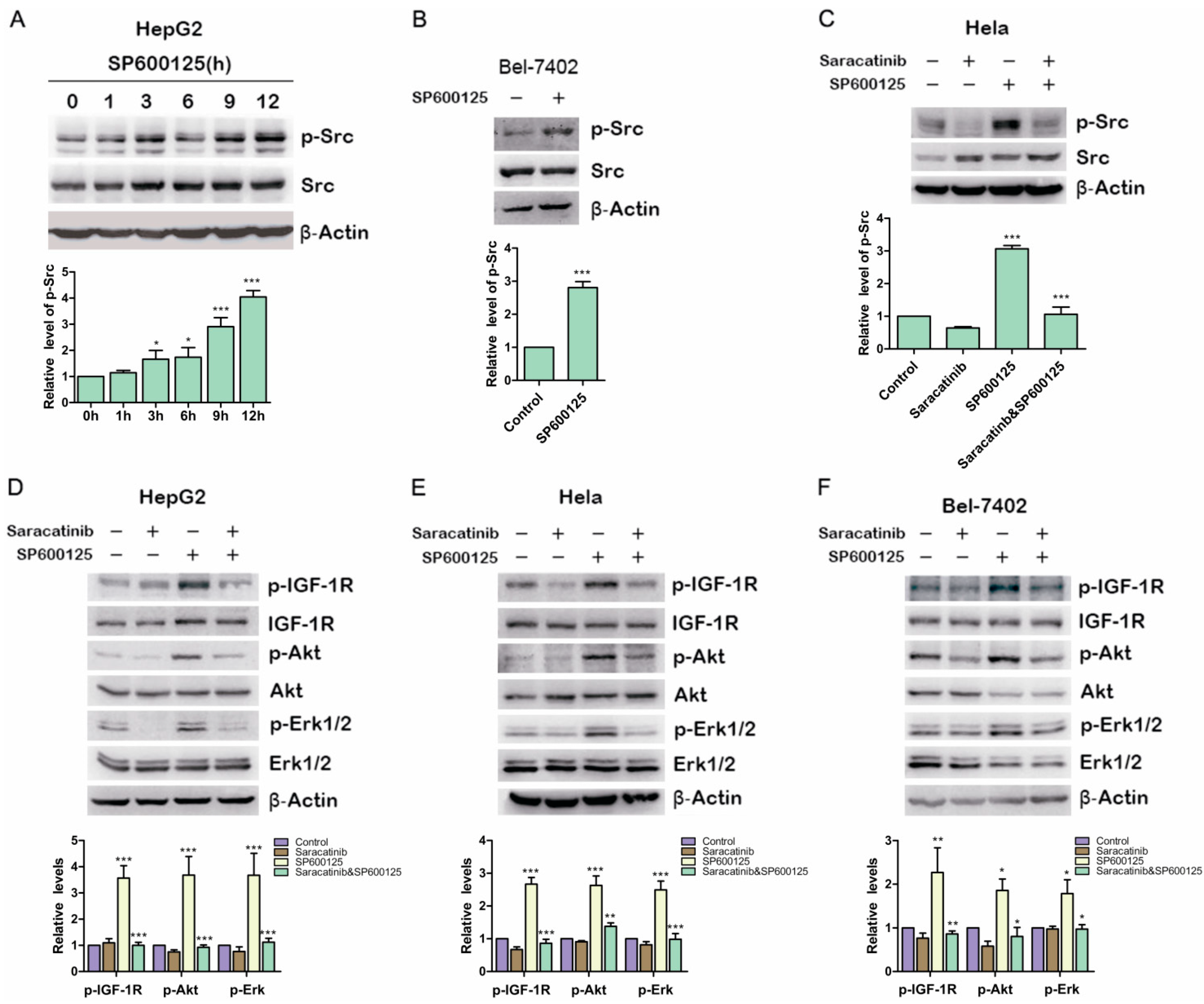
Previous studies showed that IGF-1R can be phosphorylated and then activated by Src [
17] Therefore, we examined if SP600125 could induce Src phosphorylation. HepG2 cells were treated with SP600125 for different time points. Src phosphorylation was up-regulated significantly at 12 h after SP600125 treatment (
Figure 4A). Similar results were obtained in BELl-7402 cells (
Figure 4B). To determine whether Src activity blockade can inhibit the induction of IGF-1R phosphorylation by SP600125, HepG2 cells and Hela cells were pretreated with or without saracatinib, an Src inhibitor, and then treated with or without SP600125. Saracatinib not only blocked SP600125-induced Src phosphorylation but also inhibited the effects of SP600125 on IGF-1R signaling cascades (
Figure 4C–F). These results suggest that SP600125-induced IGF-1R phosphorylation is mediated by Src. Src is a non-receptor tyrosine kinase that plays an important role in diverse cellular processes including proliferation, survival, angiogenesis and metastasis. Reciprocal regulation between Src and receptor tyrosine kinases has been intensively studied. Src may cooperate with receptor tyrosine kinase to perform its role [
18]. The activation of Src is commonly correlated with a variety of tumors [
19]. It has been reported that SP600125 induce cell cycle arrest by up-regulating p21 activity and expression. This effect is mediated by Erk/Sp1 and PI3K/Akt pathways. Inhibition of PI3K/Akt can sensitize SP600125-induced apoptosis [
7].
2.5. Combination of SP600125 and Saracatinib Synergistically Inhibits Cell Proliferation
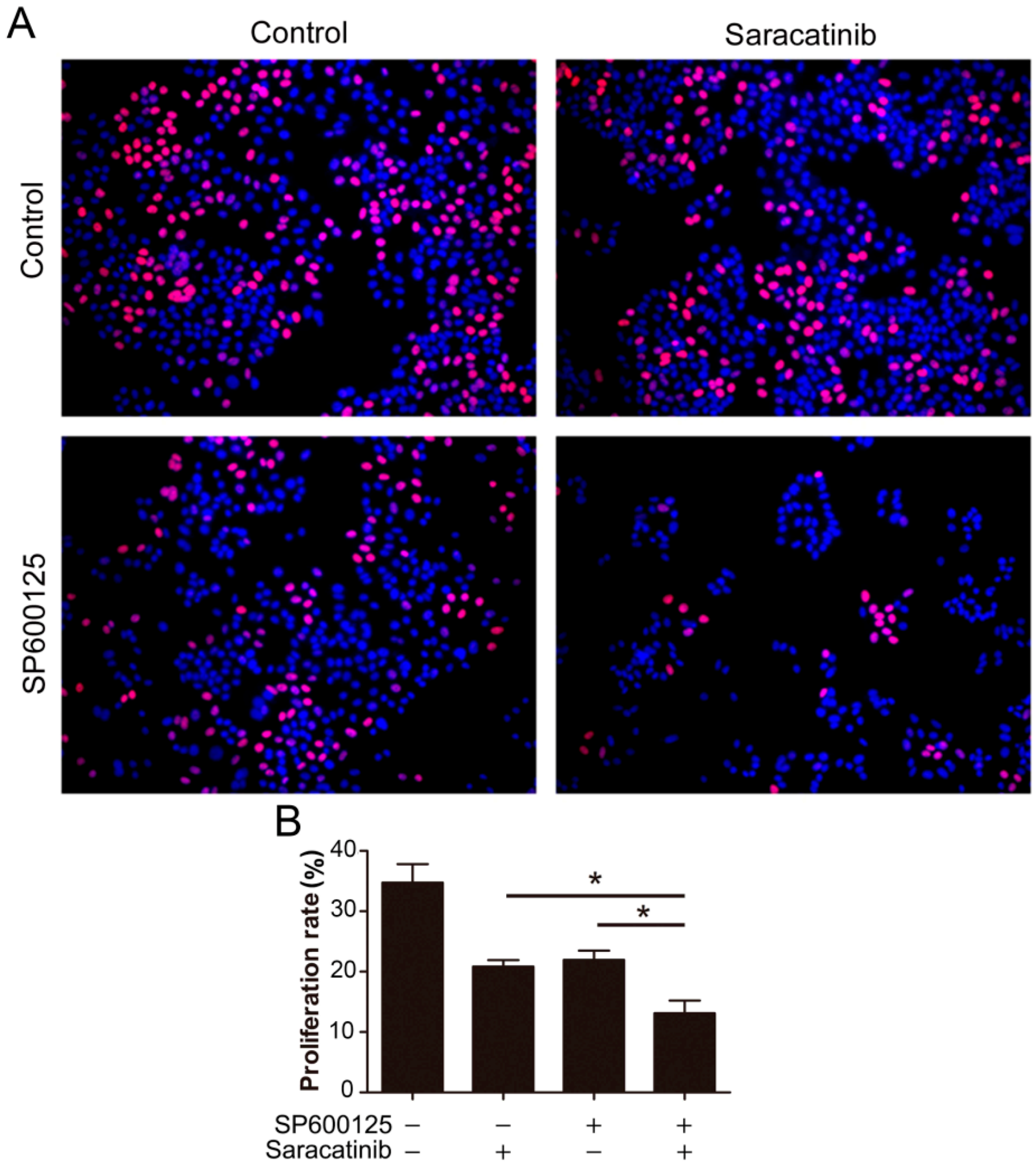
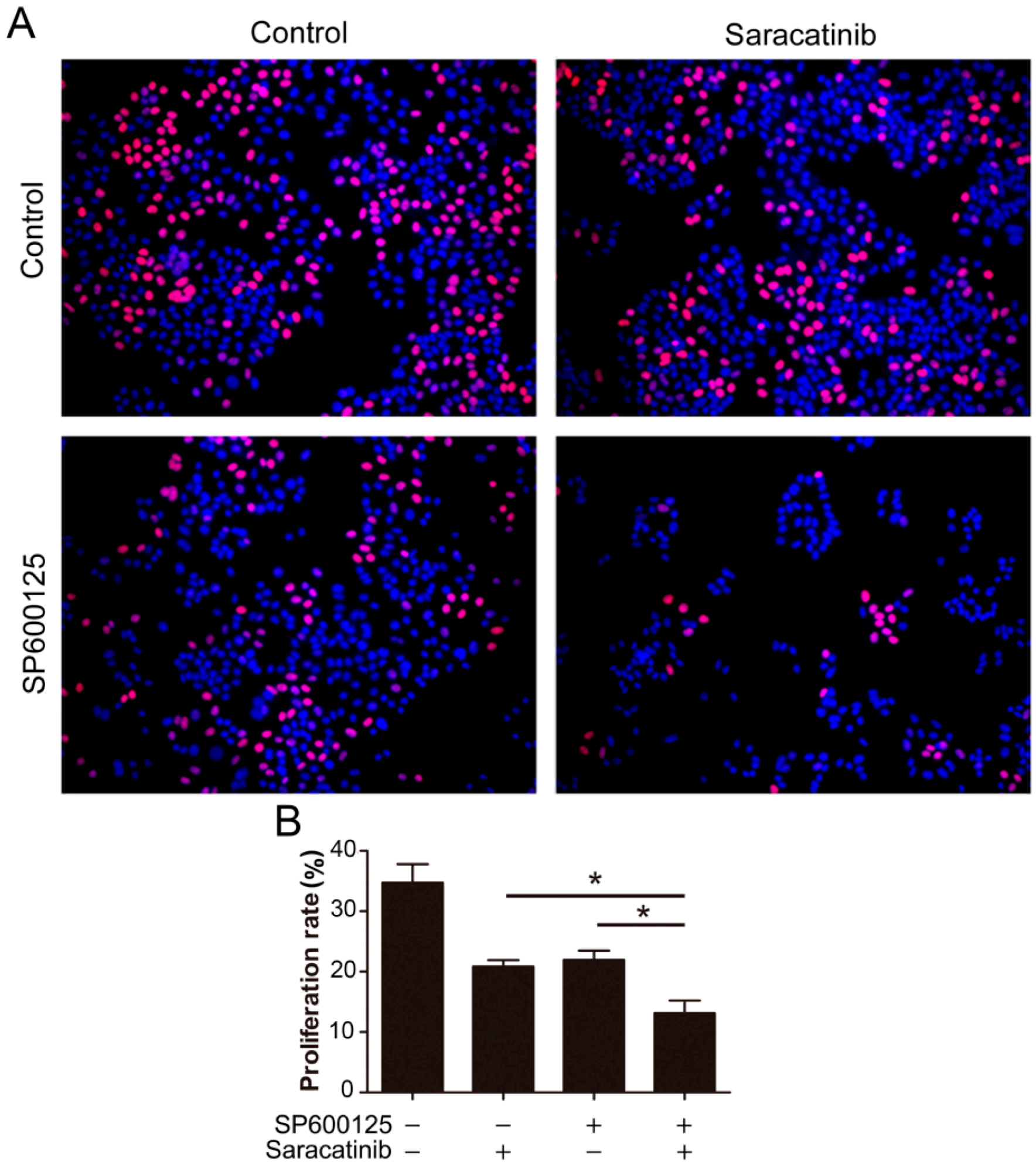
Given that SP600125 induces Src phosphorylation, we tested if inhibition of Src can potentiate the anti-cancer activity of SP600125. We utilized EdU assays to detect the effect of combination of saracatinib and SP600125 on cell proliferation. While both saracatinib and SP600125 slightly inhibited HepG2 cell proliferation, combination of saracatinib and SP600125 significantly inhibited cell proliferation (
Figure 5). These data suggest that activation of Src by SP600125 may compromise the inhibitory effect of SP600125 on cell proliferation. Also, treatment of tumor cells with SP600125 may sensitize them to Src inhibitors.
Previous studies demonstrate that SP600125 may potently inhibit voltage-dependent potassium ion channel and up-regulate REDD1 in JNK-independent manner [
11,
12]. G iven that SP600125 is used as a JNK blocker in a number of studies of the roles of JNK in signal transduction, these studies may be of importance and primary interest. Interpreting the results based on SP600125 as a JNK inhibitor should be done with extreme care. Other JNK inhibitors such as inhibitor VIII, which does not induce Akt and Erk1/2 phosphorylation, may be used in future studies on JNK to determine roles of JNK in cellular processes, rather than SP600125. Notably, JNK inhibitor VIII has been shown not to cause cellular growth arrest [
20]. Future studies are warranted to determine how SP600125 induces Src phosphorylation.
In summary, this study uncovers that SP600125 activates Src-IGF-1R-Akt/Erk pathways independent of inhibition of JNK. While JNK appears to be an attractive target for treating cancer, especially hepatocellular carcinoma [
21], activation of Src-IGF-1R-Akt/Erk pathways may compromise the anti-cancer effect of SP600125. Combination of SP600125 with Src/IGF-1R inhibitor may be a strategy for cancer treatment.
Figure 4.
Src is involved in SP600125-induced IGF-1R phosphorylation. (A) HepG2 cells were treated with 15 µM SP600125 for the indicated time. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; (B) Bel-7402 cells were treated with 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; (C) Hela cells were pretreated with 5 µM saracatinib for 3 h and then treated with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; and (D–F) HepG2, Hela and Bel-7402 cells were pretreated with 5 µM saracatinib for 3 h and then treated with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control samples.
Figure 4.
Src is involved in SP600125-induced IGF-1R phosphorylation. (A) HepG2 cells were treated with 15 µM SP600125 for the indicated time. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; (B) Bel-7402 cells were treated with 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; (C) Hela cells were pretreated with 5 µM saracatinib for 3 h and then treated with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated Src and total Src; and (D–F) HepG2, Hela and Bel-7402 cells were pretreated with 5 µM saracatinib for 3 h and then treated with or without 15 µM SP600125 for 48 h. Total protein extracts were harvested and subjected to western blot analysis of phosphorylated IGF-1R, Akt, Erk1/2 and total IGF-1R, Akt, Erk1/2. β-Actin was detected as loading control. The blots were subjected to densitometric analysis and relative quantification. Levels in control samples were set as 1. A representative of three experiments was shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control samples.
![]()
Figure 5.
Combination of SP600125 and saracatinib synergistically inhibits HepG2 cell proliferation. (A) HepG2 cells were pretreated with 5 µM SP600125 for 3 h and then treated with or without 10 µM SP600125 for 48 h, followed by EdU labeling. A representative of two independent experiments in triplicate is shown; and (B) The proliferation rate was plotted. Values represent mean ± SD (n = 3). *, p < 0.05.
Figure 5.
Combination of SP600125 and saracatinib synergistically inhibits HepG2 cell proliferation. (A) HepG2 cells were pretreated with 5 µM SP600125 for 3 h and then treated with or without 10 µM SP600125 for 48 h, followed by EdU labeling. A representative of two independent experiments in triplicate is shown; and (B) The proliferation rate was plotted. Values represent mean ± SD (n = 3). *, p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}