Potential Spermatogenesis Recovery with Bone Marrow Mesenchymal Stem Cells in an Azoospermic Rat Model


Abstract
:
1. Introduction
2. Results
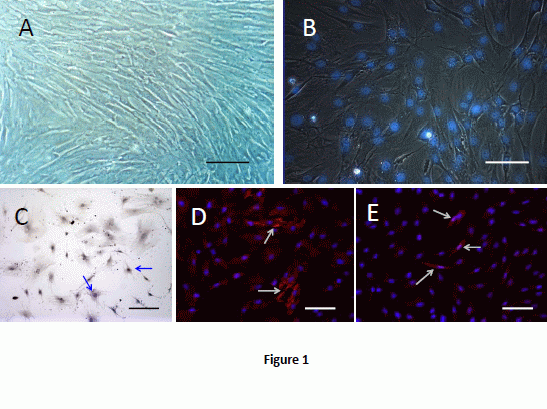
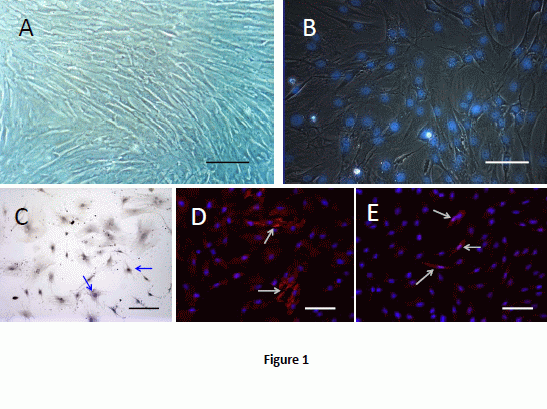
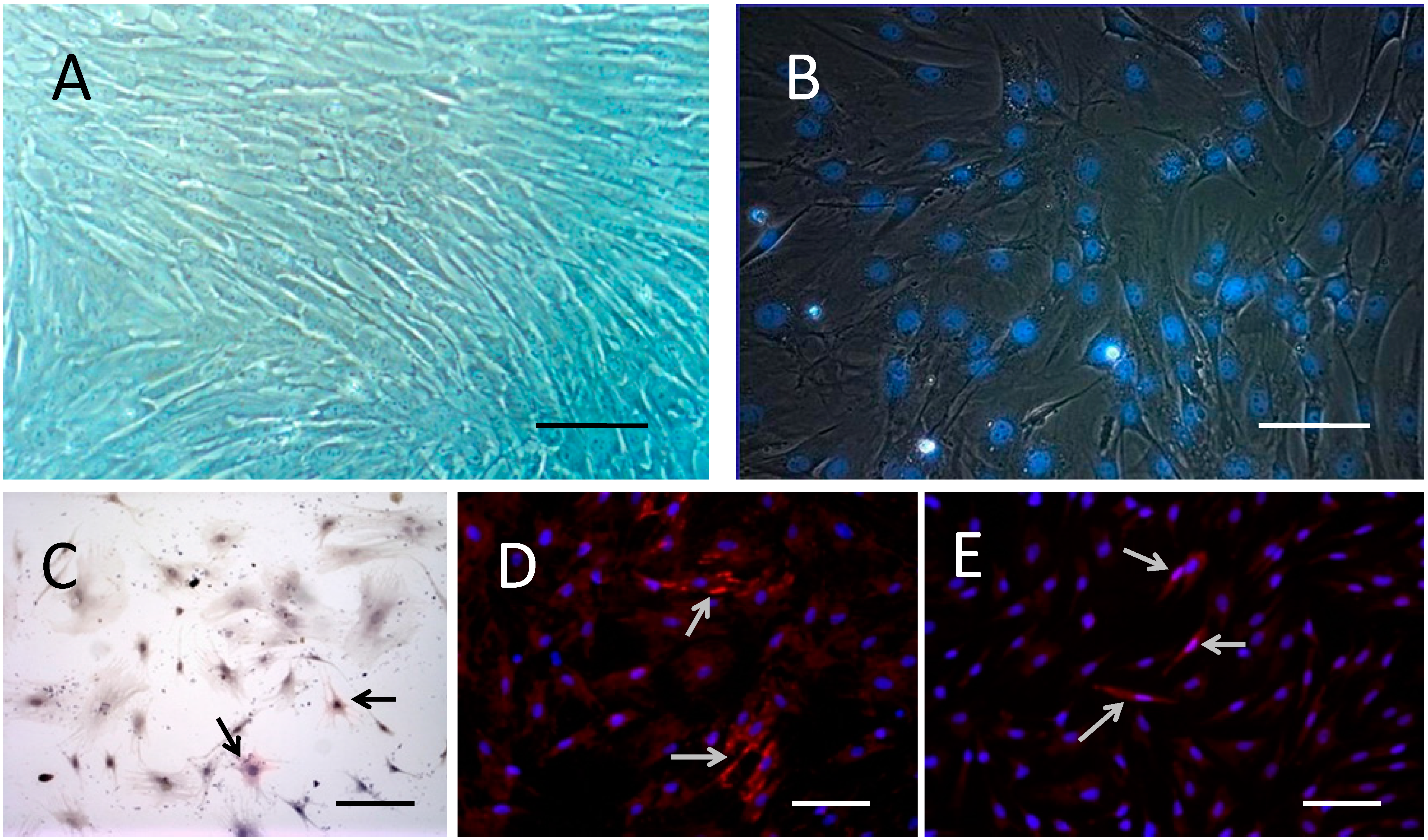
2.1. Cell Culture and Labeling
2.2. BMSCs Exhibit Multi-Lineage Differentiation Ability
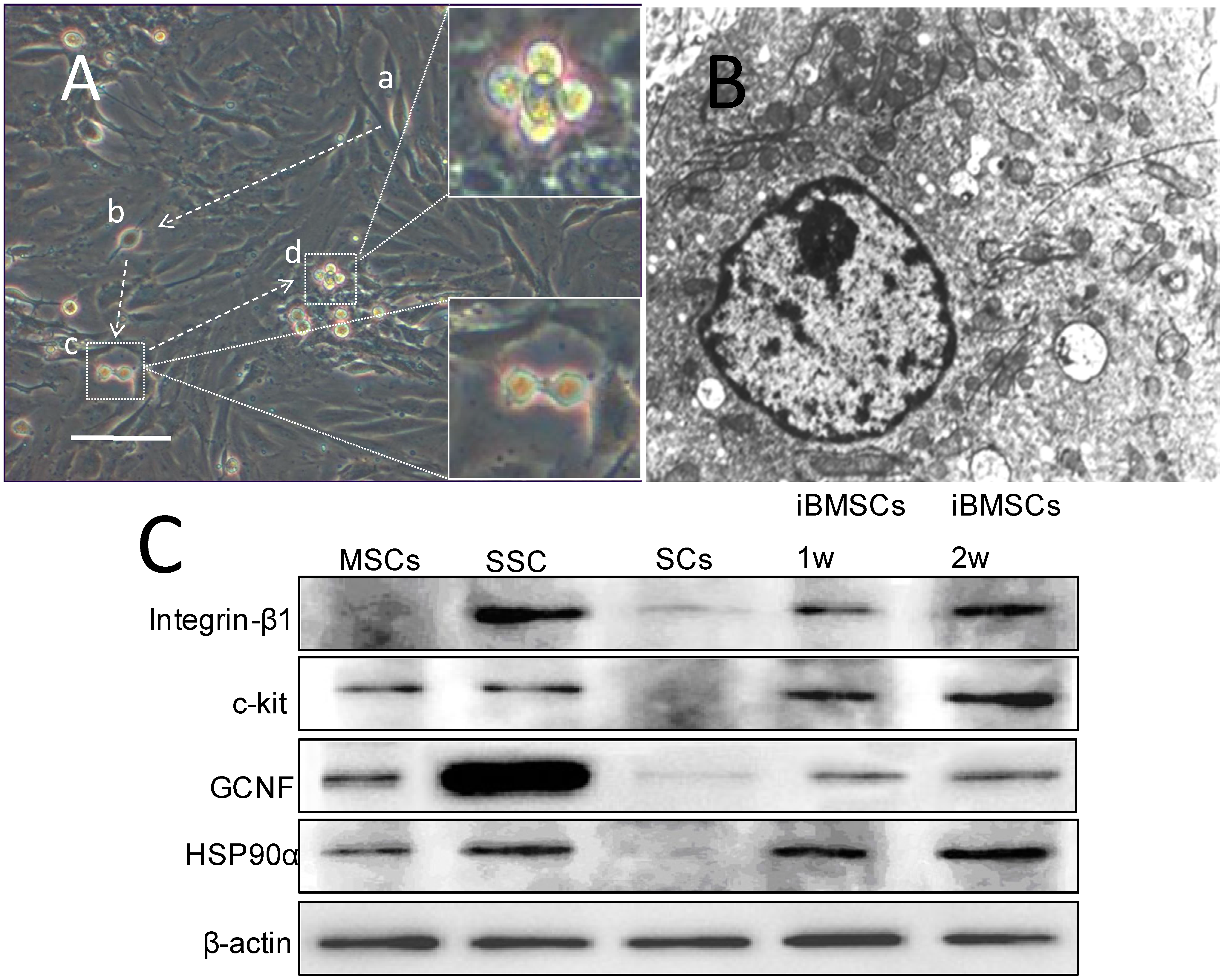
2.3. Morpholgy Changes and Spermatogenenic Protein Expression of Induced BMSCs in Vitro
2.4. Recipient Rats
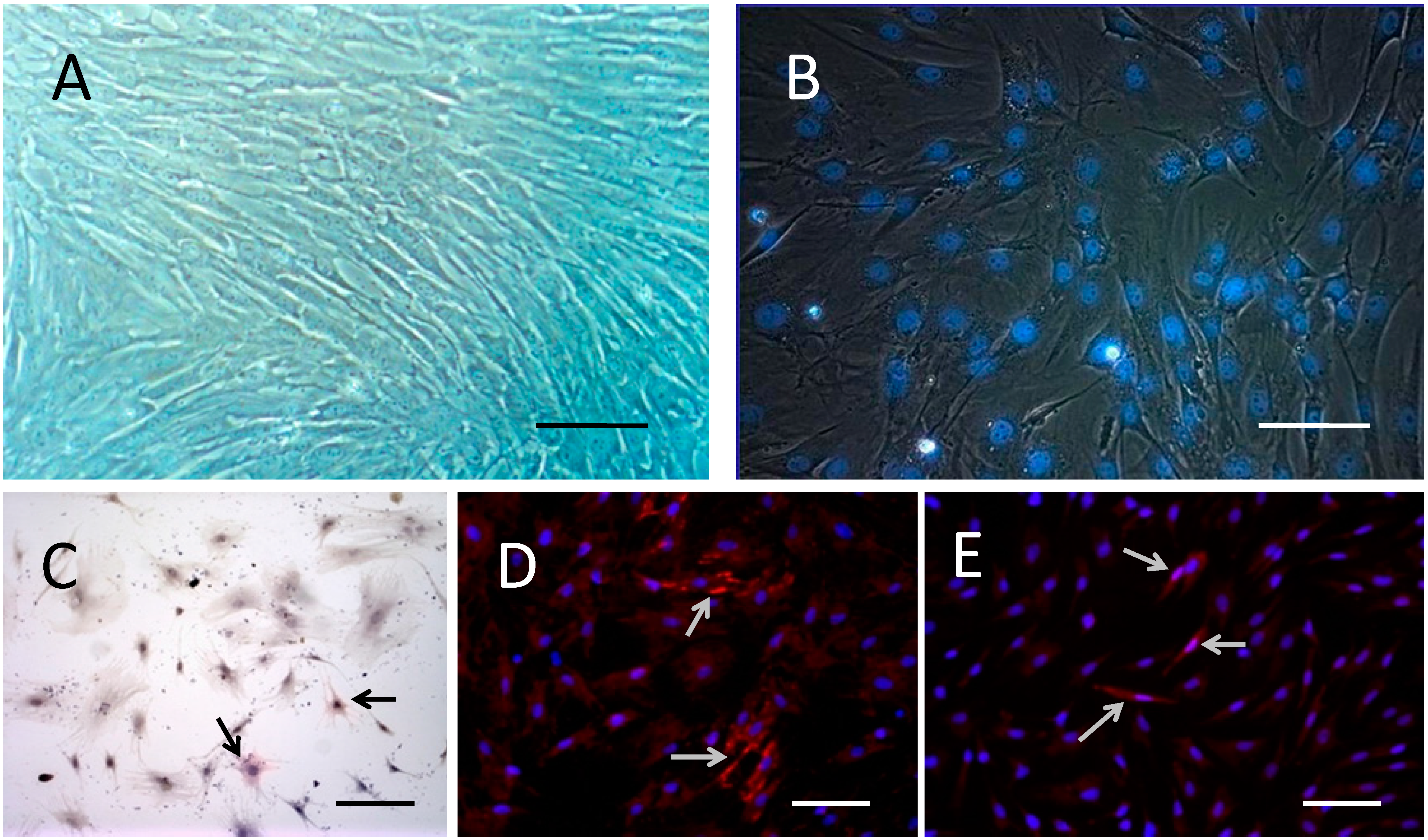
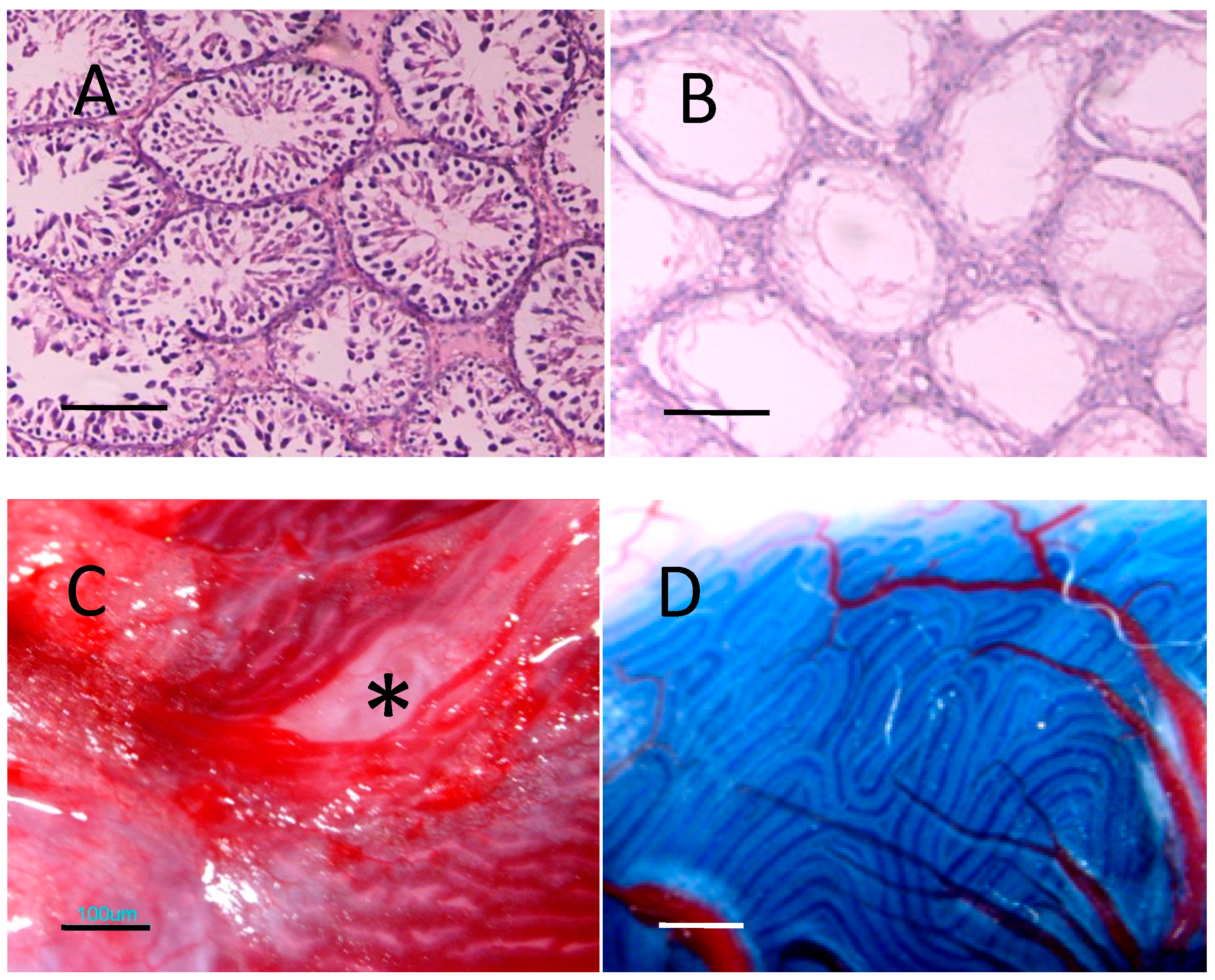
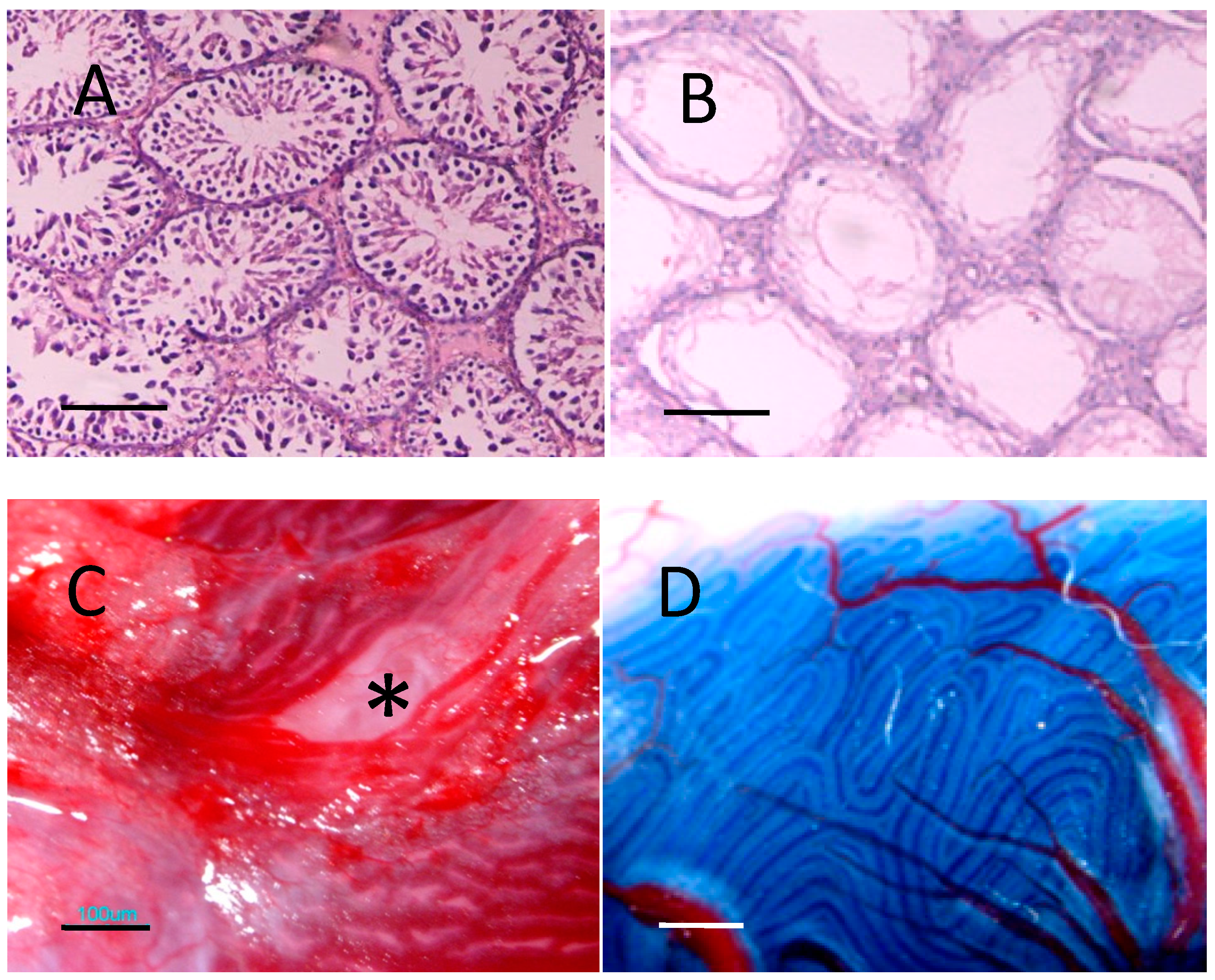
2.5. Donor Cells in Recipient Seminiferous Tubules
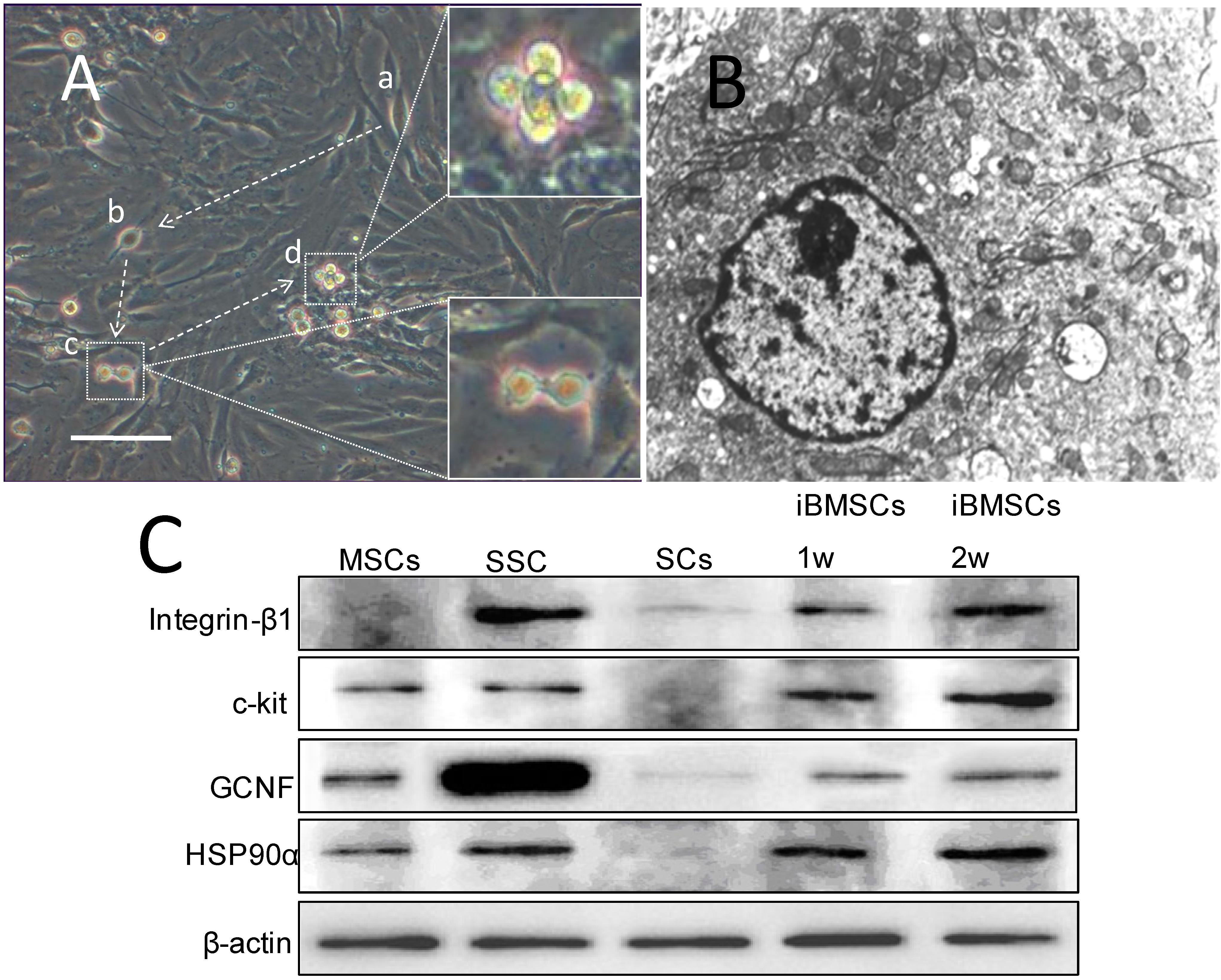
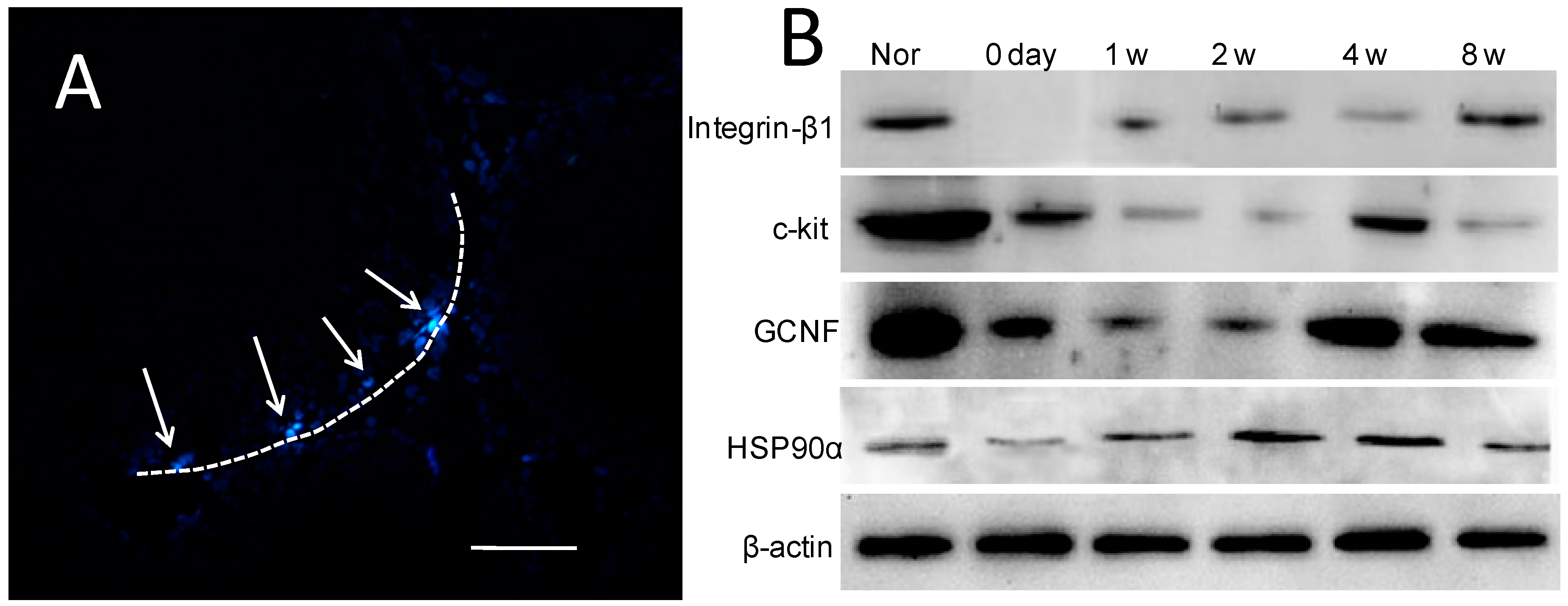
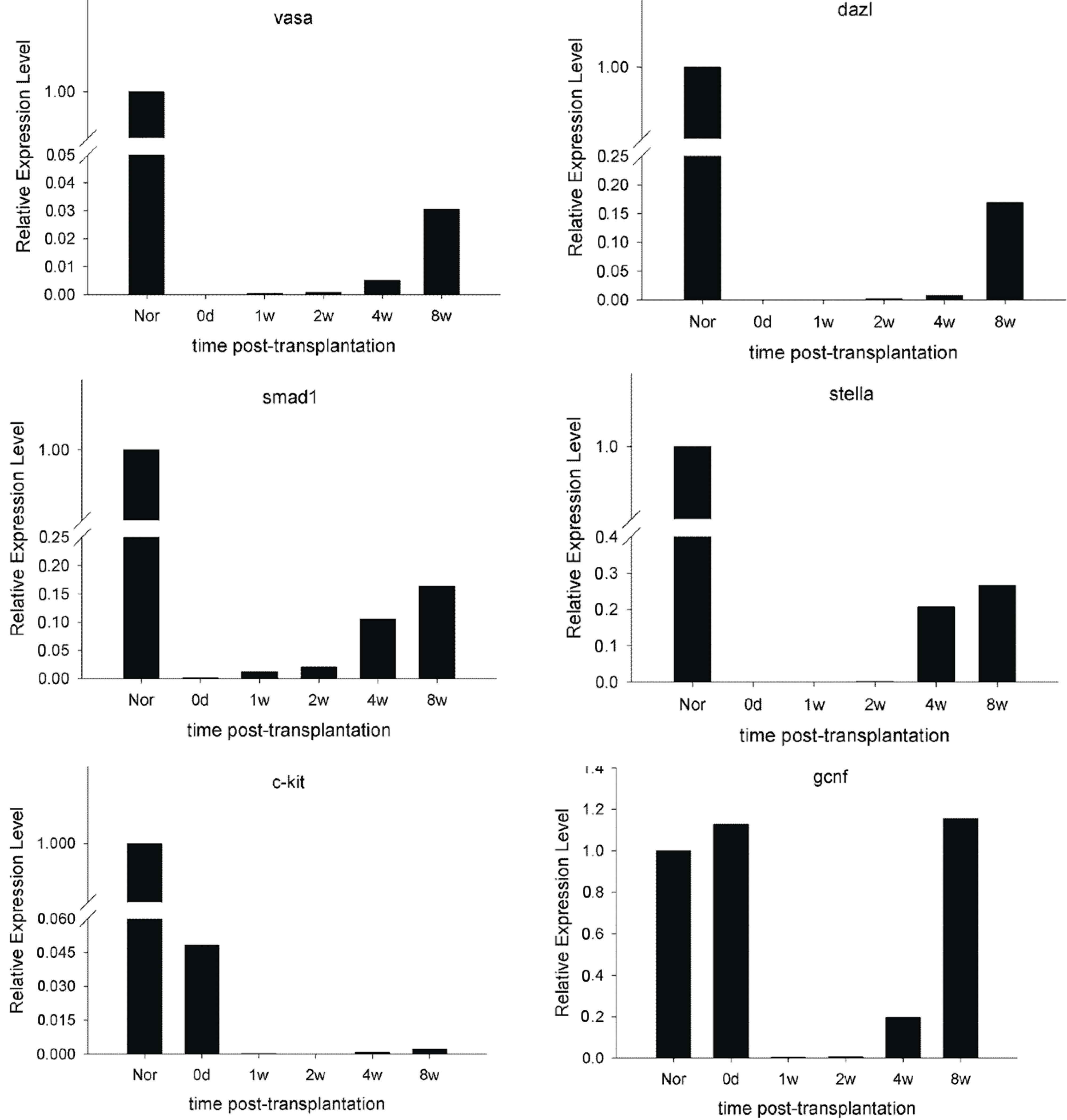
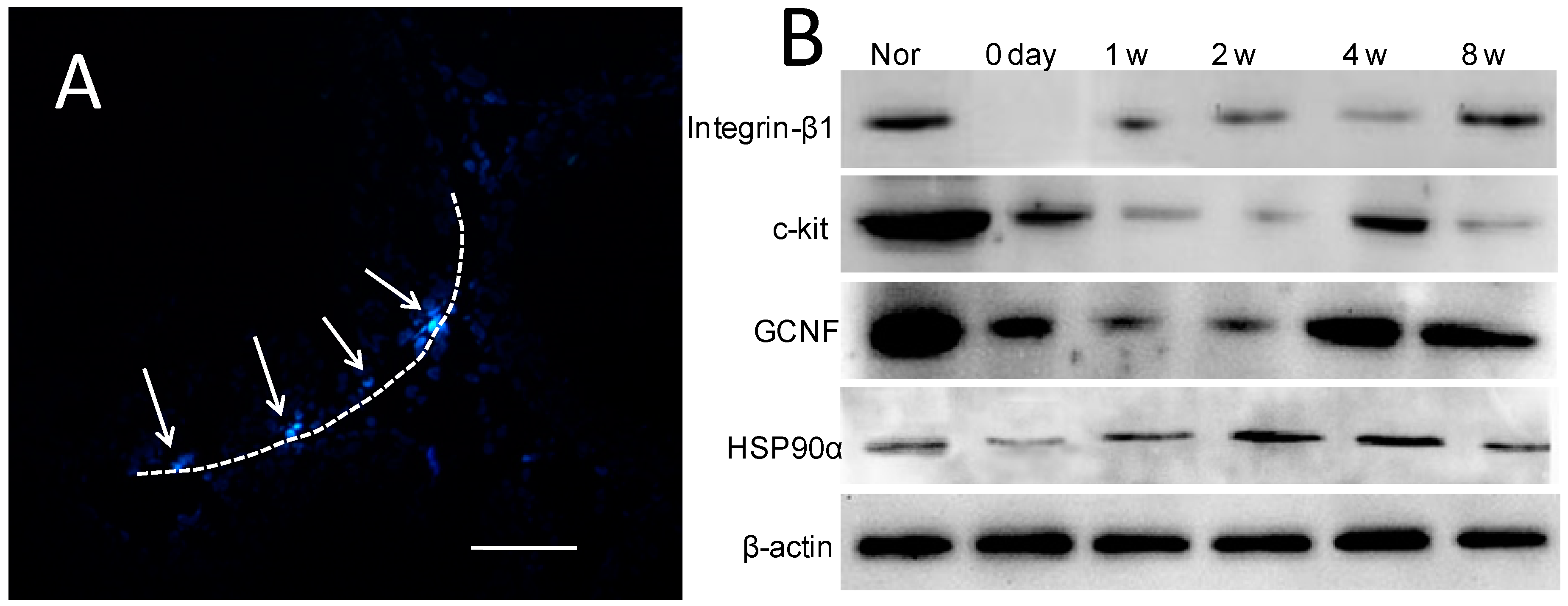
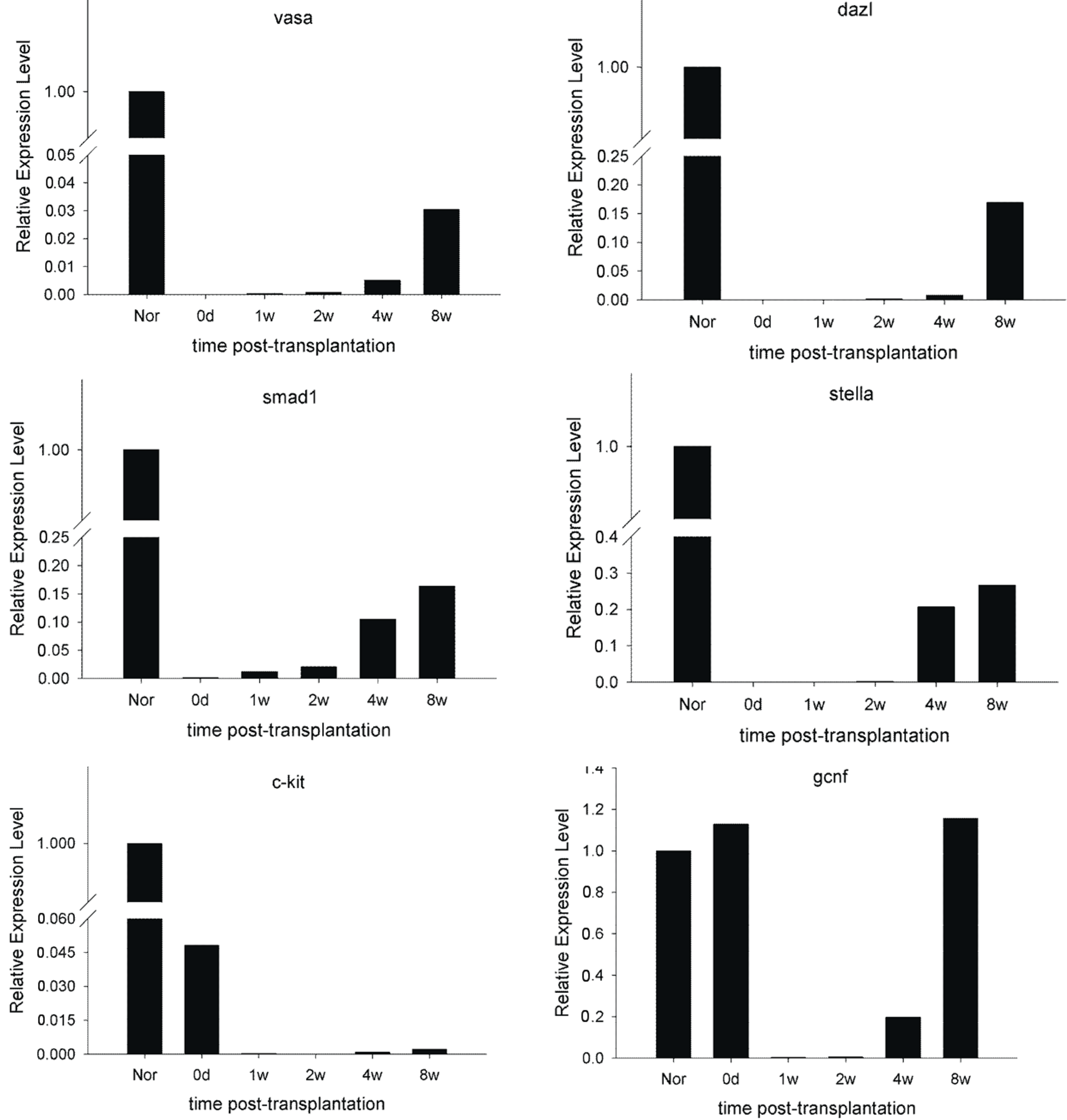
2.6. Expression of Spermatogenic Molecular Markers in Recipient Testicular Tissue

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Integrin-β1 | c-kit | GCNF | HSP90α |
|---|---|---|---|---|
| Nor | 0.77 ± 0.10 | 2.17 ± 0.50 | 1.84 ± 0.70 | 0.65 ± 0.17 |
| 0 day | 0.01 ± 0.01 * | 0.94 ± 0.19 * | 0.48 ± 0.13 * | 0.21 ± 0.08 * |
| 1 week | 0.11± 0.05 * | 0.62 ± 0.17 * | 0.27 ± 0.09 * | 0.45 ± 0.06 * |
| 2 weeks | 0.20 ± 0.08 * | 0.27 ± 0.08 * | 0.35 ± 0.09 * | 0.65 ± 0.13 |
| 4 weeks | 0.33 ± 0.12 * | 0.85 ± 0.14 * | 1.30 ± 0.23 | 0.76 ± 0.09 |
| 8 weeks | 0.69 ± 0.22 | 0.75 ± 0.24 * | 1.36 ± 0.25 | 0.71 ± 0.11 |
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. BMSCs Collection, Culture and Differentiation Potential Test
4.3. Induction of BMSCs to Spermatogenic Differentiation in Vitro by Co-Culture with Testicular Sertoli Cells in Conditioned Media
4.4. Preparation of Donor MSCs for Transplantation in Vivo
4.5. Preparation of Recipient Rats: Busulfan-Induced Azoospermatism Model
4.6. BMSCs Transplantation and Testicular Tissue Collection
4.7. RNA Extraction and Quantitative Real-Time RT-PCR
4.8. SDS-PAGE and Western Blotting
| Target Gene | Locus No. | Sequence of Primers | Product Size |
|---|---|---|---|
| Vasa | S75275 | F: 5'-GCGAGACTACATCTACAAC-3' | 135 bp |
| R: 5'-GAGTATCTTCACAGTCATTA-3' | |||
| SMAD1 | AF067727 | F: 5'-CTCATGTCATTTATTGCCG-3' | 138 bp |
| R: 5'-CTCGCTTATAGTGGTAGGGA-3' | |||
| Stella | BK001414 | F: 5'-CTATCATCGTCGTCAAAGG-3' | 177 bp |
| R: 5'-CTCTGCTCAATCCGAACAA-3' | |||
| Dazl | NM_001025742 | F: 5'-CGACGAAATCGGGAAGCTC-3' | 94 bp |
| R: 5'-CACAACCTCACCATACTGGGAAA-3' | |||
| GCNF | AJ783965 | F: 5'-CAACTGAACAAGCGGTATT-3' | 114 bp |
| R: 5'-GATGTATCGGATCTCTGGC-3' | |||
| c-kit | NM_022264 | F: 5'-TGCCCGAAACAAGTCATCTCC-3' | 112 bp |
| R: 5'-GGCTGAGGGTTCAACTTTATCCA-3' | |||
| β-actin | NM_031144 | F: 5'-GCTCGTCGTCGACAACGGCTC-3' | 353 bp |
| R: 5'-CAAACATGATCTGGGTCATCTTCTC-3' |
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pfister, O.; della Verde, G.; Liao, R.; Kuster, G.M. Regenerative therapy for cardiovascular disease. Transl. Res.: J. Lab. Clin. Med. 2014, 163, 307–320. [Google Scholar] [CrossRef]
- Leatherman, J. Stem cells supporting other stem cells. Front. Genet. 2013, 4, 257. [Google Scholar] [CrossRef]
- Brinster, R.L.; Zimmermann, J.W. Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA 1994, 91, 11298–11302. [Google Scholar] [CrossRef]
- Brinster, R.L.; Avarbock, M.R. Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. USA 1994, 91, 11303–11307. [Google Scholar] [CrossRef]
- Faes, K.; Tournaye, H.; Goethals, L.; Lahoutte, T.; Hoorens, A.; Goossens, E. Testicular cell transplantation into the human testes. Fertil. Steril. 2013, 100, 981–988. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Qu, R.; He, Y.; Tian, X.; Zeng, W. Spermatogonial stem cells from domestic animals: Progress and prospects. Reproduction 2014, 147, R65–R74. [Google Scholar] [CrossRef]
- Takehashi, M.; Kanatsu-Shinohara, M.; Shinohara, T. Generation of genetically modified animals using spermatogonial stem cells. Dev. Growth Differ. 2010, 52, 303–310. [Google Scholar] [CrossRef]
- Duggal, G.; Heindryckx, B.; Deroo, T.; de Sutter, P. Use of pluripotent stem cells for reproductive medicine: Are we there yet? Vet. Q. 2014, 34, 42–51. [Google Scholar] [CrossRef]
- Medrano, J.V.; Pera, R.A.; Simon, C. Germ cell differentiation from pluripotent cells. Semin. Reprod. Med. 2013, 31, 14–23. [Google Scholar] [CrossRef]
- Li, P.; Hu, H.; Yang, S.; Tian, R.; Zhang, Z.; Zhang, W.; Ma, M.; Zhu, Y.; Guo, X.; Huang, Y.; et al. Differentiation of induced pluripotent stem cells into male germ cells in vitro through embryoid body formation and retinoic acid or testosterone induction. BioMed Res. Int. 2013, 2013, 608728. [Google Scholar]
- Shirazi, R.; Zarnani, A.H.; Soleimani, M.; Abdolvahabi, M.A.; Nayernia, K.; Ragerdi Kashani, I. Bmp4 can generate primordial germ cells from bone-marrow-derived pluripotent stem cells. Cell Biol. Int. 2012, 36, 1185–1193. [Google Scholar] [CrossRef]
- He, Z. Derivation of male germ cells from induced pluripotent stem (ips) cells: A novel and crucial source for generating male gametes. Asian J. Androl. 2012, 14, 516–517. [Google Scholar] [CrossRef]
- Hayashi, Y.; Saitou, M.; Yamanaka, S. Germline development from human pluripotent stem cells toward disease modeling of infertility. Fertil. Steril. 2012, 97, 1250–1259. [Google Scholar] [CrossRef]
- Hayashi, K.; Ohta, H.; Kurimoto, K.; Aramaki, S.; Saitou, M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 2011, 146, 519–532. [Google Scholar] [CrossRef]
- Ensenat-Waser, R.; Pellicer, A.; Simon, C. Reprogrammed induced pluripotent stem cells: How suitable could they be in reproductive medicine? Fertil. Steril. 2009, 91, 971–974. [Google Scholar] [CrossRef]
- Kwan, M.D.; Longaker, M.T. Regenerative medicine: The next frontier. Transplantation 2008, 86, 206–207. [Google Scholar] [CrossRef]
- Gnecchi, M.; Melo, L.G. Bone marrow-derived mesenchymal stem cells: Isolation, expansion, characterization, viral transduction, and production of conditioned medium. Methods Mol. Biol. 2009, 482, 281–294. [Google Scholar] [CrossRef]
- Huselstein, C.; Li, Y.; He, X. Mesenchymal stem cells for cartilage engineering. Bio-Med. Mater. Eng. 2012, 22, 69–80. [Google Scholar]
- Nayernia, K.; Lee, J.H.; Drusenheimer, N.; Nolte, J.; Wulf, G.; Dressel, R.; Gromoll, J.; Engel, W. Derivation of male germ cells from bone marrow stem cells. Lab. Investig. J. Tech. Methods Pathol. 2006, 86, 654–663. [Google Scholar] [CrossRef]
- Cakici, C.; Buyrukcu, B.; Duruksu, G.; Haliloglu, A.H.; Aksoy, A.; Isik, A.; Uludag, O.; Ustun, H.; Subasi, C.; Karaoz, E. Recovery of fertility in azoospermia rats after injection of adipose-tissue-derived mesenchymal stem cells: The sperm generation. BioMed Res. Int. 2013, 2013, 529589. [Google Scholar]
- Gudeloglu, A.; Parekattil, S.J. Update in the evaluation of the azoospermic male. Clinics 2013, 68, 27–34. [Google Scholar] [CrossRef]
- Berookhim, B.M.; Schlegel, P.N. Azoospermia due to spermatogenic failure. Urol. Clin. N. Am. 2014, 41, 97–113. [Google Scholar] [CrossRef]
- Fraietta, R.; Zylberstejn, D.S.; Esteves, S.C. Hypogonadotropic hypogonadism revisited. Clinics 2013, 68, 81–88. [Google Scholar]
- Bibber, B.; Sinha, G.; Lobba, A.R.; Greco, S.J.; Rameshwar, P. A review of stem cell translation and potential confounds by cancer stem cells. Stem Cells Int. 2013, 2013, 241048. [Google Scholar]
- Takashima, S.; Kanatsu-Shinohara, M.; Tanaka, T.; Takehashi, M.; Morimoto, H.; Shinohara, T. Rac mediates mouse spermatogonial stem cell homing to germline niches by regulating transmigration through the blood-testis barrier. Cell Stem Cell 2011, 9, 463–475. [Google Scholar] [CrossRef]
- Kanatsu-Shinohara, M.; Shinohara, T. Spermatogonial stem cell self-renewal and development. Annu. Rev. Cell Dev. Biol. 2013, 29, 163–187. [Google Scholar] [CrossRef]
- Brinster, C.J.; Ryu, B.Y.; Avarbock, M.R.; Karagenc, L.; Brinster, R.L.; Orwig, K.E. Restoration of fertility by germ cell transplantation requires effective recipient preparation. Biol. Reprod. 2003, 69, 412–420. [Google Scholar] [CrossRef]
- Ogawa, T.; Dobrinski, I.; Brinster, R.L. Recipient preparation is critical for spermatogonial transplantation in the rat. Tissue Cell 1999, 31, 461–472. [Google Scholar] [CrossRef]
- Lassalle, B.; Mouthon, M.A.; Riou, L.; Barroca, V.; Coureuil, M.; Boussin, F.; Testart, J.; Allemand, I.; Fouchet, P. Bone marrow-derived stem cells do not reconstitute spermatogenesis in vivo. Stem Cells 2008, 26, 1385–1386. [Google Scholar] [CrossRef]
- Ren, G.; Chen, X.; Dong, F.; Li, W.; Ren, X.; Zhang, Y.; Shi, Y. Concise review: Mesenchymal stem cells and translational medicine: Emerging issues. Stem Cells Transl. Med. 2012, 1, 51–58. [Google Scholar] [CrossRef]
- Mital, P.; Kaur, G.; Dufour, J.M. Immunoprotective sertoli cells: Making allogeneic and xenogeneic transplantation feasible. Reproduction 2010, 139, 495–504. [Google Scholar] [CrossRef]
- Mital, P.; Hinton, B.T.; Dufour, J.M. The blood-testis and blood-epididymis barriers are more than just their tight junctions. Biol. Reprod. 2011, 84, 851–858. [Google Scholar] [CrossRef]
- Monsefi, M.; Fereydouni, B.; Rohani, L.; Talaei, T. Mesenchymal stem cells repair germinal cells of seminiferous tubules of sterile rats. Iran. J. Reprod. Med. 2013, 11, 537–544. [Google Scholar]
- Kucia, M.; Wu, W.; Ratajczak, M.Z. Bone marrow-derived very small embryonic-like stem cells: Their developmental origin and biological significance. Dev. Dyn. 2007, 236, 3309–3320. [Google Scholar] [CrossRef]
- Gou, S.; Liu, T.; Li, X.; Cui, J.; Wan, C.; Wang, C. Pancreatic ductal cells acquire mesenchymal characteristics through cell fusion with bone marrow-derived mesenchymal stem cells and sirt1 attenuates the apoptosis of hybrid cells. Cells Tissues Organs 2012, 196, 129–136. [Google Scholar] [CrossRef]
- Song, Y.H.; Pinkernell, K.; Alt, E. Stem cell induced cardiac regeneration: Fusion/mitochondrial exchange and/or transdifferentiation? Cell Cycle 2011, 10, 2281–2286. [Google Scholar] [CrossRef]
- Bhartiya, D. Are mesenchymal cells indeed pluripotent stem cells or just stromal cells? Oct-4 and vsels biology has led to better understanding. Stem Cells Int. 2013, 2013, 547501. [Google Scholar]
- Bhartiya, D.; Kasiviswananthan, S.; Shaikh, A. Cellular origin of testis-derived pluripotent stem cells: A case for very small embryonic-like stem cells. Stem Cells and Dev. 2012, 21, 670–674. [Google Scholar] [CrossRef]
- Bhartiya, D.; Unni, S.; Parte, S.; Anand, S. Very small embryonic-like stem cells: Implications in reproductive biology. BioMed Res. Int. 2013, 2013, 682326. [Google Scholar]
- Pereira, R.F.; Halford, K.W.; OHara, M.D.; Leeper, D.B.; Sokolov, B.P.; Pollard, M.D.; Bagasra, O.; Prockop, D.J. Cultured adherent cells from marrow can serve as long-lasting precursor cells for bone, cartilage, and lung in irradiated mice. Proc. Natl. Acad. Sci. USA 1995, 92, 4857–4861. [Google Scholar] [CrossRef]
- Grima, J.; Wong, C.C.; Zhu, L.J.; Zong, S.D.; Cheng, C.Y. Testin secreted by sertoli cells is associated with the cell surface, and its expression correlates with the disruption of sertoli-germ cell junctions but not the inter-sertoli tight junction. J. Biol. Chem. 1998, 273, 21040–21053. [Google Scholar]
- Ogawa, T.; Arechaga, J.M.; Avarbock, M.R.; Brinster, R.L. Transplantation of testis germinal cells into mouse seminiferous tubules. Int. J. Dev. Biol. 1997, 41, 111–122. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, D.; Liu, X.; Peng, J.; He, D.; Lin, T.; Zhu, J.; Li, X.; Zhang, Y.; Wei, G. Potential Spermatogenesis Recovery with Bone Marrow Mesenchymal Stem Cells in an Azoospermic Rat Model. Int. J. Mol. Sci. 2014, 15, 13151-13165. https://doi.org/10.3390/ijms150813151
Zhang D, Liu X, Peng J, He D, Lin T, Zhu J, Li X, Zhang Y, Wei G. Potential Spermatogenesis Recovery with Bone Marrow Mesenchymal Stem Cells in an Azoospermic Rat Model. International Journal of Molecular Sciences. 2014; 15(8):13151-13165. https://doi.org/10.3390/ijms150813151
Chicago/Turabian StyleZhang, Deying, Xing Liu, Jinpu Peng, Dawei He, Tao Lin, Jing Zhu, Xuliang Li, Yuanyuan Zhang, and Guanghui Wei. 2014. "Potential Spermatogenesis Recovery with Bone Marrow Mesenchymal Stem Cells in an Azoospermic Rat Model" International Journal of Molecular Sciences 15, no. 8: 13151-13165. https://doi.org/10.3390/ijms150813151
APA StyleZhang, D., Liu, X., Peng, J., He, D., Lin, T., Zhu, J., Li, X., Zhang, Y., & Wei, G. (2014). Potential Spermatogenesis Recovery with Bone Marrow Mesenchymal Stem Cells in an Azoospermic Rat Model. International Journal of Molecular Sciences, 15(8), 13151-13165. https://doi.org/10.3390/ijms150813151