BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling

Abstract
:1. Introduction
2. Results and Discussion
2.1. The Expression of Bone Morphogenetic Protein (BMP) and Its Receptors (BMPR1A and BMPR2) in Pulmonary Artery
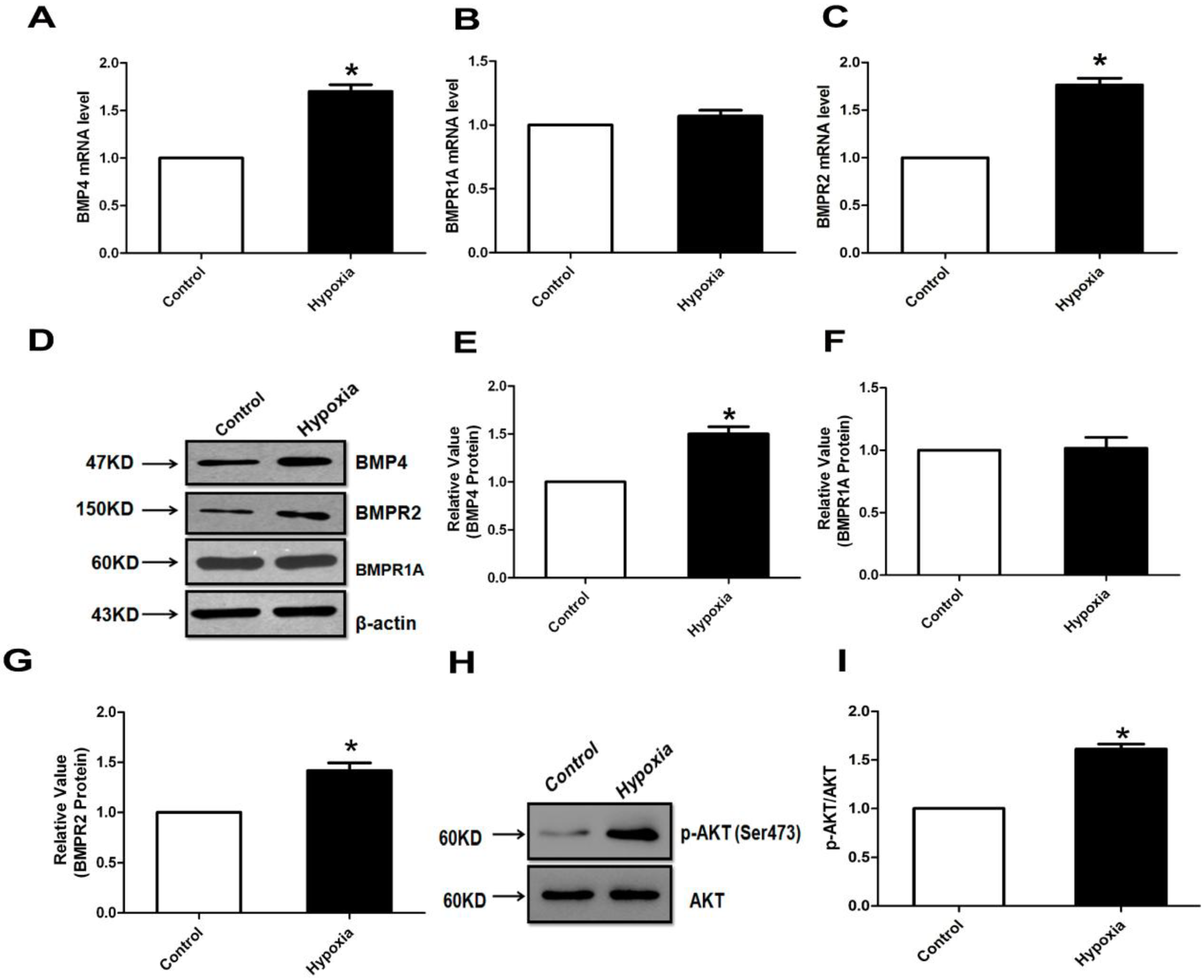
2.2. Hypoxia Induces BMPR Receptors Protein Expression in Human Pulmonary Artery Smooth Muscle Cells (HPASMCs) for Different Time Course
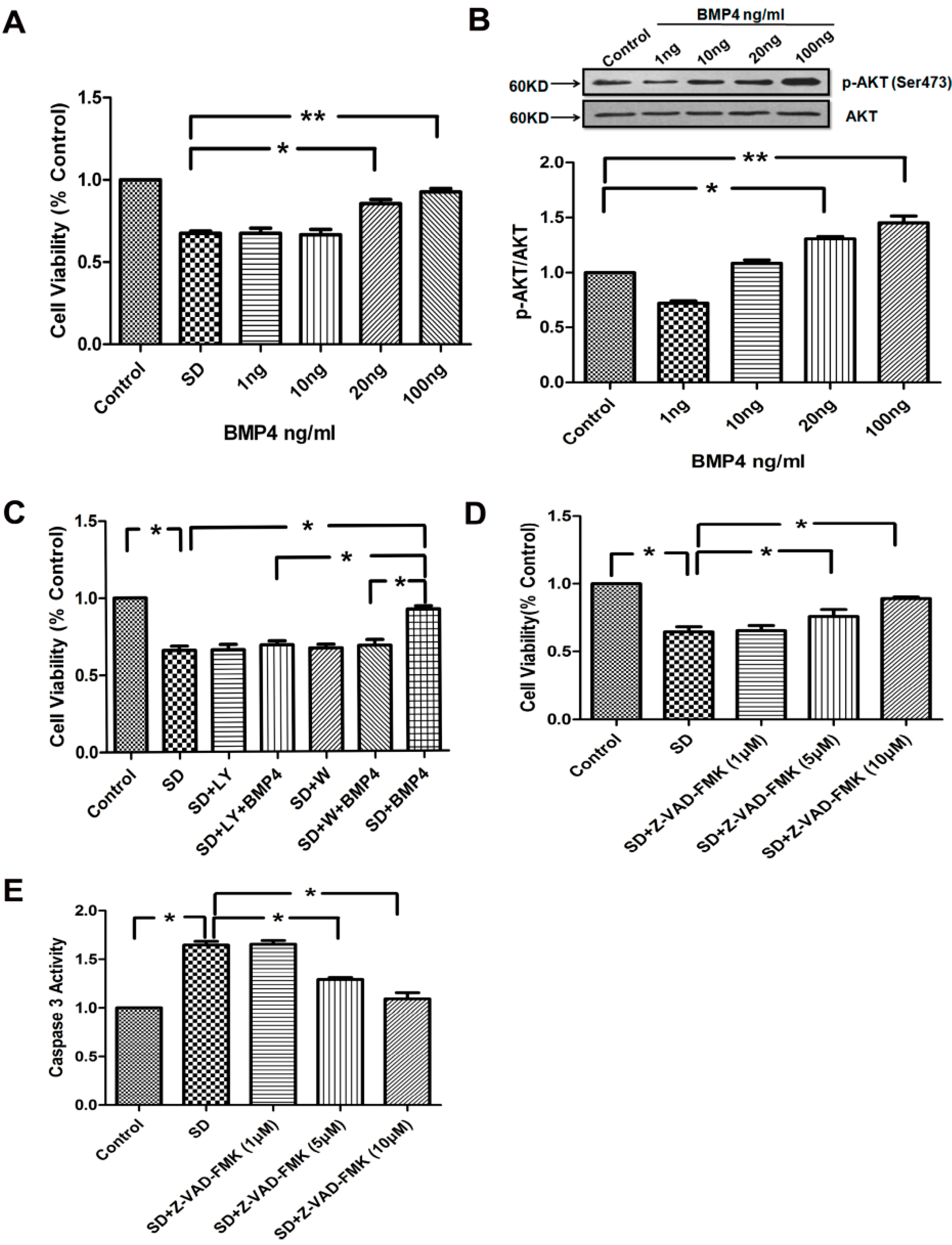
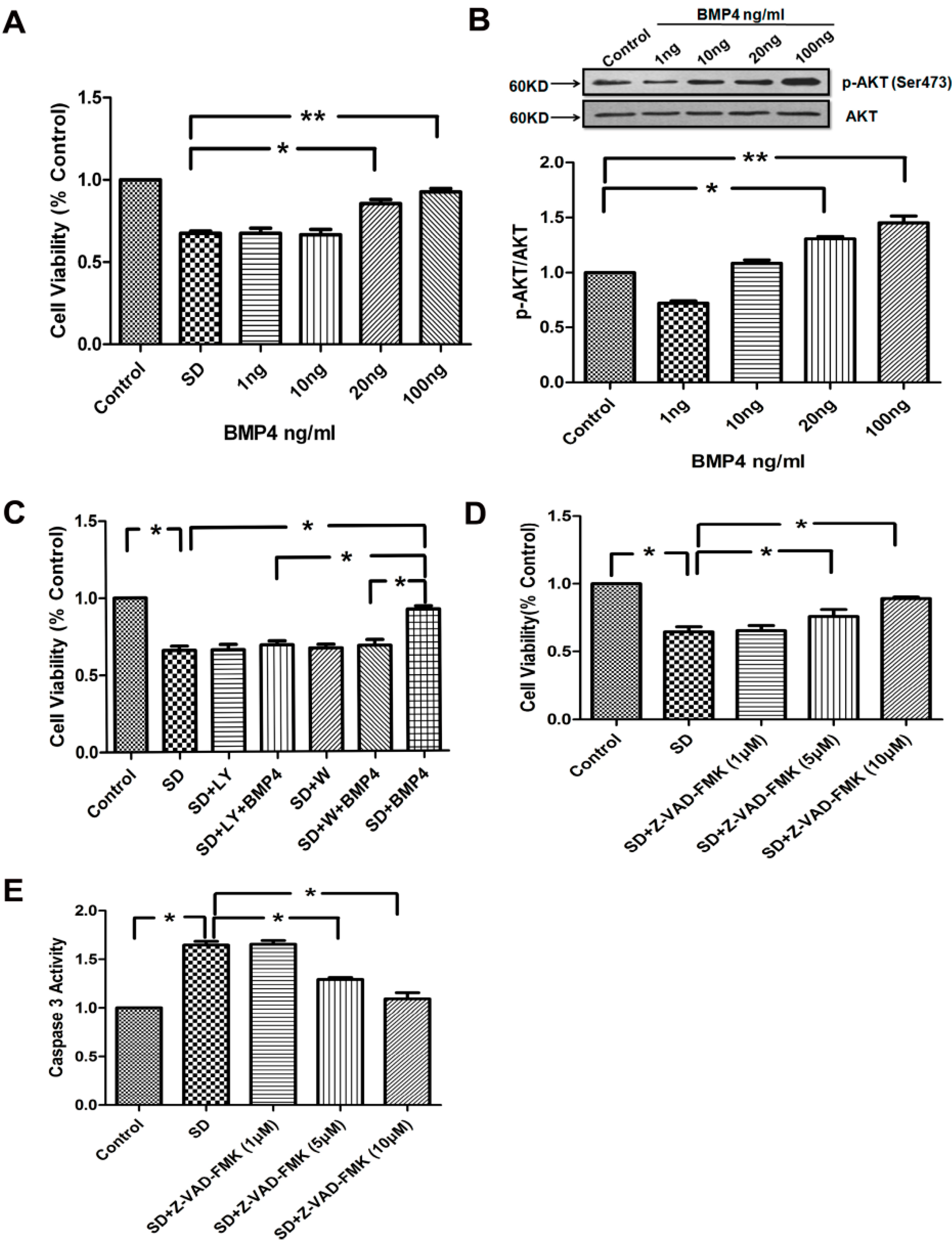
2.3. BMP4 Improved PASMC Viability via the PI3K/AKT Survival Pathway, and Caspase-3 Was Involved in SD-Induced Apoptosis
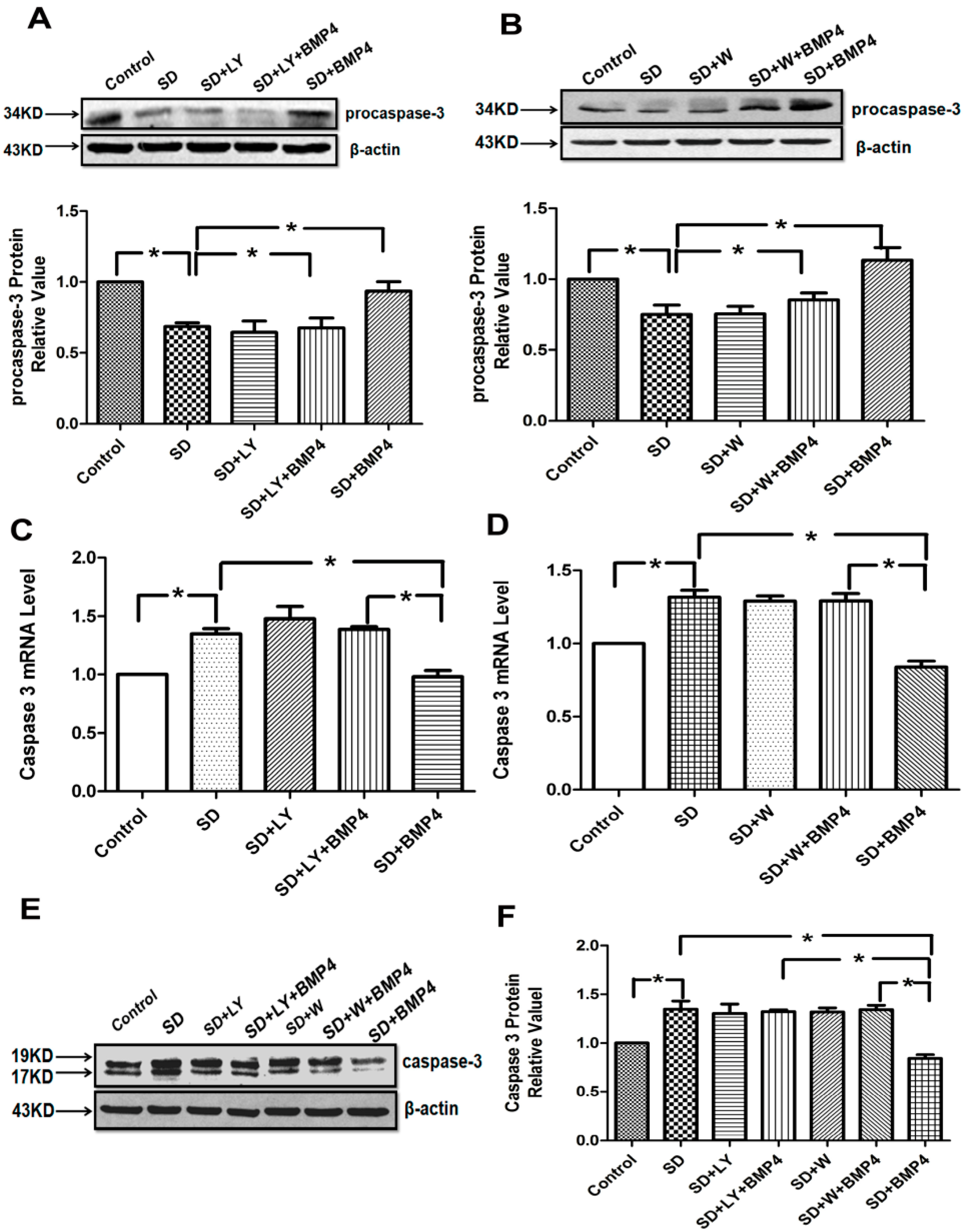
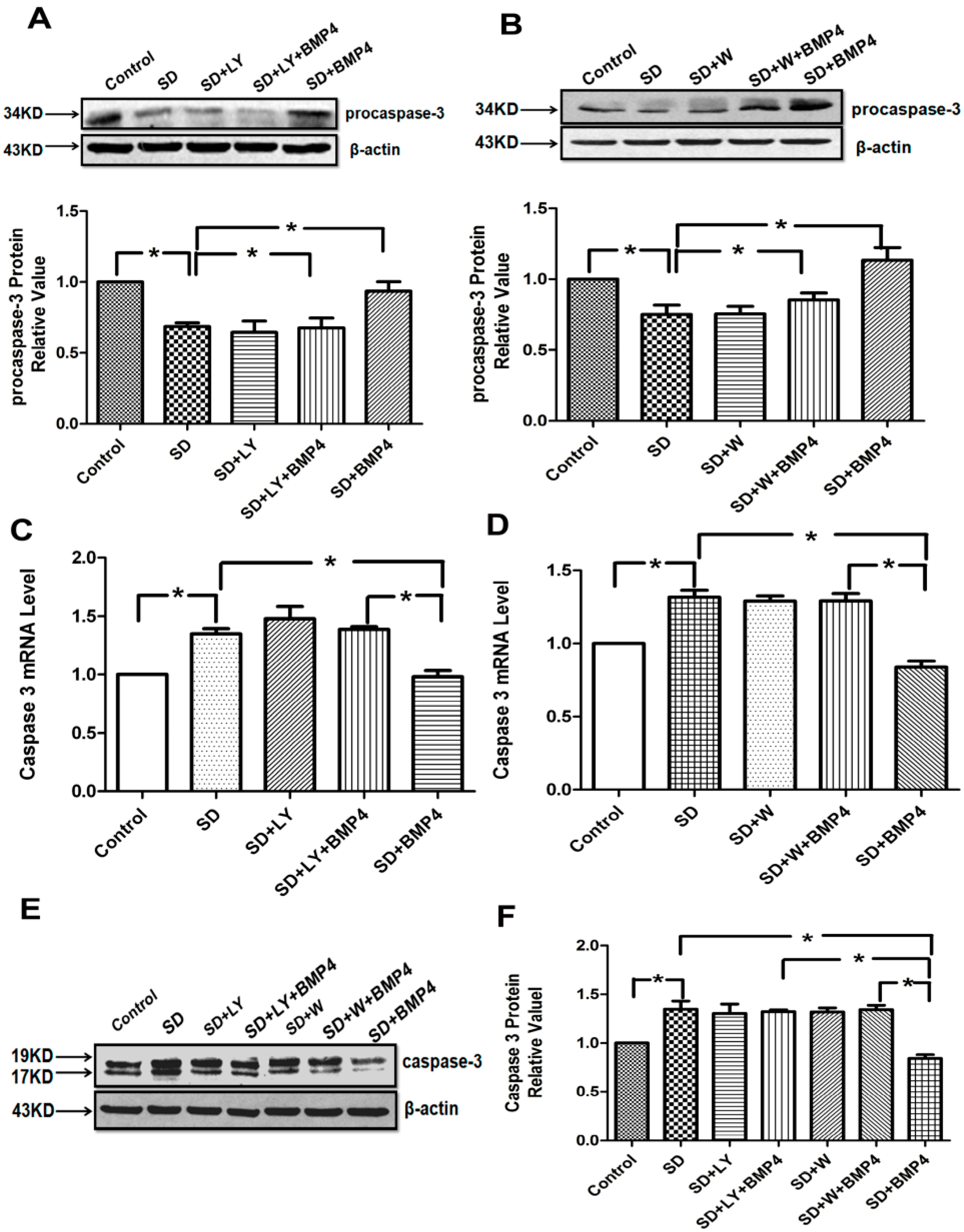
2.4. The Inhibitory Effects of BMP4 on Caspase-3 Expression and Procaspase-3 Cleavage Were Blocked by PI3K/AKT Inhibitors
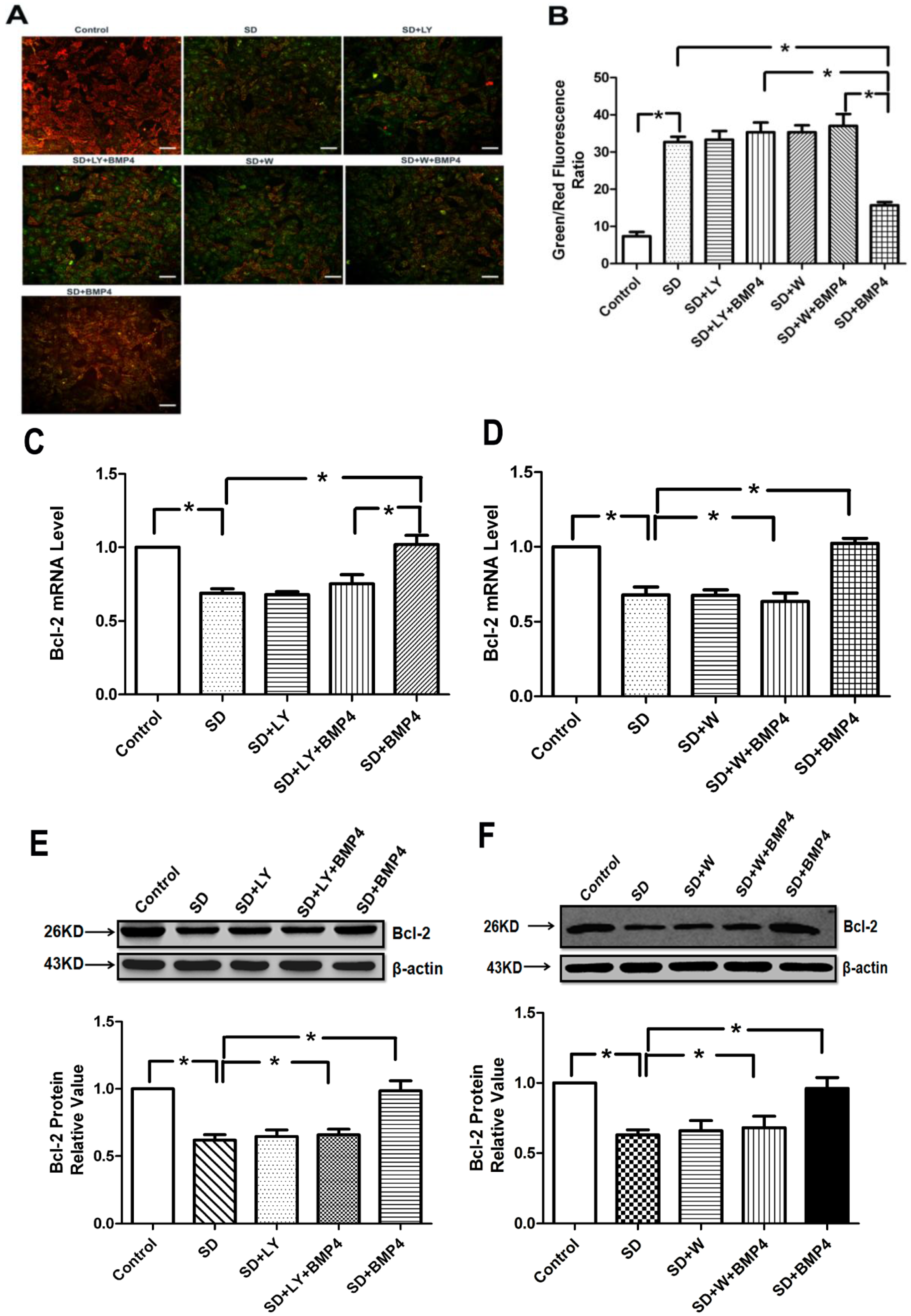
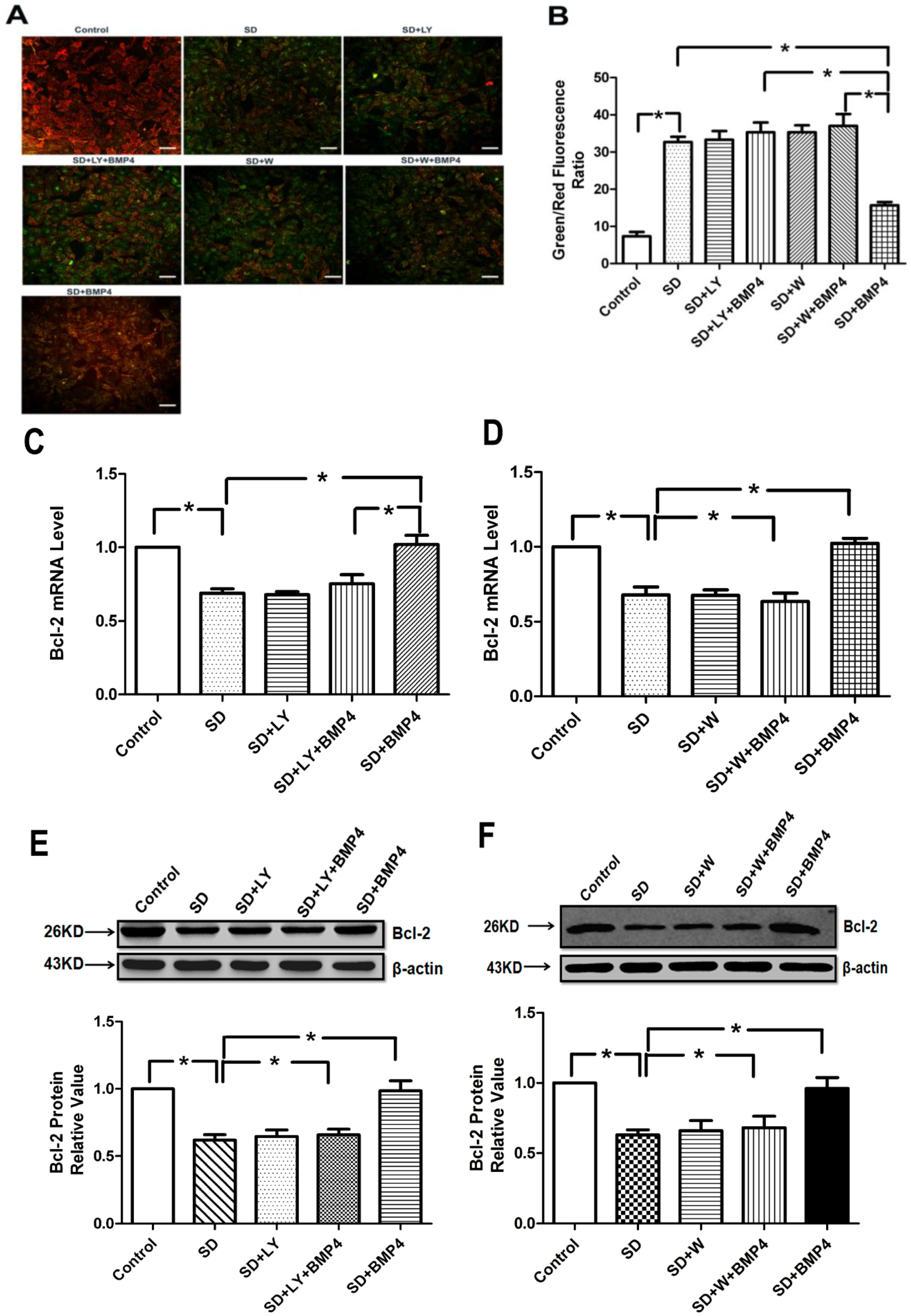
2.5. BMP4 Relieved Mitochondrial Depolarization and Induced Bcl-2 Expression in PASMCs after Serum Deprivation through PI3K/AKT Pathway
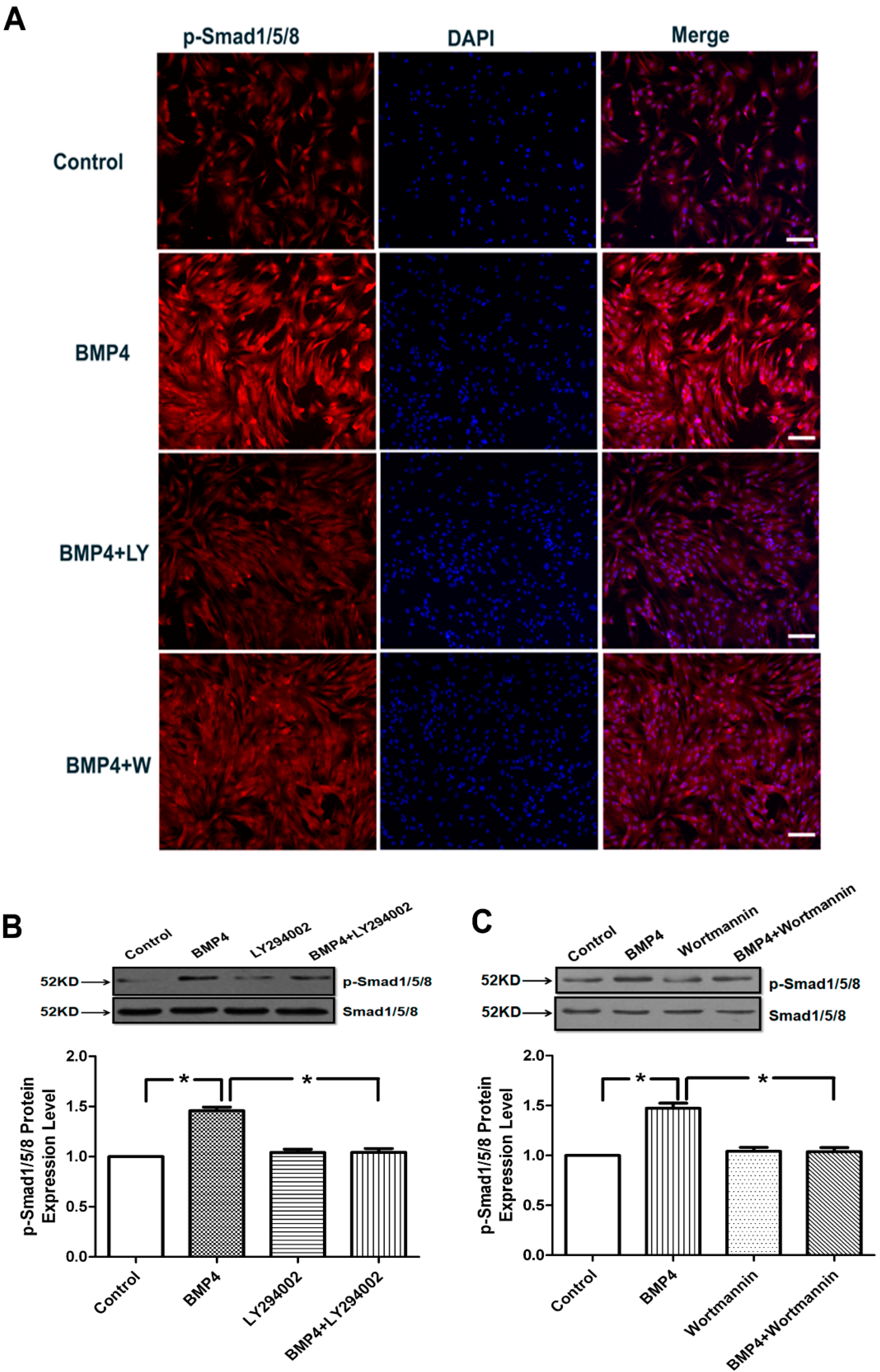
2.6. BMP4 Activates Smad1/5/8 Phosphorylation by the PI3K/AKT Signaling Pathways in Pulmonary Arterial Smooth Muscle Cells
2.7. Discussion
3. Experimental Section
3.1. Materials
3.2. Animals and Lung Tissues Preparation
3.3. Cell Culture
3.4. Real-Time Quantitative RT-PCR (qPCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequences (5' to 3') | PCR Product Size | Accession Number |
|---|---|---|---|
| Bcl-2 | Forward: 5'-CGGGAGAACAGGGTATGA-3' | 149 bp | NM: 016993 |
| Reverse: 5'-CAGGCTGGAAGGAGAAGAT-3' | |||
| Caspas3 | Forward: 5'-CTACCGCACCCGGTTACTAT-3' | 133 bp | NM: 012922.2 |
| Reverse: 5'-TTCCGGTTAACACGAGTGAG-3' | |||
| β-Actin | Forward: 5'-AGGCCCCTCTGAACCCTAAG-3' | 118 bp | EF: 156276.1 |
| Reverse: 5'-CCAGAGGCATACAGGGACAAC-3' |
3.5. Western Blot Analysis
3.6. MTT Assay
3.7. Measurement of Caspase-3 Activity
3.8. Mitochondrial Depolarization Assay
3.9. Immunofluorescence
3.10. Statistical Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chan, S.Y.; Loscalzo, J. Pathogenic mechanisms of pulmonary arterial hypertension. J. Mol. Cell. Cardiol. 2008, 44, 14–30. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef]
- Archer, S.; Rich, S. Primary pulmonary hypertension: A vascular biology and translational research “Work in progress”. Circulation 2000, 102, 2781–2791. [Google Scholar] [CrossRef]
- De Caestecker, M.; Meyrick, B. Bone morphogenetic proteins, genetics and the pathophysiology of primary pulmonary hypertension. Respir. Res. 2001, 2, 193–197. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Mecham, R.P. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu. Rev. Physiol. 1997, 59, 89–144. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Tuder, R.M. Cellular and molecular biology of vascular smooth muscle cells in pulmonary hypertension. Pulm. Pharmacol. Ther. 1997, 10, 231–241. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar]
- Rider, C.C.; Mulloy, B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 2010, 429, 1–12. [Google Scholar] [CrossRef]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar]
- Morty, R.E.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Zakrzewicz, A.; Kouri, F.M.; Peters, D.M.; Dumitrascu, R.; Seeger, W.; Knaus, P.; et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1072–1078. [Google Scholar] [CrossRef]
- Yu, P.B.; Deng, D.Y.; Beppu, H.; Hong, C.C.; Lai, C.; Hoyng, S.A.; Kawai, N.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J. Biol. Chem. 2008, 283, 3877–3888. [Google Scholar]
- Weaver, M.; Yingling, J.M.; Dunn, N.R.; Bellusci, S.; Hogan, B.L. BMP signaling regulates proximal–distal differentiation of endoderm in mouse lung development. Development 1999, 126, 4005–4015. [Google Scholar]
- Frank, D.B.; Abtahi, A.; Yamaguchi, D.J.; Manning, S.; Shyr, Y.; Pozzi, A.; Baldwin, H.S.; Johnson, J.E.; de Caestecker, M.P. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ. Res. 2005, 97, 496–504. [Google Scholar] [CrossRef]
- Weaver, M.; Dunn, N.R.; Hogan, B.L. BMP4 and FGF10 play opposing roles during lung bud morphogenesis. Development 2000, 127, 2695–2704. [Google Scholar]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β (1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Li, X.; Lu, W.; Fu, X.; Zhang, Y.; Yang, K.; Zhong, N.; Ran, P.; Wang, J. BMP4 increases canonical transient receptor potential protein expression by activating p38 MAPK and ERK1/2 signaling pathways in pulmonary arterial smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 212–220. [Google Scholar] [CrossRef]
- Lu, W.; Ran, P.; Zhang, D.; Lai, N.; Zhong, N.; Wang, J. Bone morphogenetic protein 4 enhances canonical transient receptor potential expression, store-operated Ca2+ entry, and basal [Ca2+]i in rat distal pulmonary arterial smooth muscle cells. Cell Physiol. 2010, 299, C1370–C1378. [Google Scholar]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Li, Y.; Song, Y.H.; Mohler, J.; Delafontaine, P. ANG II induces apoptosis of human vascular smooth muscle via extrinsic pathway involving inhibition of Akt phosphorylation and increased FasL expression. Heart Circ. Physiol. 2006, 290, H2116–H2123. [Google Scholar]
- Wang, X.Q.; Sun, P.; Paller, A.S. Inhibition of integrin-linked kinase/protein kinase B/Akt signaling: Mechanism for ganglioside-induced apoptosis. J. Biol. Chem. 2001, 276, 44504–44511. [Google Scholar] [CrossRef]
- Garat, C.V.; Fankell, D.; Erickson, P.F.; Reusch, J.E.; Bauer, N.N.; McMurtry, I.F.; Klemm, D.J. Platelet-derived growth factor BB induces nuclear export and proteasomal degradation of CREB via phosphatidylinositol 3-kinase/Akt signaling in pulmonary artery smooth muscle cells. Mol. Cell. Biol. 2006, 26, 4934–4948. [Google Scholar]
- Sheares, K.K.; Jeffery, T.K.; Long, L.; Yang, X.; Morrell, N.W. Differential effects of TGF-β1 and BMP-4 on the hypoxic induction of cyclooxygenase-2 in human pulmonary artery smooth muscle cells. Lung Cell. Mol. Physiol. 2004, 287, L919–L927. [Google Scholar]
- Kobayashi, T.; Masumoto, J.; Tada, T.; Nomiyama, T.; Hongo, K.; Nakayama, J. Prognostic significance of the immunohistochemical staining of cleaved caspase-3, an activated form of caspase-3, in gliomas. Clin. Cancer Res. 2007, 13, 3868–3874. [Google Scholar] [CrossRef]
- Cook, S.A.; Sugden, P.H.; Clerk, A. Regulation of Bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: Association with changes in mitochondrial membrane potential. Circ. Res. 1999, 85, 940–949. [Google Scholar] [CrossRef]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef]
- Gerasimovskaya, E.V.; Tucker, D.A.; Stenmark, K.R. Activation of phosphatidylinositol 3-kinase, Akt, and mammalian target of rapamycin is necessary for hypoxia-induced pulmonary artery adventitial fibroblast proliferation. J. Appl. Physiol. 2005, 98, 722–731. [Google Scholar]
- Yang, Z.Z.; Tschopp, O.; Hemmings-Mieszczak, M.; Feng, J.; Brodbeck, D.; Perentes, E.; Hemmings, B.A. Protein kinase B α/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 2003, 278, 32124–32131. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Fernandez-Hernando, C.; Cirino, G.; Sessa, W.C. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc. Natl. Acad. Sci. USA 2009, 106, 14552–14557. [Google Scholar]
- Li, J.; Kim, K.; Hahm, E.; Molokie, R.; Hay, N.; Gordeuk, V.R.; Du, X.; Cho, J. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J. Clin. Investig. 2014, 124, 1483–1496. [Google Scholar]
- Ding, L.; Biswas, S.; Morton, R.E.; Smith, J.D.; Hay, N.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab. 2012, 15, 861–872. [Google Scholar] [CrossRef]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar]
- Whitman, M. Smads and early developmental signaling by the TGFβ superfamily. Genes Dev. 1998, 12, 2445–2462. [Google Scholar] [CrossRef]
- Salido, M.; Gonzalez, J.L.; Vilches, J. Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol. Cancer Ther. 2007, 6, 1292–1299. [Google Scholar] [CrossRef]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.E.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef]
- Zhang, L.; Pu, Z.; Wang, J.; Zhang, Z.; Hu, D.; Wang, J. Baicalin inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via the AKT/HIF-1α/p27-associated pathway. Int. J. Mol. Sci. 2014, 15, 8153–8168. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, J.; Yu, Z.; Su, D. BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling. Int. J. Mol. Sci. 2014, 15, 13738-13754. https://doi.org/10.3390/ijms150813738
Wu J, Yu Z, Su D. BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling. International Journal of Molecular Sciences. 2014; 15(8):13738-13754. https://doi.org/10.3390/ijms150813738
Chicago/Turabian StyleWu, Jian, Zhigang Yu, and Dechun Su. 2014. "BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling" International Journal of Molecular Sciences 15, no. 8: 13738-13754. https://doi.org/10.3390/ijms150813738
APA StyleWu, J., Yu, Z., & Su, D. (2014). BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling. International Journal of Molecular Sciences, 15(8), 13738-13754. https://doi.org/10.3390/ijms150813738