The Cross Talk between cGMP Signal Pathway and PKC in Pulmonary Endothelial Cell Angiogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. TGF-β1 Induces PEVC (Pulmonary Vascular Endothelial Cell) Proliferation
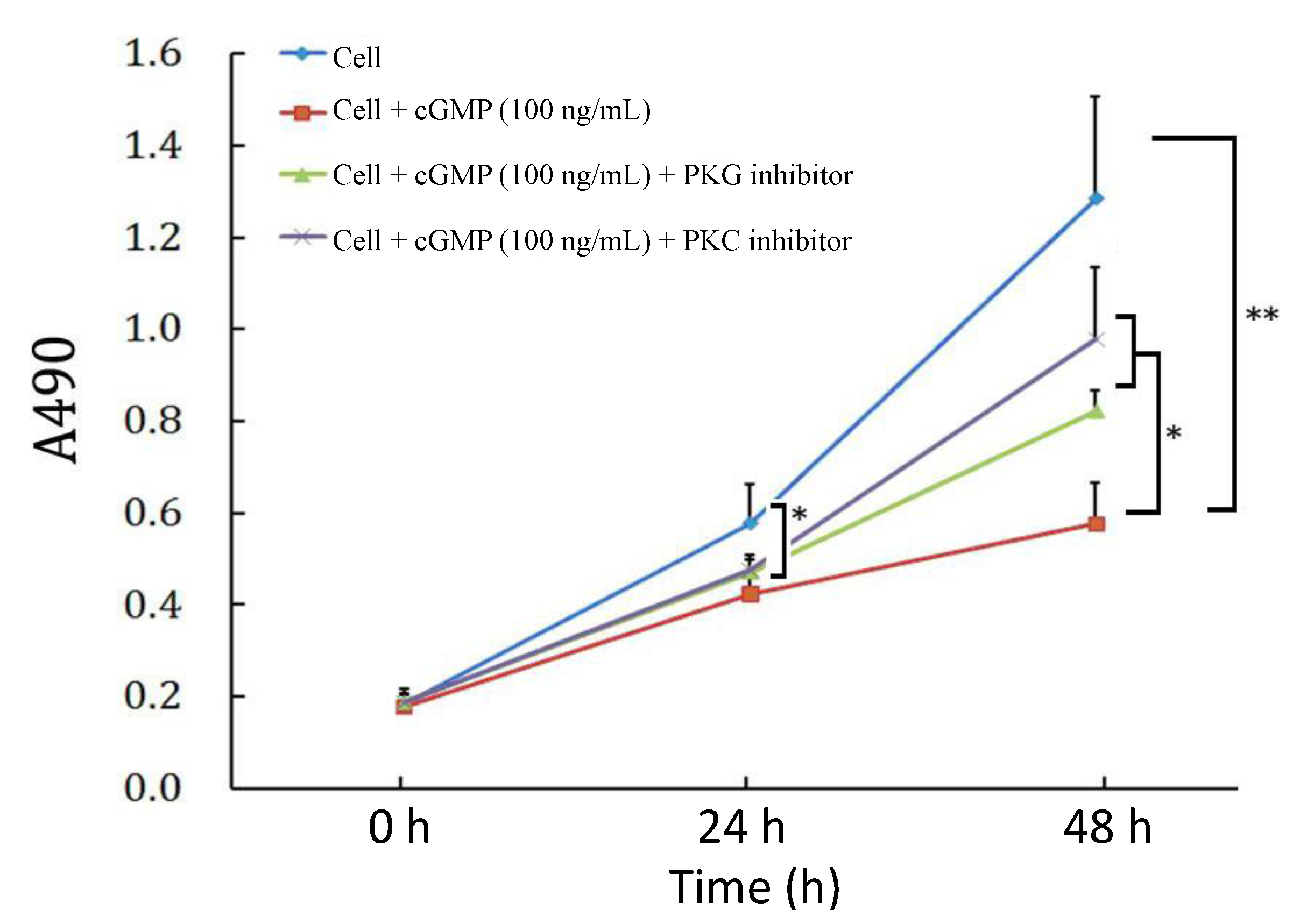
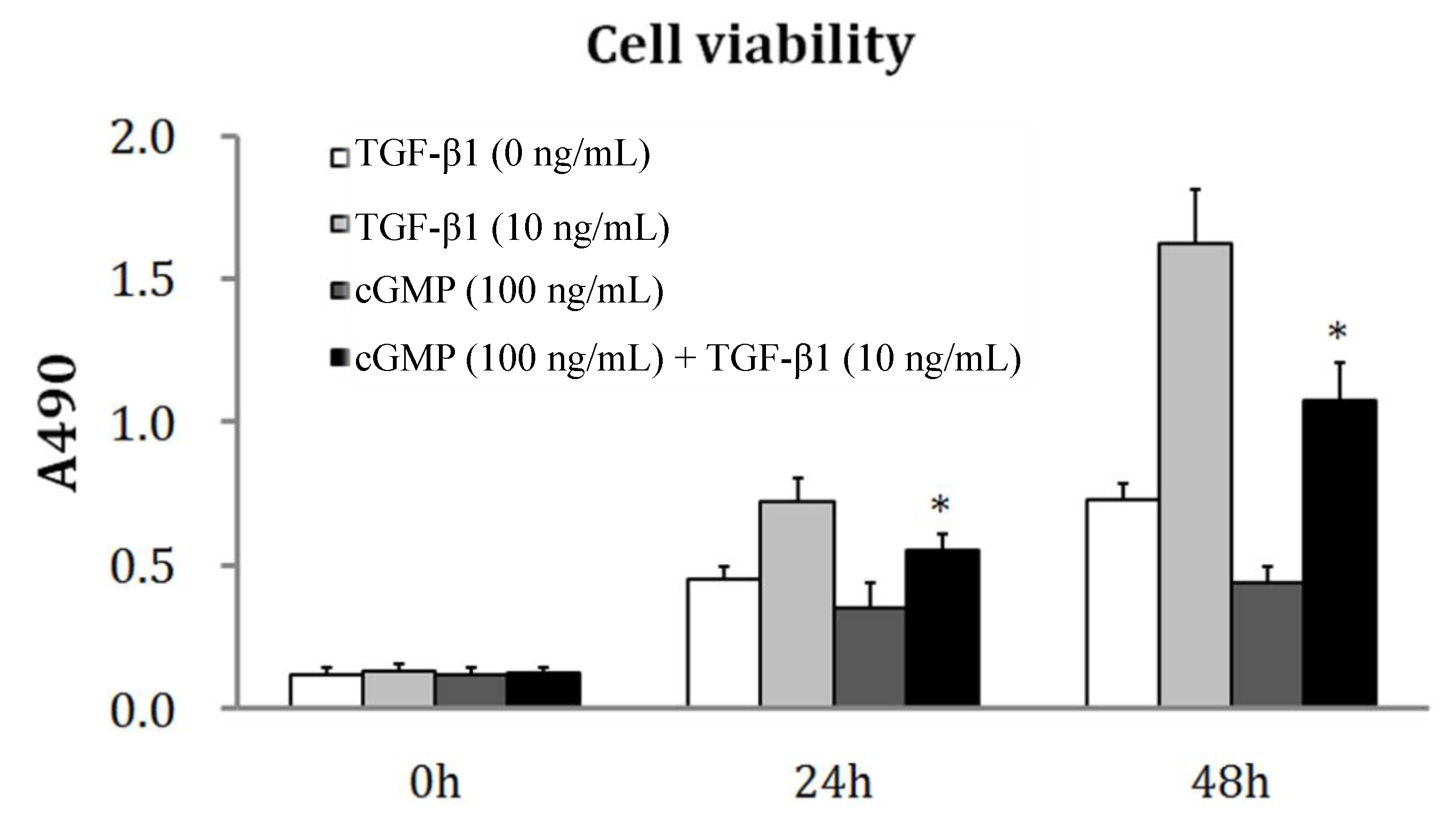
2.2. The Reversibility of Cell Proliferation by cGMP (Cyclic Guanosine Monophosphate)
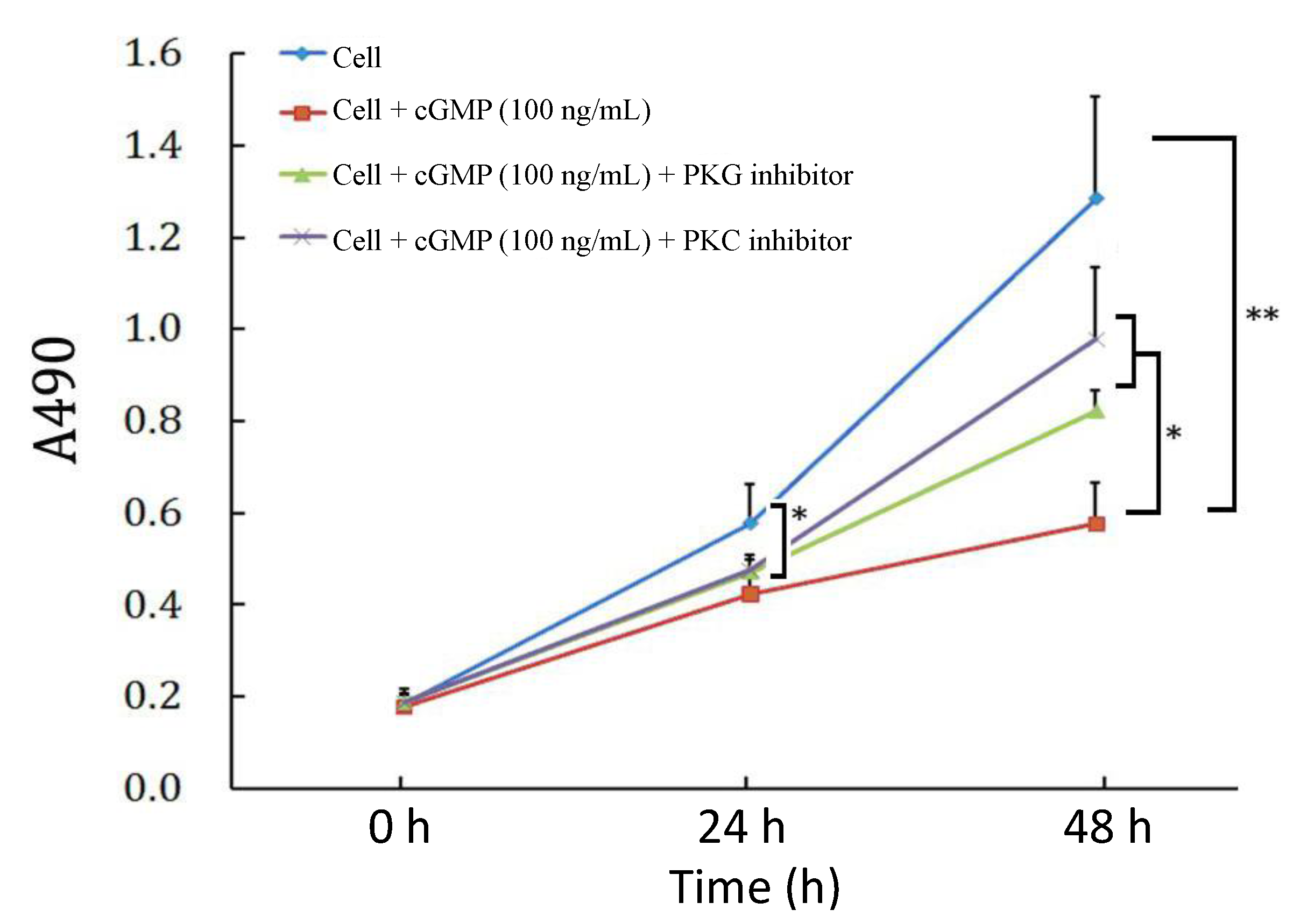
2.3. The Effects of PKC (Protein Kinase C) Inhibitor on Cell Proliferation Regulated by the cGMP-PKG (Protein Kinase G) Pathway
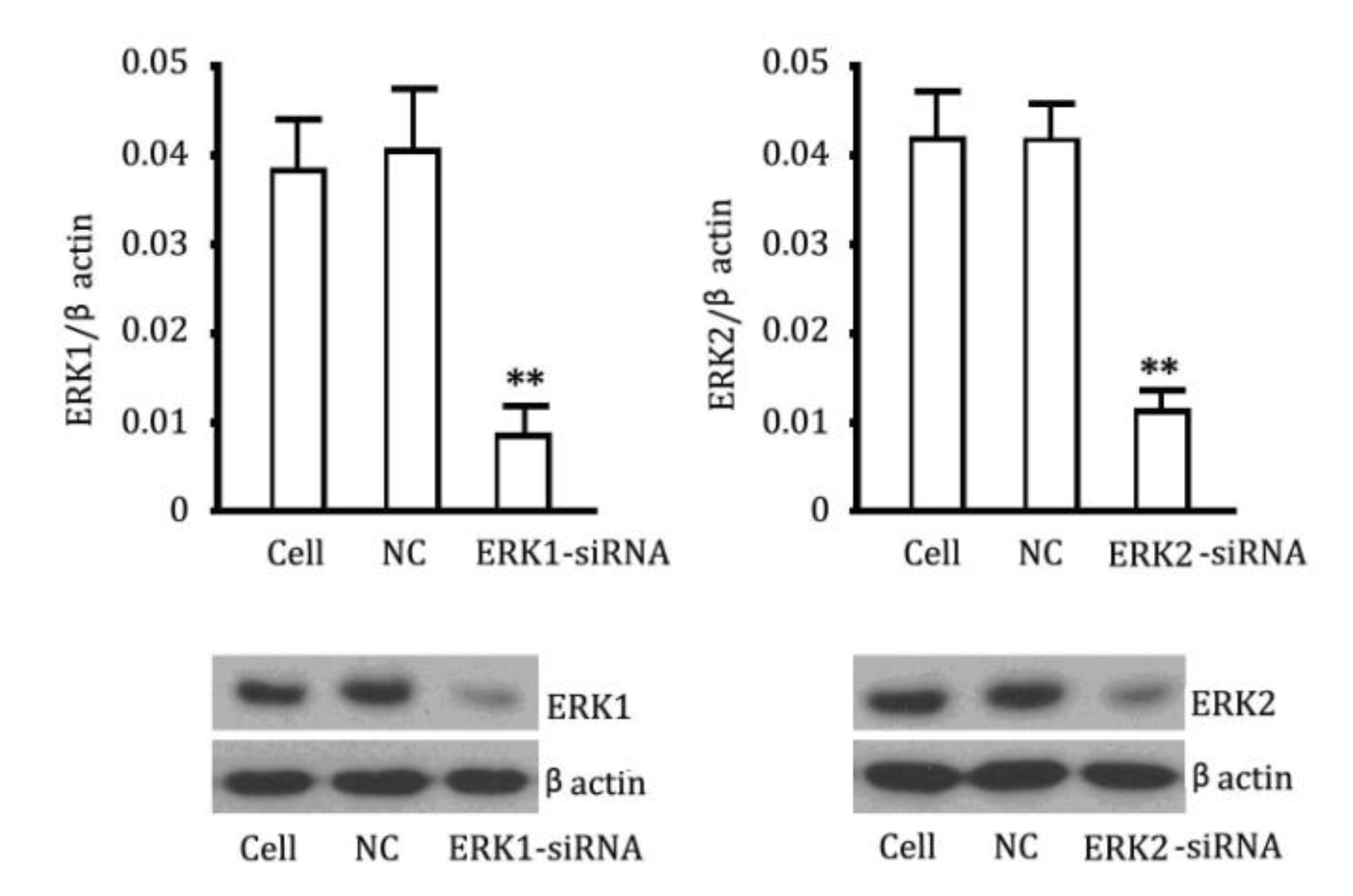
2.4. The Reversibility of Cell Proliferation of ERK siRNA
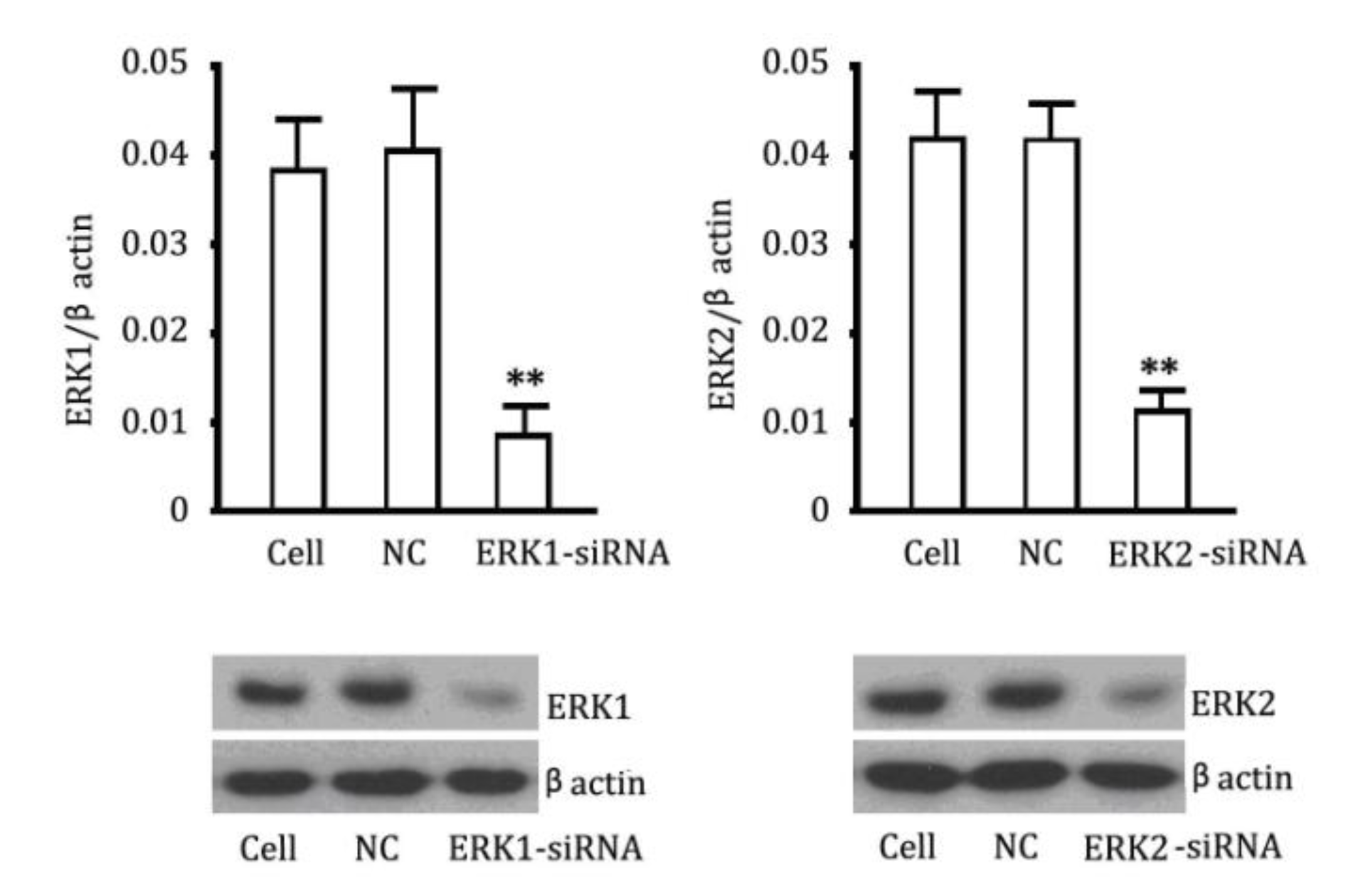
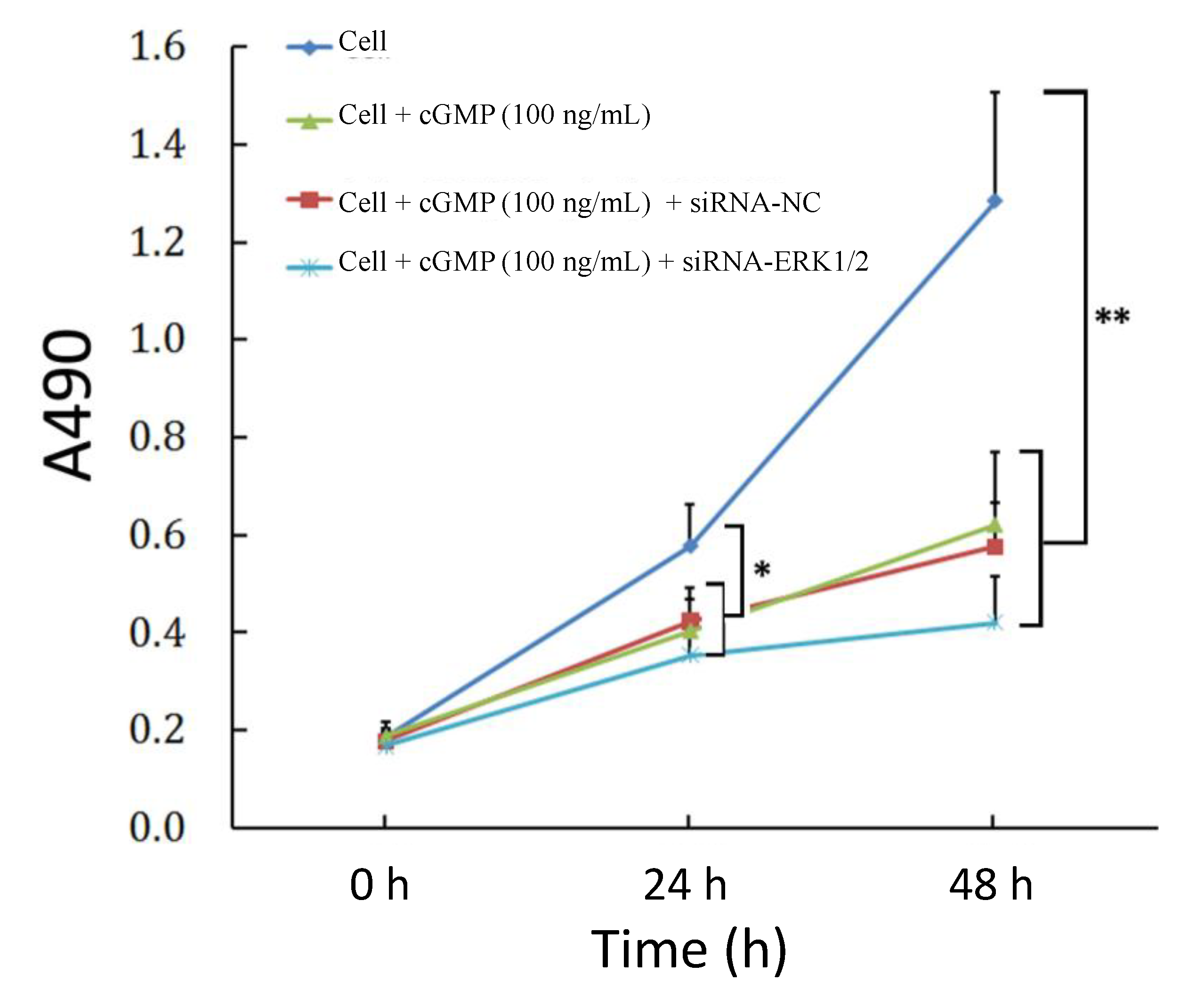
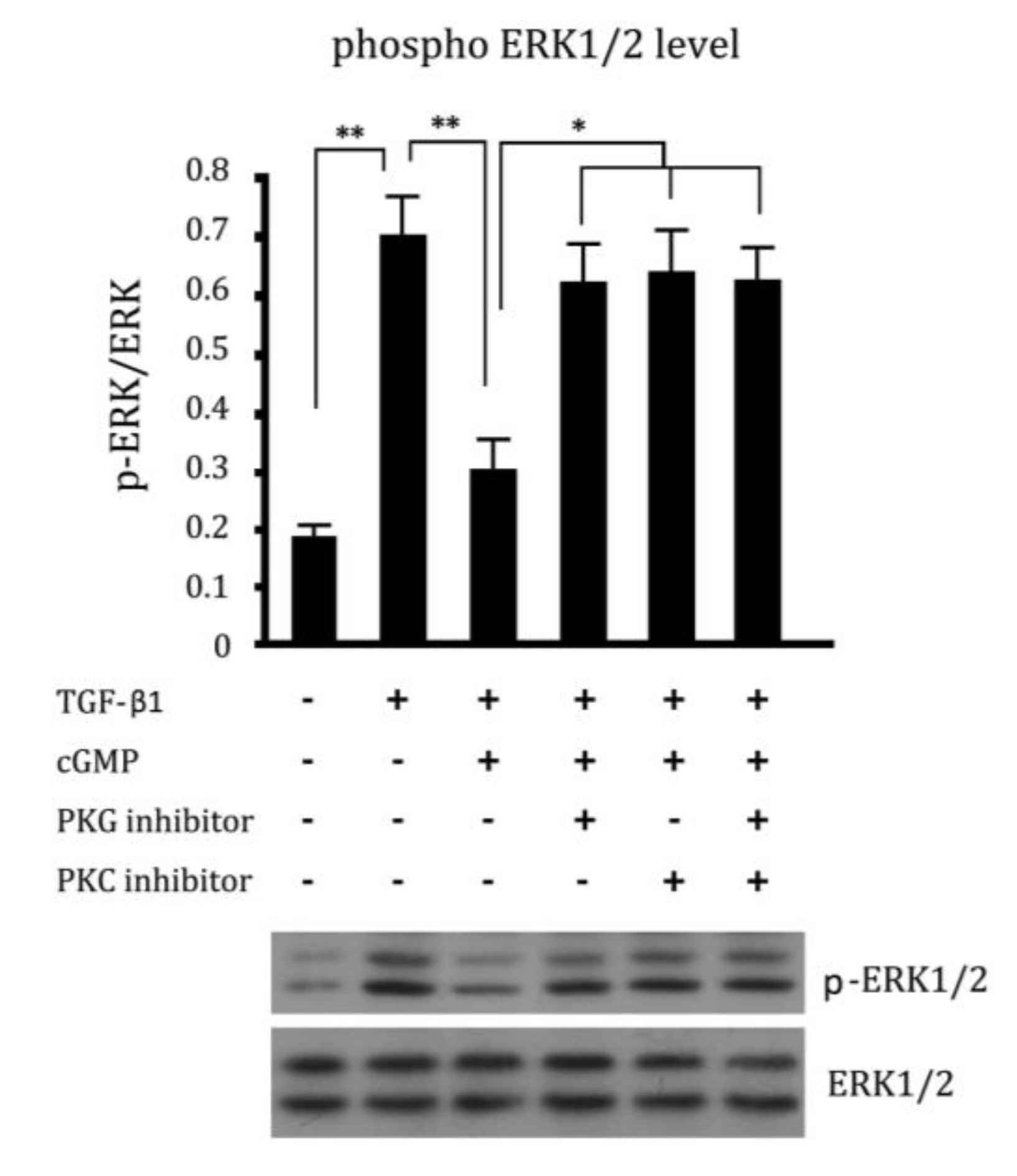
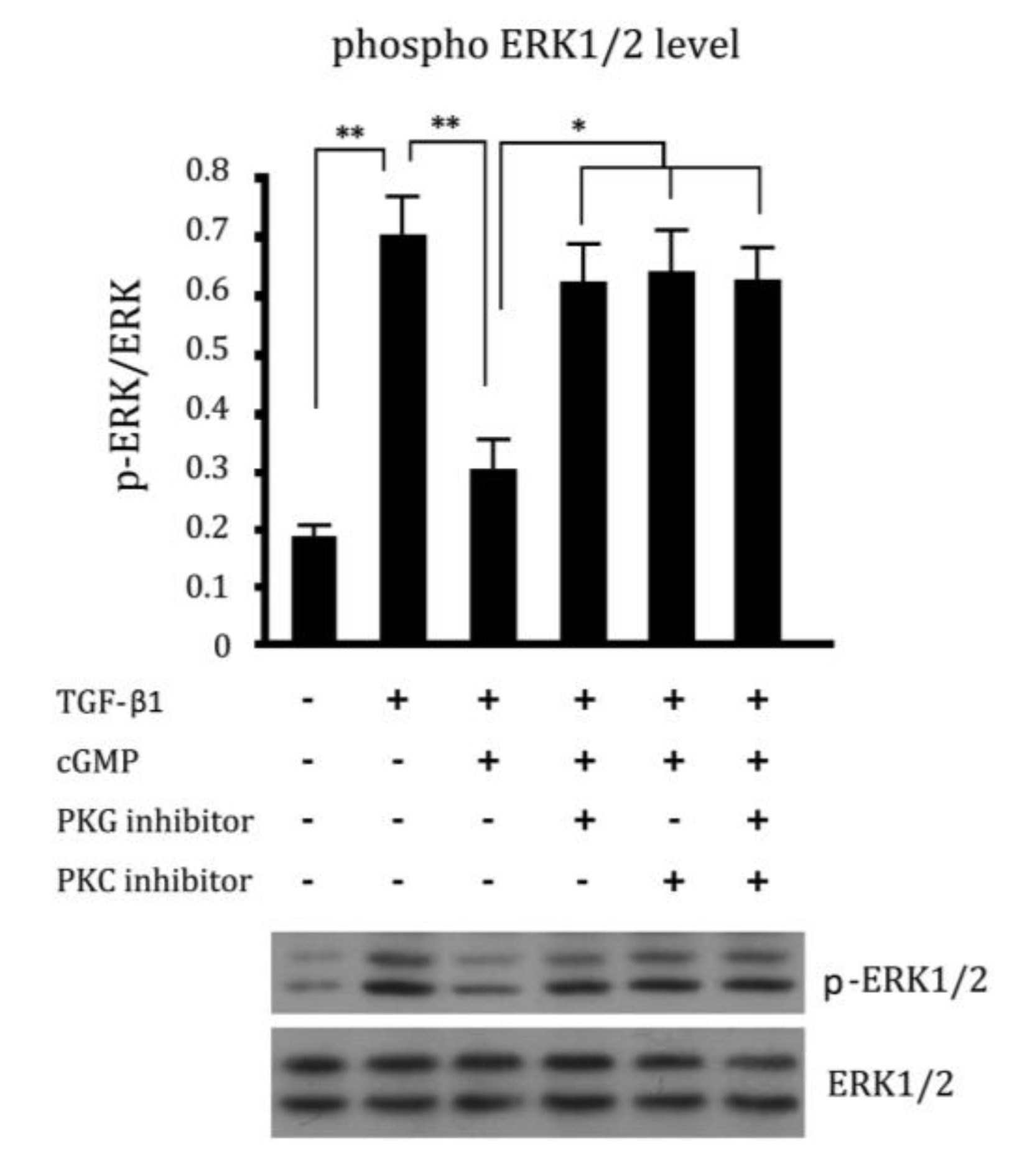
2.5. Cross Talk Effects between PKC Inhibitor and cGMP Down-Regulated ERK Pathway
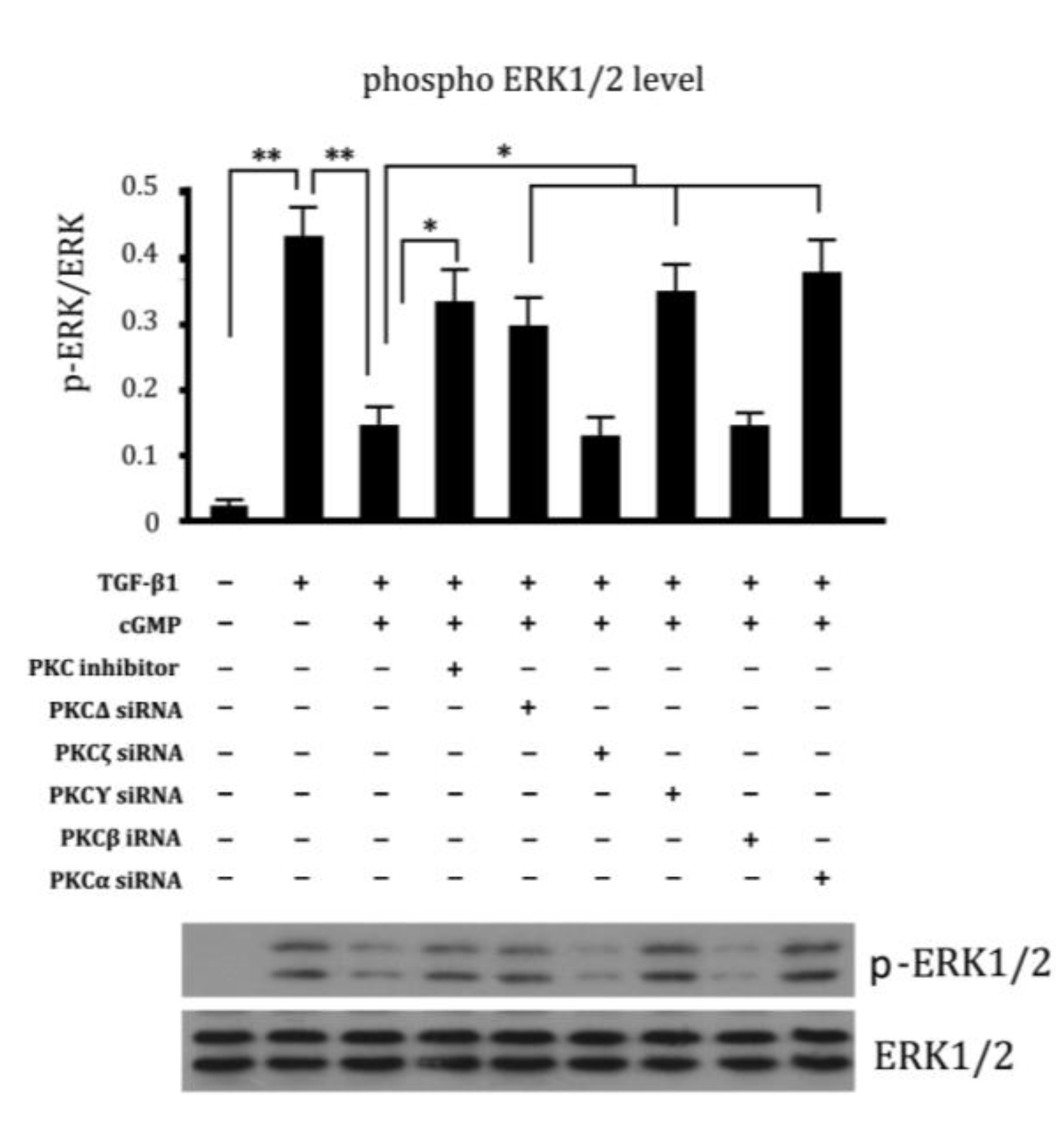
2.6. Involvement of Different Subtypes of PKC in Cell Proliferation Regulated by cGMP
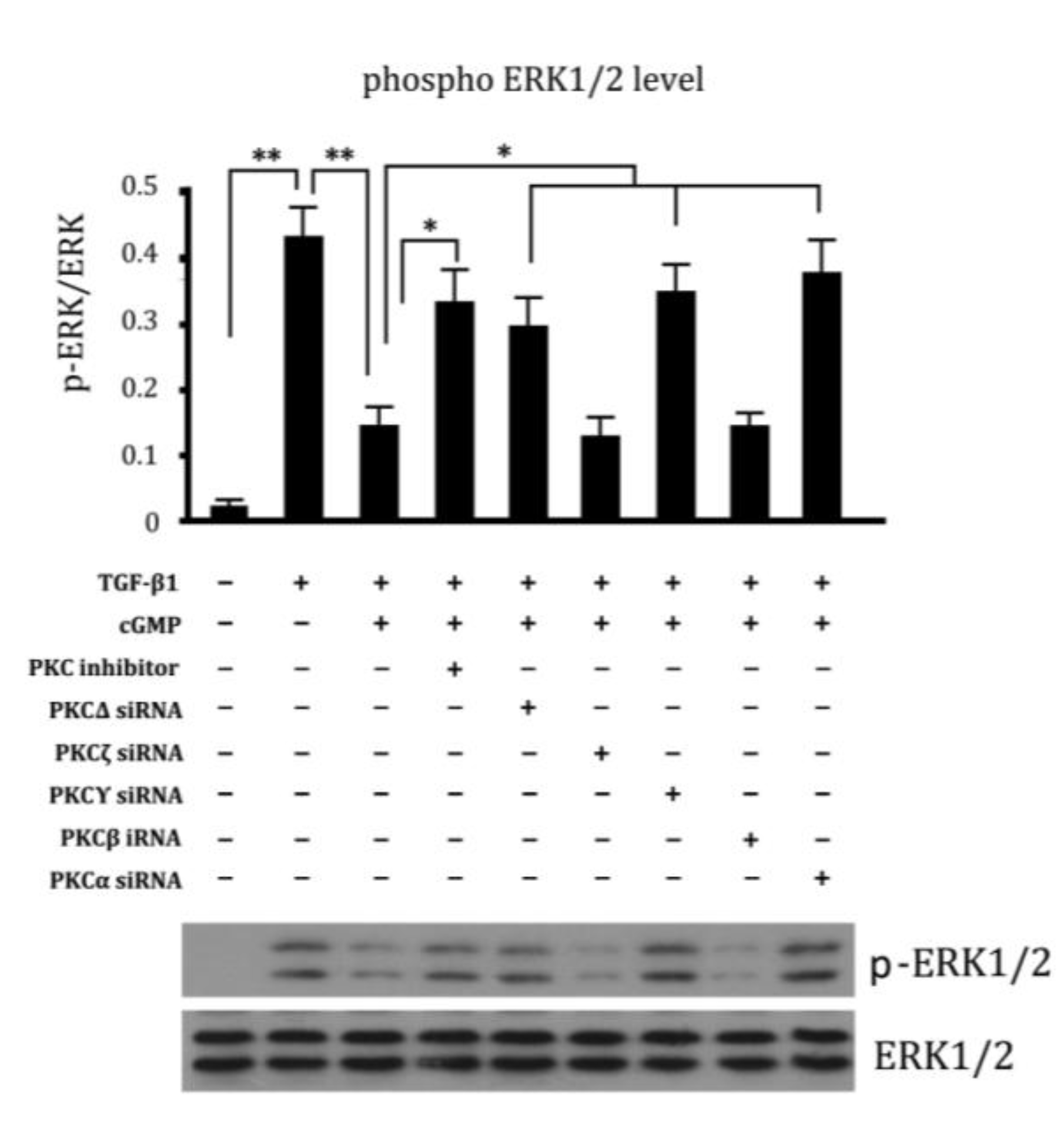
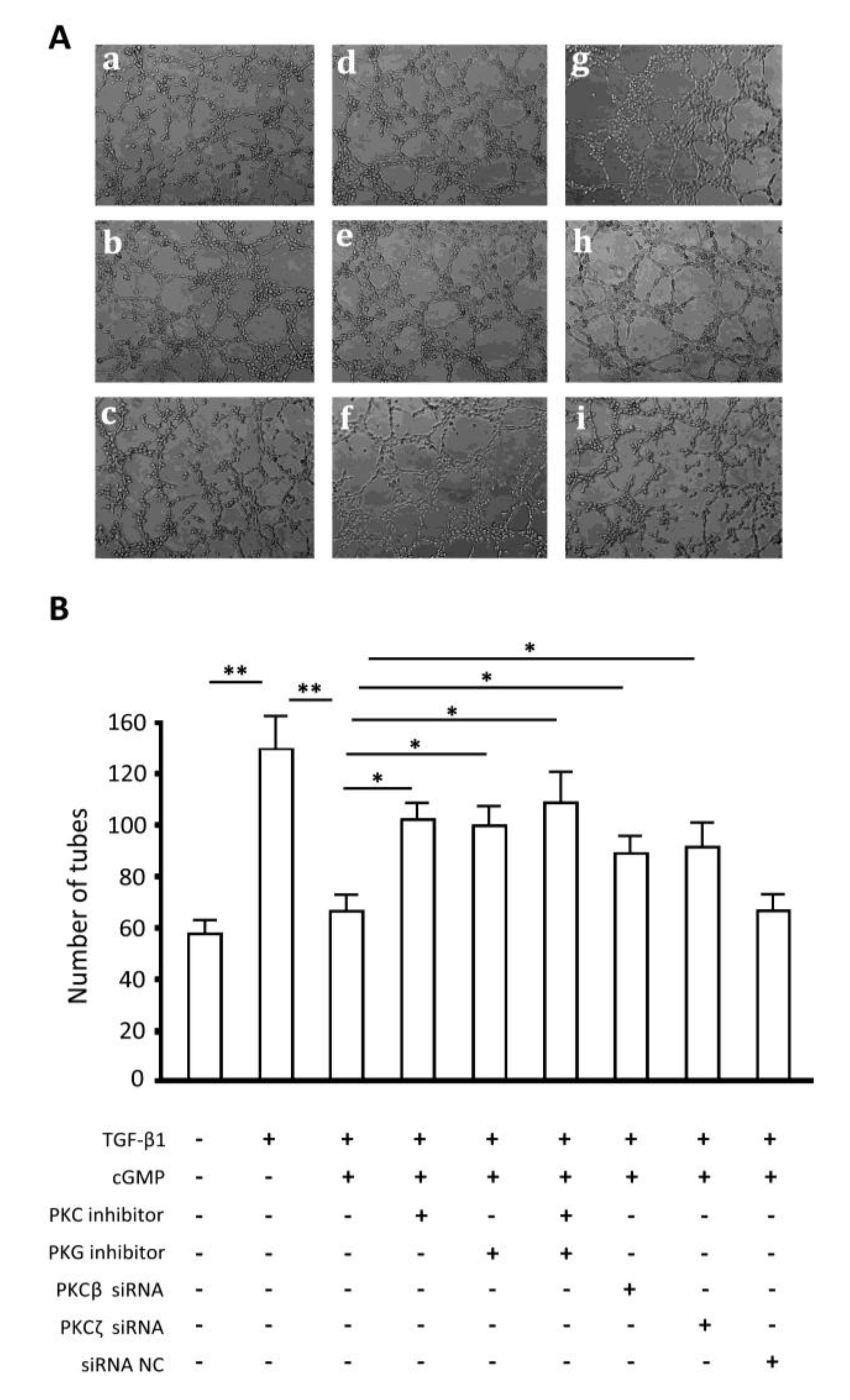
2.7. Endothelial Tube Formation Assay
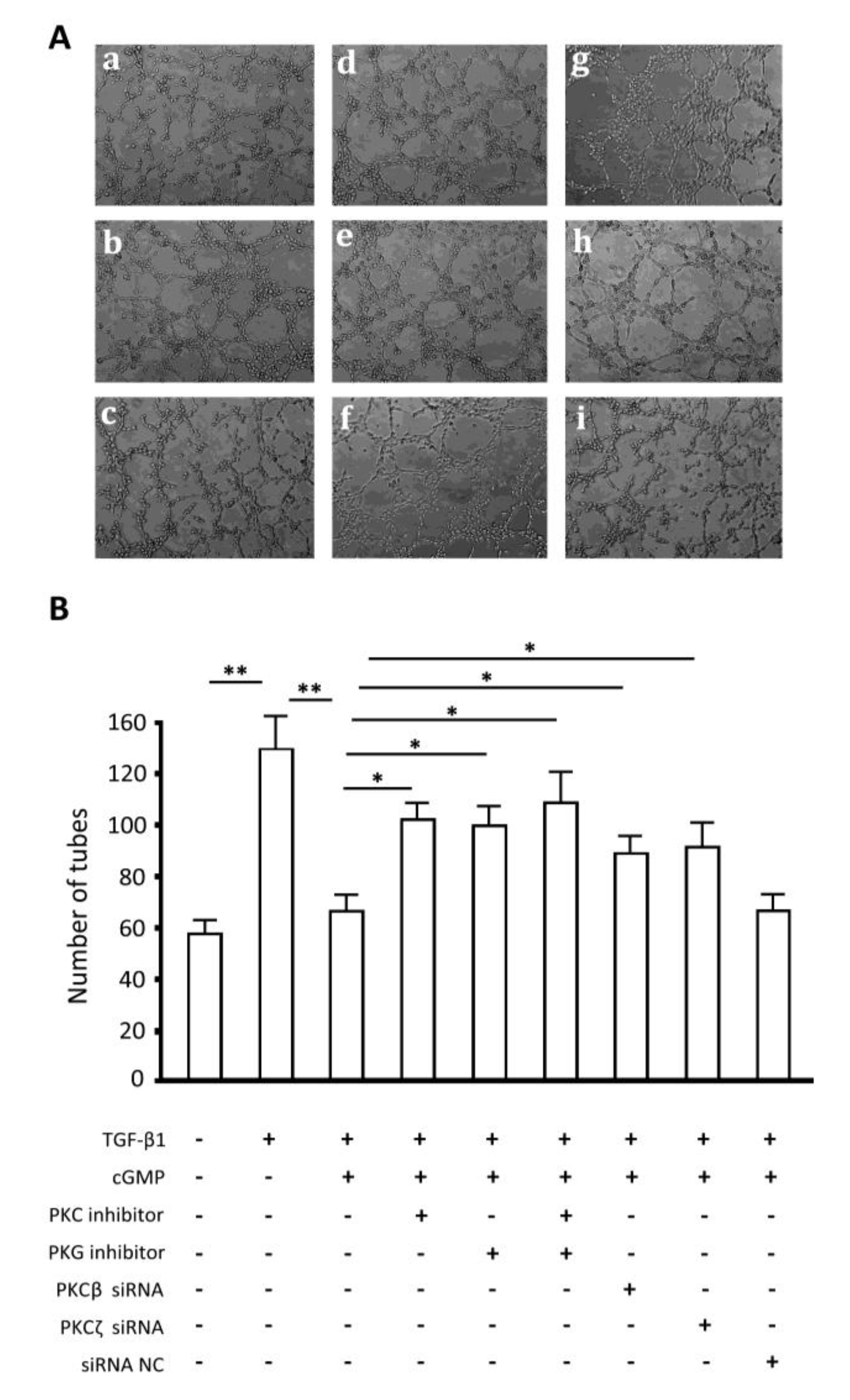
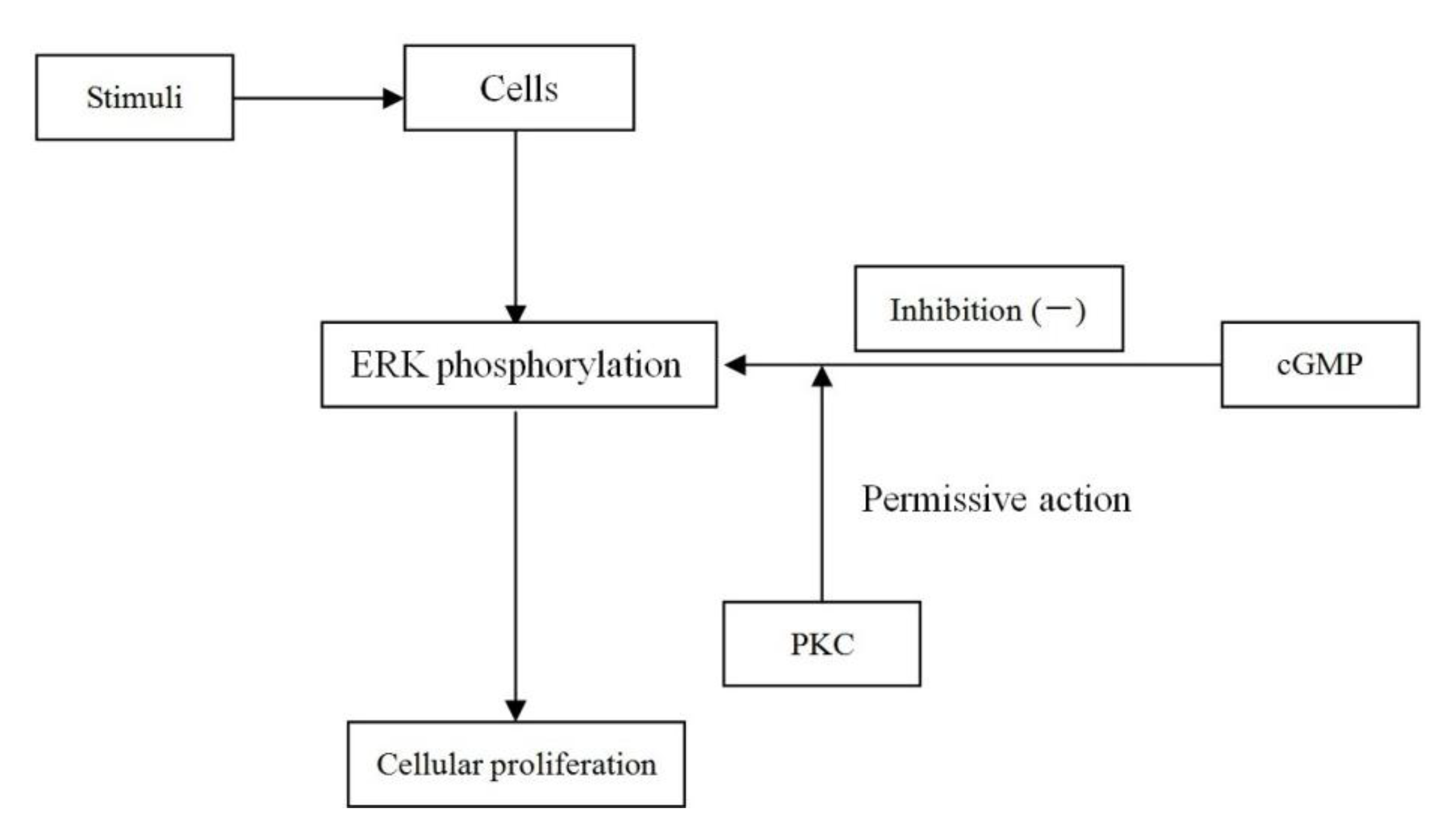
2.8. Discussion
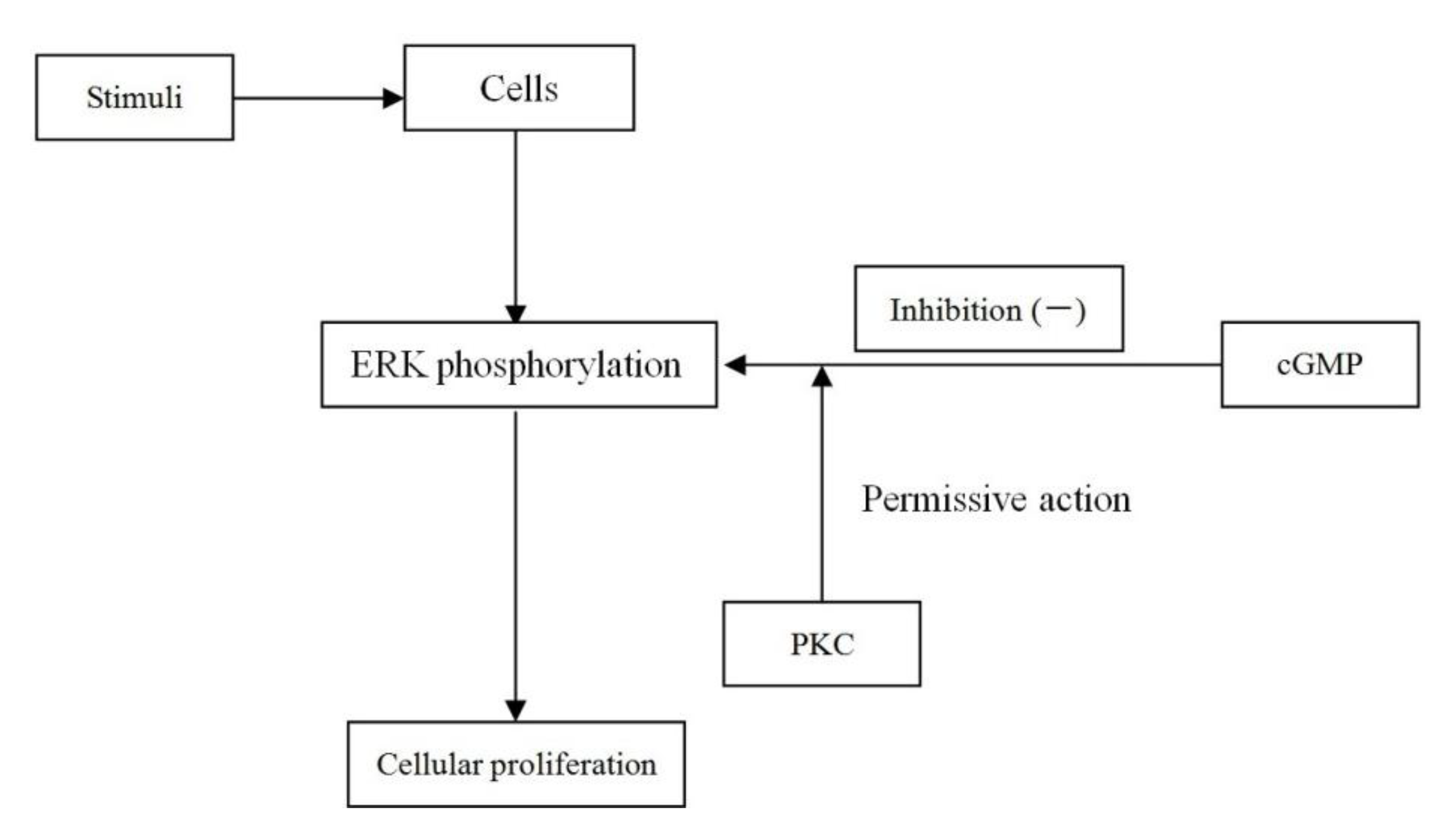
3. Experimental Section
3.1. Antibody and Regents
3.2. Cell Culture
3.2.1. Isolation and Culture of Rat Pulmonary Microvascular Endothelial Cells
3.2.2. Isolation and Identification of Rat Peripheral Blood Endothelial Progenitor Cells and Differentiation of Endothelial Cells
3.3. Cell Treatment
3.3.1. Treatment A
3.3.2. Treatment B
3.3.3. Treatment C
3.3.4. Treatment D
3.3.5. Treatment E
3.4. MTT (3-4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) Assay
3.5. Design of siRNA Targeting ERK1/2 Genes
3.6. Western Blotting
3.7. In Vitro Tube Formation Assay
3.8. Statistics
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Hirose, S.; Hosoda, Y.; Furuya, S.; Otsuki, T.; Ikeda, E. Expression of vascular endothelial growth factor and its receptors correlates closely with formation of the plexiform lesion in human pulmonary hypertension. Pathol. Int. 2000, 50, 472–479. [Google Scholar]
- Quarck, R.; Wynants, M.; Ronisz, A.; Sepulveda, M.R.; Wuytack, F.; van Raemdonck, D.; Meyns, B.; Delcroix, M. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir. Res. 2012, 13, 27:1–27:10. [Google Scholar]
- Fadini, G.P.; Schiavon, M.; Avogaro, A.; Agostini, C. The emerging role of endothelial progenitor cells in pulmonary hypertension and diffuse lung diseases. Sarcoidosis Vasc. Diffuse Lung Dis. 2007, 24, 85–93. [Google Scholar]
- Ghofrani, H.A.; Wiedemann, R.; Rose, F.; Olschewski, H.; Schermuly, R.T.; Weissmann, N.; Seeger, W.; Grimminger, F. Combination therapy with oral sildenafil and inhaled iloprost for severe pulmonary hypertension. Ann. Intern. Med. 2002, 136, 515–522. [Google Scholar] [CrossRef]
- Zhao, L.; Mason, N.A.; Strange, J.W.; Walker, H.; Wilkins, M.R. Beneficial effects of phosphodiesterase 5 inhibition in pulmonary hypertension are influenced by natriuretic peptide activity. Circulation 2003, 107, 234–237. [Google Scholar]
- Zeng, Z.; Li, Y.; Jiang, Z.; Wang, C.S.; Li, B.B.; Jiang, W. The extracellular signal-regulated kinase was involved in the effects of sildenafil on pulmonary vascular remodeling. Cardiovasc. Ther. 2010, 28, 23–29. [Google Scholar] [CrossRef]
- Li, B.B.; Yang, L.C.; Jiang, Z.; Shen, J.Y.; Wang, C.S. The antiproliferative effect of sildenafil on pulmonary artery smooth muscle cells is mediated via upregulation of mitogen-activated protein kinase phosphatase-1 and degradation of extracellular signal-regulated kinase 1/2 phosphorylation. Anesth. Analg. 2007, 105, 1034–1041. [Google Scholar]
- Jacob, A; Molkentin, J.D.; Smolenski, A.; Lohmann, S.M.; Begum, N. Insulin inhibits PDGF-directed VSMC migration via NO/cGMP increase of MKP-1 and its inactivation of MAPKs. Am. J. Physiol. Cell Physiol. 2002, 283, 704–713. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.-J.; Li, J.-C.; Sun, W.D.; Huang, G.Q.; Wang, X.L. Activation of PKCa and pulmonary vascular remodeling in broilers. Res. Vet. Sci. 2005, 79, 131–137. [Google Scholar] [CrossRef]
- Das, A.; Ockaili, R.; Salloum, F.; Kukreja, R.C. Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, 1455–1460. [Google Scholar]
- Hyvelin, J.M.; Howell, K.; Nichol, A.; Costello, C.M.; Preston, R.J.; McLoughlin, P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ. Res. 2005, 97, 185–191. [Google Scholar] [CrossRef]
- Howell, K.; Costello, C.M.; Sands, M.; Doolry, I.; McLoughlin, P. l-Arginine promotes angiogenesis in the chronically hypoxic lung: A novel mechanism ameliorating pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, 1042–1050. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Chesler, N.; Molthen, R.C. Point: Counterpoint: Chronic hypoxia-induced pulmonary hypertension does/does not lead to loss of pulmonary vasculature. J. Appl. Physiol. 2007, 103, 1449–1451. [Google Scholar] [CrossRef]
- Ichinose, F.; Erana-Garcia, J.; Hromi, J.; Raveh, Y.; Jones, R.; Krim, L.; Clark, M.W.; Winkler, J.D.; Bloch, K.D.; Zapol, W.M. Nebulized sildenafil is a selective pulmonary vasodilator in lambs with acute pulmonary hypertension. Crit. Care Med. 2001, 29, 1000–1005. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Z.Y.; Guan, Q. Oral sildenafil prevents and reverses the development of pulmonary hypertension in monocrotaline-treated rats. Interact. Cardiovasc. Thorac. Surg. 2007, 6, 608–613. [Google Scholar] [CrossRef]
- Caino, M.C.; Meshki, J.; Kazanietz, M.G. Hallmarks for senescence in carcinogenesis: Novel signaling players. Apoptosis 2009, 14, 392–408. [Google Scholar]
- Balasubramanian, S.; Johnston, R.K.; Moschella, P.C.; Mani, S.K.; Tuxworth, W.J., Jr.; Kuppuswamy, D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 52–63. [Google Scholar] [CrossRef]
- Puente, L.G.; He, J.S.; Ostergaard, H.L. A novel PKC regulates ERK activation and degranulation of cytotoxic T lymphocytes: Plasticity in PKC regulation of ERK. Eur. J. Immunol. 2006, 36, 1009–1018. [Google Scholar] [CrossRef]
- Robertson, L.; Mireau, L.; Ostergaard, H. A role for phosphatidylinositol 3-kinase in TCR-stimulated ERK activation leading to paxillin phosphorylation and CTL degranulation. J. Immunol. 2005, 175, 8138–8145. [Google Scholar] [CrossRef]
- Grammer, T.; Blenis, J. Evidence for MEK-independent pathways regulating the prolonged activation of the ERK-MAP kinases. Oncogene 1997, 14, 1635–1642. [Google Scholar]
- Michell, B.J.; Chen, Z.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar]
- Meoli, D.F.; White, R.J. Endothelin-1 induces pulmonary but not aortic smooth muscle cell migration by activating ERK1/2 MAP kinase. Can. J. Physiol. Pharamacol. 2010, 88, 830–839. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zeng, Z.; Li, Y.-C.; Jiao, Z.-H.; Yao, J.; Xue, Y. The Cross Talk between cGMP Signal Pathway and PKC in Pulmonary Endothelial Cell Angiogenesis. Int. J. Mol. Sci. 2014, 15, 10185-10198. https://doi.org/10.3390/ijms150610185
Zeng Z, Li Y-C, Jiao Z-H, Yao J, Xue Y. The Cross Talk between cGMP Signal Pathway and PKC in Pulmonary Endothelial Cell Angiogenesis. International Journal of Molecular Sciences. 2014; 15(6):10185-10198. https://doi.org/10.3390/ijms150610185
Chicago/Turabian StyleZeng, Zhen, Ying-Chuan Li, Zhi-Hua Jiao, Jun Yao, and Ying Xue. 2014. "The Cross Talk between cGMP Signal Pathway and PKC in Pulmonary Endothelial Cell Angiogenesis" International Journal of Molecular Sciences 15, no. 6: 10185-10198. https://doi.org/10.3390/ijms150610185
APA StyleZeng, Z., Li, Y.-C., Jiao, Z.-H., Yao, J., & Xue, Y. (2014). The Cross Talk between cGMP Signal Pathway and PKC in Pulmonary Endothelial Cell Angiogenesis. International Journal of Molecular Sciences, 15(6), 10185-10198. https://doi.org/10.3390/ijms150610185