The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology

 ,
, 
Abstract
:
1. Introduction
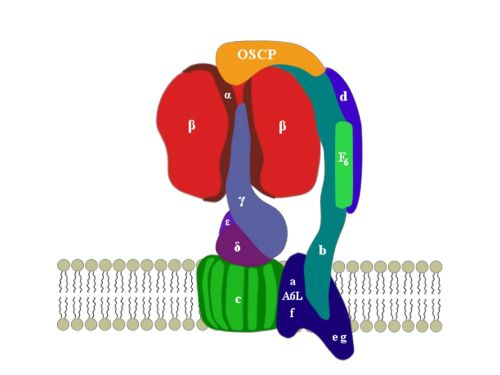
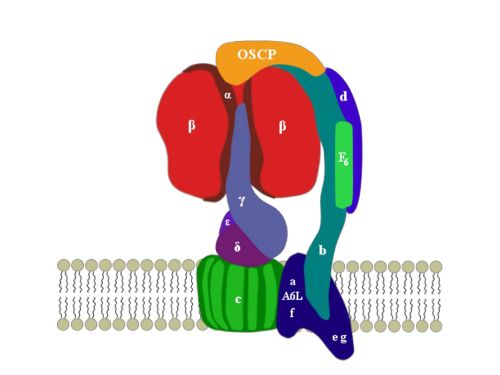
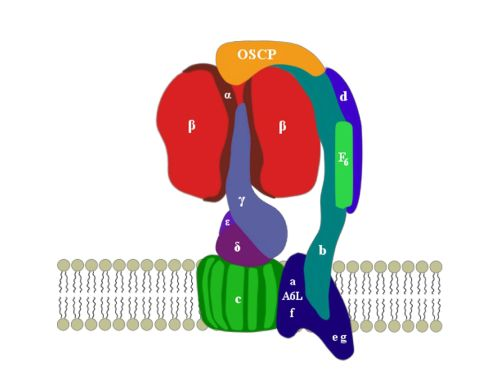
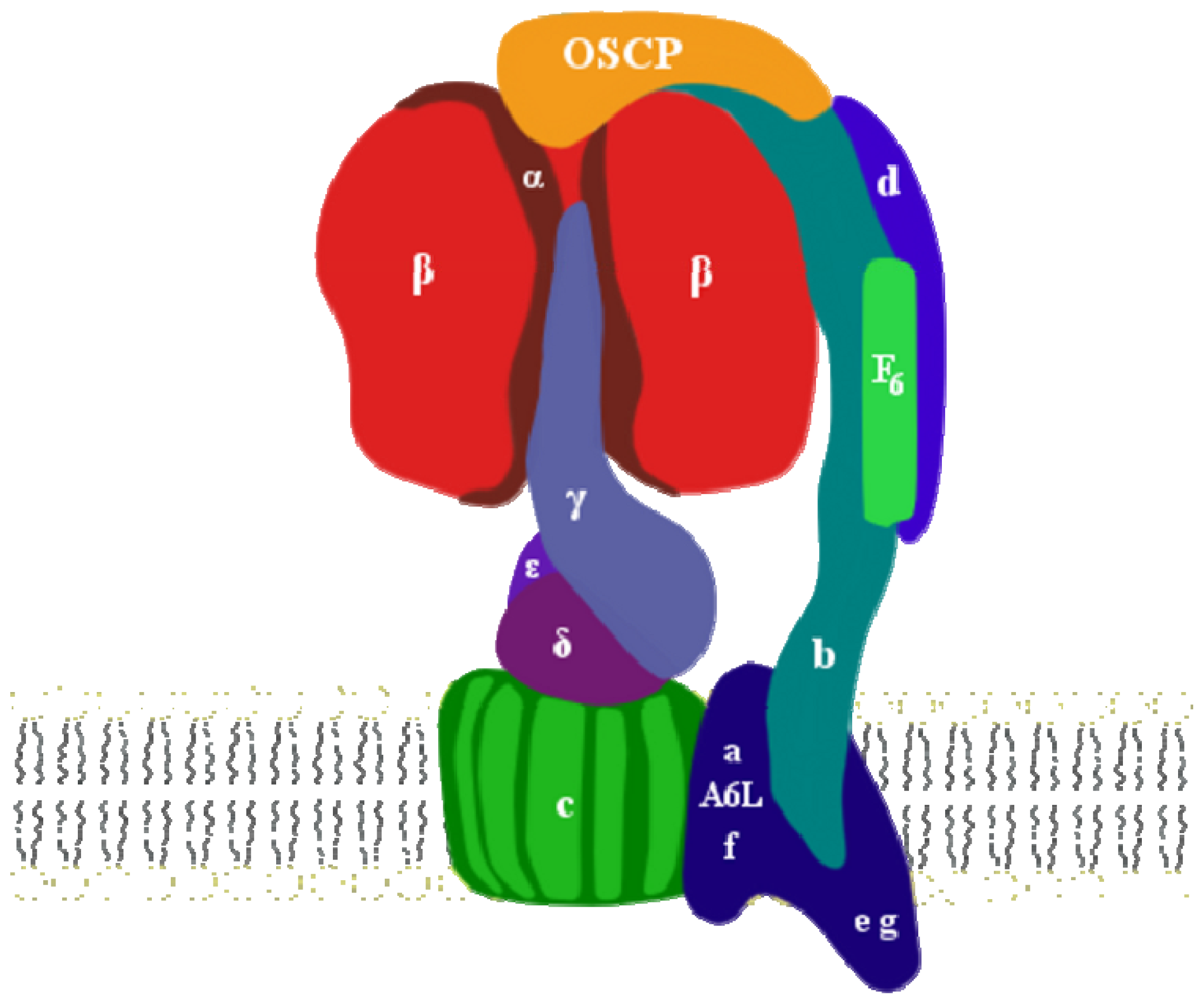
2. ATP Synthase Structure
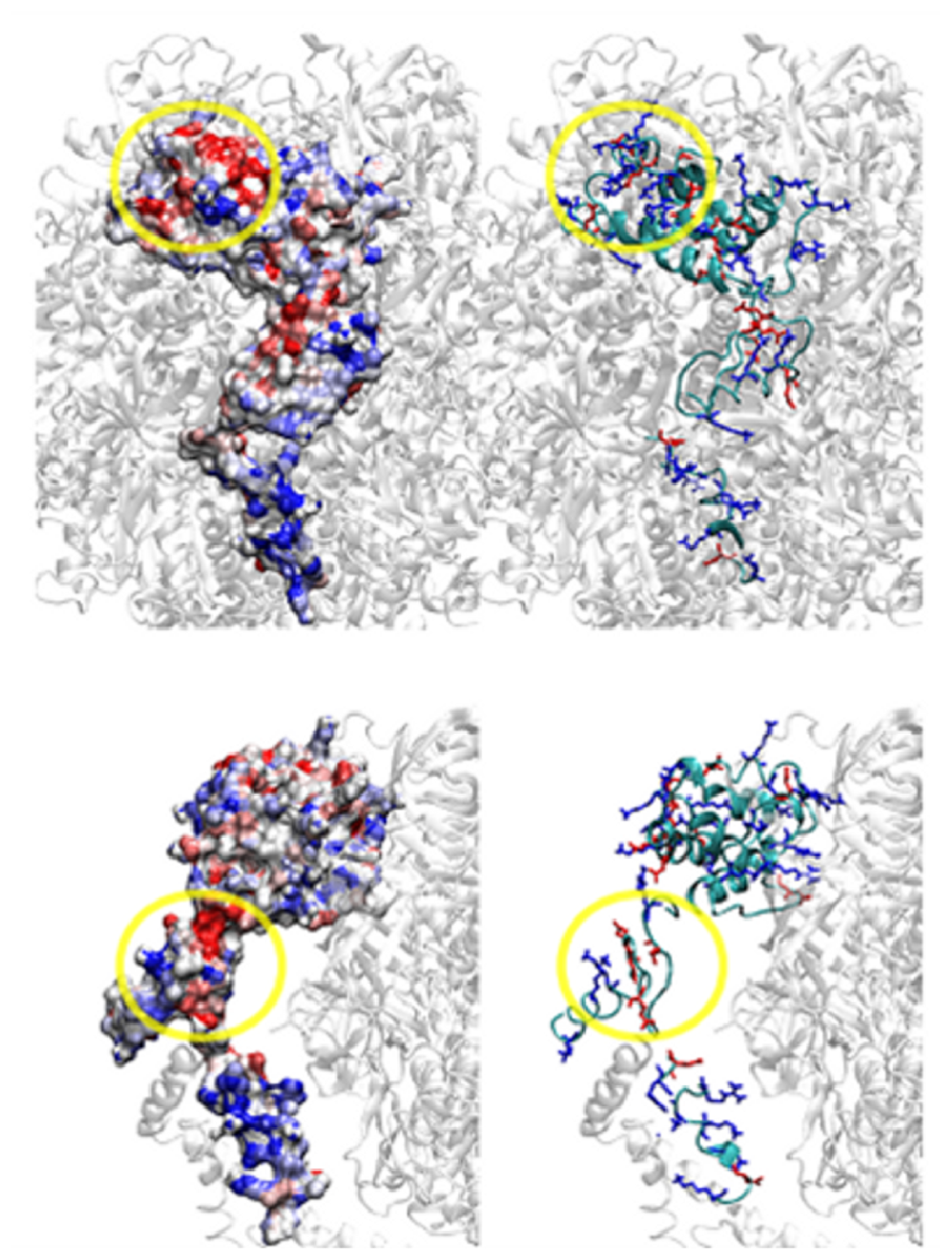
3. OSCP: Location and Structure
4. Post Translational Modifications of OSCP Subunit
5. OSCP Interactors: CyPD and PTP Formation
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig 2013, 43, 886–893. [Google Scholar]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol 2013, 4, 95. [Google Scholar]
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kühlbrandt, W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabò, I.; et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [Green Version]
- Rasola, A.; Sciacovelli, M.; Chiara, F.; Pantic, B.; Brusilow, W.S.; Bernardi, P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. USA 2010, 107, 726–731. [Google Scholar]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar]
- Burnham-Marusich, A.; Berninsone, P. Multiple proteins with essential mitochondrial functions have glycosylated isoforms. Mitochondrion 2012, 12, 423–427. [Google Scholar]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial F(o)F(1)ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977 bp deletion of mitochondrial DNA. Biochim. Biophys. Acta 2013, 1832, 216–227. [Google Scholar]
- Elstner, M.; Andreoli, C.; Ahting, U.; Tetko, I.; Klopstock, T.; Meitinger, T.; Prokisch, H. MitoP2: An integrative tool for the analysis of the mitochondrial proteome. Mol. Biotechnol 2008, 40, 306–315. [Google Scholar]
- Lotz, C.; Lin, A.J.; Black, C.M.; Zhang, J.; Lau, E.; Deng, N.; Wang, Y.; Zong, N.C.; Choi, J.H.; Xu, T.; et al. Characterization, Design, and Function of the Mitochondrial Proteome: From Organs to Organisms. J. Proteome Res 2013, 13, 433–446. [Google Scholar]
- Baker, M.J.; Frazier, A.E.; Gulbis, J.M.; Ryan, M.T. Mitochondrial protein-import machinery: Correlating structure with function. Trends Cell Biol 2007, 17, 456–464. [Google Scholar]
- Wallace, D.C.; Brown, M.D.; Lott, M.T. Mitochondrial DNA variation in human evolution and disease. Gene 1999, 238, 211–230. [Google Scholar]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis 2012, 35, 211–225. [Google Scholar]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet 2004, 13, 869–879. [Google Scholar]
- Mrácek, T.; Pecina, P.; Vojtísková, A.; Kalous, M.; Sebesta, O.; Houstek, J. Two components in pathogenic mechanism of mitochondrial ATPase deficiency: Energy deprivation and ROS production. Exp. Gerontol 2006, 41, 683–687. [Google Scholar]
- Mourier, A.; Ruzzenente, B.; Brandt, T.; Ku, W. Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet 2014, 23, 1–13. [Google Scholar]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar]
- Mitchell, P. Keilin’s respiratory chain concept and its chemiosmotic consequences. Science 1979, 206, 1148–1159. [Google Scholar]
- Boyer, P.D. The A.T.P. synthase A splendid molecular machine. Annu. Rev. Biochem 1997, 66, 717–749. [Google Scholar]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar]
- Vantourout, P.; Radojkovic, C.; Lichtenstein, L.; Pons, V.; Champagne, E.; Martinez, L.O. Ecto-F1-ATPase: A moonlighting protein complex and an unexpected apoA-I receptor. World J. Gastroenterol 2010, 16, 5925–5935. [Google Scholar]
- Rai, A.K.; Spolaore, B.; Harris, D.A.; Dabbeni-Sala, F.; Lippe, G. Ectopic FOF1 ATP synthase contains both nuclear and mitochondrially-encoded subunits. J. Bioenerg. Biomembr 2013, 45, 569–579. [Google Scholar]
- Futai, M.; Nakanishi-Matsui, M.; Okamoto, H.; Sekiya, M.; Nakamoto, R.K. Rotational catalysis in proton pumping ATPases: From E. coli F-ATPase to mammalian V-ATPase. Biochim. Biophys. Acta 2012, 1817, 1711–1721. [Google Scholar]
- Stock, D.; Gibbons, C.; Arechaga, I.; Leslie, A.G.; Walker, J.E. The rotary mechanism of ATP synthase. Curr. Opin. Struct. Biol 2000, 10, 672–679. [Google Scholar]
- Weber, J.; Senior, A.E. ATP synthesis driven by proton transport in F1FO-ATP synthase. FEBS Lett 2003, 545, 61–70. [Google Scholar]
- Martin, J.L.; Ishmukhametov, R.; Hornung, T.; Ahmad, Z.; Frasch, W.D. Anatomy of F1-ATPase powered rotation. Proc. Natl. Acad. Sci. USA 2014, 111, 3715–3720. [Google Scholar]
- Sielaff, H.; Börsch, M. Twisting and subunit rotation in single F(O)(F1)-ATP synthase. Philos. Trans. R. Soc. Lond. Ser. B, Biol. Sci 368, 0024.
- Nathanson, L.; Gromet-Elhanan, Z. Mutations in the beta-subunit Thr(159) and Glu(184) of the Rhodospirillum rubrum F(O)F(1) ATP synthase reveal differences in ligands for the coupled Mg(2+)- and decoupled Ca(2+)-dependent F(0)F(1) activities. J. Biol. Chem 2000, 275, 901–905. [Google Scholar]
- Papageorgiou, S.; Melandri, A.B.; Solaini, G. Relevance of divalent cations to ATP-driven proton pumping in beef heart mitochondrial FOF1-ATPase. J. Bioenerg. Biomembr 1998, 30, 533–541. [Google Scholar]
- Junge, W.; Sielaff, H.; Engelbrecht, S. Torque generation and elastic power transmission in the rotary F(O)F(1)-ATPase. Nature 2009, 459, 364–370. [Google Scholar]
- Bason, J.V.; Runswick, M.J.; Fearnley, I.M.; Walker, J.E. Binding of the inhibitor protein IF(1) to bovine F(1)-ATPase. J. Mol. Biol 2011, 406, 443–453. [Google Scholar]
- Campanella, M.; Parker, N.; Tan, C.; Hall, A.; Duchen, M. IF(1): Setting the pace of the F(1)F(O)-ATP synthase. Trends Biochem. Sci 2009, 34, 343–350. [Google Scholar]
- Formentini, L.; Pereira, M.P.; Sánchez-Cenizo, L.; Santacatterina, F.; Lucas, J.J.; Navarro, C.; Martínez-Serrano, A.; Cuezva, J.M. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J 2014, 33, 762–778. [Google Scholar]
- Di Pancrazio, F.; Mavelli, I.; Isola, M.; Losano, G.; Pagliaro, P.; Harris, D.A.; Lippe, G. In vitro and in vivo studies of F(O)F(1)ATP synthase regulation by inhibitor protein IF(1) in goat heart. Biochim. Biophys. Acta 2004, 1659, 52–62. [Google Scholar]
- Nakamura, J.; Fujikawa, M.; Yoshida, M. IF1, a natural inhibitor of mitochondrial ATP synthase, is not essential for the normal growth and breeding of mice. Biosci. Rep 2013, 33, e00067. [Google Scholar]
- Collinson, I.R.; Fearnley, I.M.; Skehel, J.M.; Runswick, M.J.; Walker, J.E. ATP synthase from bovine heart mitochondria: Identification by proteolysis of sites in FO exposed by removal of F1 and the oligomycin-sensitivity conferral protein. Biochem. J. 1994, 303, 639–645. [Google Scholar]
- Watt, I.N.; Montgomery, M.G.; Runswick, M.J.; Leslie, A.G.W.; Walker, J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 16823–16827. [Google Scholar]
- Baker, L.A.; Watt, I.N.; Runswick, M.J.; Walker, J.E.; Rubinstein, J.L. Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM. Proc. Natl. Acad. Sci. USA 2012, 109, 11675–11680. [Google Scholar]
- Devenish, R.J.; Prescott, M.; Boyle, G.M.; Nagley, P. The oligomycin axis of mitochondrial ATP synthase: OSCP and the proton channel. J. Bioenerg. Biomembr 2000, 32, 507–515. [Google Scholar]
- Chen, R.; Runswick, M.J.; Carroll, J.; Fearnley, I.M.; Walker, J.E. Association of two proteolipids of unknown function with ATP synthase from bovine heart mitochondria. FEBS Lett 2007, 581, 3145–3148. [Google Scholar]
- Meyer, B.; Wittig, I.; Trifilieff, E.; Karas, M.; Schägger, H. Identification of two proteins associated with mammalian ATP synthase. Mol. Cell. Proteomics 2007, 6, 1690–1699. [Google Scholar]
- Ohsakaya, S.; Fujikawa, M.; Hisabori, T.; Yoshida, M. Knockdown of DAPIT (diabetes-associated protein in insulin-sensitive tissue) results in loss of ATP synthase in mitochondria. J. Biol. Chem 2011, 286, 20292–20296. [Google Scholar]
- Carroll, J.; Fearnley, I.M.; Wang, Q.; Walker, J.E. Measurement of the molecular masses of hydrophilic and hydrophobic subunits of ATP synthase and complex I in a single experiment. Anal. Biochem 2009, 395, 249–255. [Google Scholar]
- Habersetzer, J.; Larrieu, I.; Priault, M.; Salin, B.; Rossignol, R.; Brèthes, D.; Paumard, P. Human F1FO ATP synthase, mitochondrial ultrastructure and OXPHOS impairment: A (super-) complex Matter? PLoS One 2013, 8, e75429. [Google Scholar]
- Habersetzer, J.; Ziani, W.; Larrieu, I.; Stines-Chaumeil, C.; Giraud, M.-F.; Brèthes, D.; Dautant, A.; Paumard, P. ATP synthase oligomerization: From the enzyme models to the mitochondrial morphology. Int. J. Biochem. Cell Biol 2013, 45, 99–105. [Google Scholar]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J 2002, 21, 221–230. [Google Scholar]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gomez, J.D.; Kuhlbrandt, W. Structure of the yeast F1FO-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar]
- Thomas, D.; Bron, P.; Weimann, T.; Dautant, A.; Giraud, M.-F.; Paumard, P.; Salin, B.; Cavalier, A.; Velours, J.; Brèthes, D. Supramolecular organization of the yeast F1FO-ATP synthase. Biol. Cell 2008, 100, 591–601. [Google Scholar]
- Bisetto, E.; di Pancrazio, F.; Simula, M.P.; Mavelli, I.; Lippe, G. Mammalian ATPsynthase monomer versus dimer profiled by blue native PAGE and activity stain. Electrophoresis 2007, 28, 3178–3185. [Google Scholar]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kühlbrandt, W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar]
- Wittig, I.; Velours, J.; Stuart, R.; Schägger, H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Mol. Cell. Proteomics 2008, 7, 995–1004. [Google Scholar]
- Spannagel, C.; Vaillier, J.; Arselin, G.; Graves, P.V.; Grandier-Vazeille, X.; Velours, J. Evidence of a subunit 4 (subunit b) dimer in favor of the proximity of ATP synthase complexes in yeast inner mitochondrial membrane. Biochim. Biophys. Acta 1998, 1414, 260–264. [Google Scholar]
- Everard-gigot, V.; Dunn, C.D.; Dolan, B.M.; Brunner, S.; Jensen, R.E.; Stuart, R.A. Functional Analysis of Subunit e of the F1FO-ATP Synthase of the Yeast Saccharomyces cerevisiae: Importance of the N-Terminal Membrane Anchor Region. Eucaryot Cell 2005, 4, 346–55. [Google Scholar]
- Bustos, D.M.; Velours, J. The modification of the conserved GXXXG motif of the membrane-spanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. J. Biol. Chem 2005, 280, 29004–29010. [Google Scholar]
- Fronzes, R.; Weimann, T.; Vaillier, J.; Velours, J.; Brèthes, D. The peripheral stalk participates in the yeast ATP synthase dimerization independently of e and g subunits. Biochemistry 2006, 45, 6715–6723. [Google Scholar]
- Wittig, I.; Meyer, B.; Heide, H.; Steger, M.; Bleier, L.; Wumaier, Z.; Karas, M.; Schägger, H. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta 2010, 1797, 1004–1011. [Google Scholar]
- Bisetto, E.; Picotti, P.; Giorgio, V.; Alverdi, V.; Mavelli, I.; Lippe, G. Functional and stoichiometric analysis of subunit e in bovine heart mitochondrial F(O)F(1)ATP synthase. J. Bioenerg. Biomembr 2008, 40, 257–267. [Google Scholar]
- Couoh-Cardel, S.J.; Uribe-Carvajal, S.; Wilkens, S.; García-Trejo, J.J. Structure of dimeric F1FO-ATP synthase. J. Biol. Chem 2010, 285, 36447–36455. [Google Scholar]
- Minauro-Sanmiguel, F.; Wilkens, S.; García, J.J. Structure of dimeric mitochondrial ATP synthase: Novel FO bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 12356–12358. [Google Scholar]
- Dudkina, N.V.; Sunderhaus, S.; Braun, H.-P.; Boekema, E.J. Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett 2006, 580, 3427–3432. [Google Scholar]
- Dienhart, M.; Pfeiffer, K.; Schagger, H.; Stuart, R.A. Formation of the yeast F1FO-ATP synthase dimeric complex does not require the ATPase inhibitor protein, Inh1. J. Biol. Chem 2002, 277, 39289–39295. [Google Scholar]
- Tomasetig, L.; di Pancrazio, F.; Harris, D.A.; Mavelli, I.; Lippe, G. Dimerization of FOF1ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim. Biophys. Acta 2002, 1556, 133–141. [Google Scholar]
- Campanella, M.; Seraphim, A.; Abeti, R.; Casswell, E.; Echave, P.; Duchen, M.R. IF1, the endogenous regulator of the F(1)F(O)-ATPsynthase, defines mitochondrial volume fraction in HeLa cells by regulating autophagy. Biochim. Biophys. Acta 2009, 1787, 393–401. [Google Scholar]
- Bisetto, E.; Comelli, M.; Salzano, A.M.; Picotti, P.; Scaloni, A.; Lippe, G.; Mavelli, I. Proteomic analysis of F1FO-ATP synthase super-assembly in mitochondria of cardiomyoblasts undergoing differentiation to the cardiac lineage. Biochim. Biophys. Acta 2013, 1827, 807–816. [Google Scholar]
- Zanotti, F.; Raho, G.; Gaballo, A.; Papa, S. Inhibitory and anchoring domains in the ATPase inhibitor protein IF1 of bovine heart mitochondrial ATP synthase. J. Bioenerg. Biomembr 2004, 36, 447–457. [Google Scholar]
- Lee, J.K.; Belogrudov, G.I.; Stroud, R.M. Crystal structure of bovine mitochondrial factor B at 0.96-A resolution. Proc. Natl. Acad. Sci. USA 2008, 105, 13379–13384. [Google Scholar]
- Slater, E.C. An evaluation of the Mitchell hypothesis of chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Eur. J. Biochem. FEBS 1967, 1, 317–326. [Google Scholar]
- Tzagoloff, A. Assembly of the Mitochondrial Membrane System. J. Biol. Chem 1970, 245, 1545–1551. [Google Scholar]
- Dupuis, A.; Issartel, J.P.; Lunardi, J.; Satre, M.; Vignais, P.V. Interactions between the oligomycin sensitivity conferring protein (OSCP) and beef heart mitochondrial F1-ATPase. 1. Study of the binding parameters with a chemically radiolabeled OSCP. Biochemistry 1985, 24, 728–733. [Google Scholar]
- Mukhopadhyay, A.; Zhou, X.Q.; Uh, M.; Mueller, D.M. Heterologous expression, purification, and biochemistry of the oligomycin sensitivity conferring protein (OSCP) from yeast. J. Biol. Chem 1992, 267, 25690–25696. [Google Scholar]
- Joshi, S.; Cao, G.J.; Nath, C.; Shah, J. Oligomycin sensitivity conferring protein (OSCP) of bovine heart mitochondrial ATP synthase: High-affinity OSCP-FO interactions require a local α-helix at the C-terminal end of the subunit. Biochemistry 1997, 36, 10936–10943. [Google Scholar]
- Golden, T.R.; Pedersen, P.L. The oligomycin sensitivity conferring protein of rat liver mitochondrial ATP synthase: Arginine 94 is important for the binding of OSCP to F1. Biochemistry 1998, 37, 13871–13881. [Google Scholar]
- Prescott, M.; Bush, N.C.; Nagley, P.; Devenish, R.J. Properties of yeast cells depleted of the OSCP subunit of mitochondrial ATP synthase by regulated expression of the ATP5 gene. Biochem. Mol. Biol. Int 1994, 34, 789–799. [Google Scholar]
- Devenish, R.; Prescott, M.; Roucou, X.; Nagley, P. Insights into ATP synthase assembly and function through the molecular genetic manipulation of subunits of the yeast mitochondrial enzyme complex. Biochim. Biophys. Acta 2000, 1458, 428–442. [Google Scholar]
- Boyle, G.M.; Roucou, X.; Nagley, P.; Devenish, R.J.; Prescott, M. Modulation at a distance of proton conductance through the Saccharomyces cerevisiae mitochondrial F1FO-ATP synthase by variants of the oligomycin sensitivity-conferring protein containing substitutions near the C-terminus. J. Bioenerg. Biomembr 2000, 32, 595–607. [Google Scholar]
- Moreno, A.J.M.; Moreira, P.I.; Custódio, J.B.A.; Santos, M.S. Mechanism of inhibition of mitochondrial ATP synthase by 17β-estradiol. J. Bioenerg. Biomembr 2013, 45, 261–270. [Google Scholar]
- Gavin, P.D.; Devenish, R.J.; Prescott, M. FRET reveals changes in the F1–stator stalk interaction during activity of F1F0-ATP synthase. Biochim. Biophys. Acta 2003, 1607, 167–179. [Google Scholar]
- Johnson, K.M.; Chen, X.; Boitano, A.; Swenson, L.; Opipari, A.W.; Glick, G.D. Identification and validation of the mitochondrial F1FO-ATPase as the molecular target of the immunomodulatory benzodiazepine Bz-423. Chem. Biol 2005, 12, 485–496. [Google Scholar]
- Rak, M.; Gokova, S.; Tzagoloff, A. Modular assembly of yeast mitochondrial ATP synthase. EMBO J 2011, 30, 920–930. [Google Scholar]
- Rees, D.M.; Leslie, A.G.W.; Walker, J.E. The structure of the membrane extrinsic region of bovine ATP synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 21597–21601. [Google Scholar]
- Carbajo, R.J.; Kellas, F.A.; Yang, J.C.; Runswick, M.J.; Montgomery, M.G.; Walker, J.E.; Neuhaus, D. How the N-terminal domain of the OSCP subunit of bovine F1FO-ATP synthase interacts with the N-terminal region of an α subunit. J. Mol. Biol 2007, 368, 310–318. [Google Scholar]
- Stelzer, A.C.; Frazee, R.W.; van Huis, C.; Cleary, J.; Opipari, A.W.; Glick, G.D.; Al-Hashimi, H.M. NMR studies of an immunomodulatory benzodiazepine binding to its molecular target on the mitochondrial F(1)F(O)-ATPase. Biopolymers 2010, 93, 85–92. [Google Scholar]
- Wilkens, S.; Borchardt, D.; Weber, J.; Senior, A.E. Structural characterization of the interaction of the delta and alpha subunits of the Escherichia coli F1FO-ATP synthase by NMR spectroscopy. Biochemistry 2005, 44, 11786–11794. [Google Scholar]
- Zhang, F.X.; Pan, W.; Hutchins, J. Phosphorylation of F1FO ATPase delta-subunit is regulated by platelet-derived growth factor in mouse cortical neurons in vitro. J. Neurochem 1995, 65, 2812–2815. [Google Scholar]
- Ko, Y.H.; Pan, W.; Inoue, C.; Pedersen, P.L. Signal transduction to mitochondrial ATP synthase: Evidence that PDGF-dependent phosphorylation of the delta-subunit occurs in several cell lines, involves tyrosine, and is modulated by lysophosphatidic acid. Mitochondrion 2002, 1, 339–348. [Google Scholar]
- Højlund, K.; Wrzesinski, K.; Larsen, P.M.; Fey, S.J.; Roepstorff, P.; Handberg, A.; Dela, F.; Vinten, J.; McCormack, J.G.; Reynet, C.; et al. Proteome analysis reveals phosphorylation of ATP synthase β-subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J. Biol. Chem 2003, 278, 10436–10442. [Google Scholar]
- Aponte, A.M.; Phillips, D.; Harris, R.A.; Blinova, K.; French, S.; Johnson, D.T.; Balaban, R.S. 32P labeling of protein phosphorylation and metabolite association in the mitochondria matrix. Methods Enzymol 2009, 457, 63–80. [Google Scholar]
- Zhao, X.; León, I.R.; Bak, S.; Mogensen, M.; Wrzesinski, K.; Højlund, K.; Jensen, O.N. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol. Cell. Proteomics 2011, 10, 1–14. [Google Scholar]
- Covian, R.; Balaban, R.S. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol 2012, 303, 940–966. [Google Scholar]
- Phillips, D.; Aponte, A.M.; Covian, R.; Balaban, R.S. Intrinsic protein kinase activity in mitochondrial oxidative phosphorylation complexes. Biochemistry 2011, 50, 2515–2529. [Google Scholar]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar]
- Margineantu, D.H.; Emerson, C.B.; Diaz, D.; Hockenbery, D.M. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One 2007, 2, e1066. [Google Scholar]
- Chandra, N.C. Identification of a Glycoprotein from rat liver mitochondrial inner membrane and demonstration of its origin in the endoplasmic reticulum. J. Biol. Chem 1998, 273, 19715–19721. [Google Scholar]
- Levrat, C.; Louisot, P.; Morelis, R. Distribution of glycosyltransferase activities in different compartments of mitochondria. Biochem. Int 1989, 18, 813–823. [Google Scholar]
- Clark, P.M.; Dweck, J.F.; Mason, D.E.; Hart, C.R.; Buck, S.B.; Peters, E.C.; Agnew, B.J.; Hsieh-Wilson, L.C. Direct in-gel fluorescence detection and cellular imaging of O-GlcNAc-modified proteins. J. Am. Chem. Soc 2008, 130, 11576–11577. [Google Scholar]
- Hu, Y.; Suarez, J.; Fricovsky, E.; Wang, H.; Scott, B.T.; Trauger, S.A.; Han, W.; Hu, Y.; Oyeleye, M.O.; Dillmann, W.H. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J. Biol. Chem 2009, 284, 547–555. [Google Scholar]
- Kung, L.A.; Tao, S.C.; Qian, J.; Smith, M.G.; Snyder, M.; Zhu, H. Global analysis of the glycoproteome in Saccharomyces cerevisiae reveals new roles for protein glycosylation in eukaryotes. Mol. Syst. Biol 2009, 5, 308. [Google Scholar]
- Teo, C.F.; Wollaston-Hayden, E.E.; Wells, L. Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes. Mol. Cell. Endocrinol 2010, 318, 44–53. [Google Scholar]
- Burnham-Marusich, A.R.; Snodgrass, C.J.; Johnson, A.M.; Kiyoshi, C.M.; Buzby, S.E.; Gruner, M.R.; Berninsone, P.M. Metabolic labeling of Caenorhabditis elegans primary embryonic cells with azido-sugars as a tool for glycoprotein discovery. PLoS One 2012, 7, e49020. [Google Scholar]
- Steentoft, C.; Vakhrushev, S.Y.; Joshi, H.J.; Kong, Y.; Vester-Christensen, M.B.; Schjoldager, K.T.B.G.; Lavrsen, K.; Dabelsteen, S.; Pedersen, N.B.; Marcos-Silva, L.; et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 2013, 32, 1478–1488. [Google Scholar]
- Boja, E.S.; Phillips, D.; French, S.A.; Harris, R.A.; Robert, S. Quantitative mitochondrial phosphoproteomics using iTRAQ on an LTQ-Orbitrap with high energy collision dissociation. J. Proteome Res 2009, 8, 4665–4675. [Google Scholar]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar]
- Jeon, H.B.; Choi, E.S.; Yoon, J.H.; Hwang, J.H.; Chang, J.W.; Lee, E.K.; Choi, H.W.; Park, Z.Y.; Yoo, Y.J. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun 2007, 357, 731–736. [Google Scholar]
- Nguyen, T.T.; Ogbi, M.; Yu, Q.; Fishman, J.B.; Thomas, W.; Harvey, B.J.; Fulton, D.; Johnson, J.A. Modulation of the protein kinase Cdelta interaction with the “d” subunit of F1F0-ATP synthase in neonatal cardiac myocytes: Development of cell-permeable, mitochondrially targeted inhibitor and facilitator peptides. J. Biol. Chem 2010, 285, 22164–22173. [Google Scholar]
- Boerries, M.; Most, P.; Gledhill, J.R.; Walker, J.E.; Katus, H.A.; Koch, W.J.; Aebi, U.; Schoenenberger, C.-A. Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol. Cell. Biol 2007, 27, 4365–4373. [Google Scholar]
- Alavian, K.N.; Li, H.; Collis, L.; Bonanni, L.; Zeng, L.; Sacchetti, S.; Lazrove, E.; Nabili, P.; Flaherty, B.; Graham, M.A.; et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat. Cell Biol 2011, 13, 1224–1233. [Google Scholar]
- Belogrudov, G.I. Factor B is essential for ATP synthesis by mitochondria. Arch. Biochem. Biophys 2002, 406, 271–274. [Google Scholar]
- Bergeaud, M.; Mathieu, L.; Guillaume, A.; Moll, U.M.; Mignotte, B.; Le Floch, N.; Vayssière, J.L.; Rincheval, V. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F1FO-ATP synthase. Cell Cycle 2013, 12, 2781–2793. [Google Scholar]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar]
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol 2007, 27, 8807–8814. [Google Scholar]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet 2012, 28, 128–136. [Google Scholar]
- Borel, J.F.; Feurer, C.; Magnée, C.; Stähelin, H. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology 1977, 32, 1017–1025. [Google Scholar]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol 2005, 6, 226. [Google Scholar]
- Dolinski, K.; Muir, S.; Cardenas, M.; Heitman, J. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 13093–13098. [Google Scholar]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev 1999, 79, 1127–1155. [Google Scholar]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar]
- Barsukova, A.; Komarov, A.; Hajnóczky, G.; Bernardi, P.; Bourdette, D.; Forte, M. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci 2011, 33, 831–842. [Google Scholar]
- Fournier, N.; Ducet, G.; Crevat, A. Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr 1987, 19, 297–303. [Google Scholar]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem 2005, 280, 18558–18561. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar]
- Kajitani, K.; Fujihashi, M.; Kobayashi, Y.; Shimizu, S.; Tsujimoto, Y.; Miki, K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-A resolution. Proteins 2008, 70, 1635–1639. [Google Scholar]
- Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P. Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem 2008, 283, 26307–26311. [Google Scholar]
- Shulga, N.; Wilson-Smith, R.; Pastorino, J.G. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J. Cell Sci 2010, 123, 894–902. [Google Scholar]
- Kohr, M.J.; Aponte, A.M.; Sun, J.; Wang, G.; Murphy, E.; Gucek, M.; Steenbergen, C. Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart Circ. Physiol 2011, 300, 1327–1335. [Google Scholar]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem 2009, 284, 9692–9699. [Google Scholar]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell 2012, 149, 1536–1548. [Google Scholar]
- Zharova, T.V.; Vinogradov, A.D. Energy-linked binding of Pi is required for continuous steady-state proton-translocating ATP hydrolysis catalyzed by FOF1 ATP synthase. Biochemistry 2006, 45, 14552–14558. [Google Scholar]
- Nicolli, A.; Basso, E.; Petronilli, V.; Wenger, R.M.; Bernardi, P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin A-sensitive channel. J. Biol. Chem 1996, 271, 2185–2192. [Google Scholar]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem 2009, 284, 33982–33988. [Google Scholar]
- Szabó, I.; Zoratti, M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem 1991, 266, 3376–3379. [Google Scholar]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Blalchy-Dyson, E.; di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J 2006, 273, 2077–2099. [Google Scholar]
- Nicolli, A.; Petronilli, V.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation. Biochemistry 1993, 32, 4461–4465. [Google Scholar]
- Petronilli, V.; Sileikyte, J.; Zulian, A.; Dabbeni-Sala, F.; Jori, G.; Gobbo, S.; Tognon, G.; Nikolov, P.; Bernardi, P.; Ricchelli, F. Switch from inhibition to activation of the mitochondrial permeability transition during hematoporphyrin-mediated photooxidative stress. Unmasking pore-regulating external thiols. Biochim. Biophys. Acta 2009, 1787, 897–904. [Google Scholar]
- Zhang, Y.; Marcillat, O.; Giulivi, C.; Ernster, L.; Davies, K.J. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J. Biol. Chem 1990, 265, 16330–16336. [Google Scholar]
- Lippe, G.; Comelli, M.; Mazzilis, D.; Sala, F.D.; Mavelli, I. The inactivation of mitochondrial F1 ATPase by H2O2 is mediated by iron ions not tightly bound in the protein. Biochem. Biophys. Res. Commun 1991, 181, 764–770. [Google Scholar]
- Comelli, M.; Lippe, G.; Mavelli, I. Differentiation potentiates oxidant injury to mitochondria by hydrogen peroxide in Friend’s erythroleukemia cells. FEBS Lett 1994, 352, 71–75. [Google Scholar]
- Poon, H.F.; Calabrese, V.; Calvani, M.; Butterfield, A. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by l-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stres. Antioxid. Redox Signal 2006, 8, 381–394. [Google Scholar]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis 2006, 22, 76–87. [Google Scholar]
- Poon, H.F.; Shepherd, H.M.; Reed, T.T.; Calabrese, V.; Stella, A.-M.G.; Pennisi, G.; Cai, J.; Pierce, W.M.; Klein, J.B.; Butterfield, D.A. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol. Aging 2006, 27, 1020–1034. [Google Scholar]
- Groebe, K.; Krause, F.; Kunstmann, B.; Unterluggauer, H.; Reifschneider, N.H.; Scheckhuber, C.Q.; Sastri, C.; Stegmann, W.; Wozny, W.; Schwall, G.P.; et al. Differential proteomic profiling of mitochondria from Podospora anserina, rat and human reveals distinct patterns of age-related oxidative changes. Exp. Gerontol 2007, 42, 887–898. [Google Scholar]
- Haynes, V.; Traaseth, N. Nitration of specific tyrosines in F0F1 ATP synthase and activity loss in aging. Am. J. Physiol. Endocrinol. Metab 2010, 95616, 978–987. [Google Scholar]
- Buchert, F.; Schober, Y.; Römpp, A.; Richter, M.L.; Forreiter, C. Reactive oxygen species affect ATP hydrolysis by targeting a highly conserved amino acid cluster in the thylakoid ATP synthase γ subunit. Biochim. Biophys. Acta 2012, 1817, 2038–2048. [Google Scholar]
- Taylor, S.W.; Fahy, E.; Murray, J.; Capaldi, R.A.; Ghosh, S.S. Oxidative post-translational modification of tryptophan residues in cardiac mitochondrial proteins. J. Biol. Chem 2003, 278, 19587–19590. [Google Scholar]
- Rexroth, S.; Poetsch, A.; Rögner, M.; Hamann, A.; Werner, A.; Osiewacz, H.D.; Schäfer, E.R.; Seelert, H.; Dencher, N.A. Reactive oxygen species target specific tryptophan site in the mitochondrial ATP synthase. Biochim. Biophys. Acta 2012, 1817, 381–387. [Google Scholar]
- Fogolari, F.; Corazza, A.; Yarra, V.; Jalaru, A.; Viglino, P.; Esposito, G. Bluues: A program for the analysis of the electrostatic properties of proteins based on generalized Born radii. BMC Bioinform 2012, 13, S18. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondria | E. coli, P. modestum and A. woodii | Chloroplast, cyanobacteria 2 and rhodobacteria 3 | |
|---|---|---|---|
| Bovine | Yeast | ||
| α | α | α | α |
| β | β | β | β |
| γ | γ | γ | γ |
| δ | δ | ɛ | ɛ |
| ɛ | ɛ | - | - |
| OSCP | OSCP | δ | δ |
| b | 4 or b | B 4 | b and b′ (I and II) |
| A6L | 8 | - | - |
| F6 | h | - | - |
| a | 6 or a | a | a (IV) |
| c | 9 or c | c | c (III) |
| d | d | - | - |
| e | e | - | - |
| f | f | - | - |
| g | g | - | - |
| - | i/j | - | - |
| - | k | - | - |
| MLQ | - | - | - |
| AGP/DAPIT | - | - | - |
| Modification | Method | Residue | Organism/Tissue | Reference |
|---|---|---|---|---|
| Acetylation | MS 1 | K60, K70, K159, K162, K172, K176, K192 | Mouse/Liver | [91] |
| K Ab | Human 143B osteosarcoma cells | [8] | ||
| Phosphorylation | ProQ | Pig/Heart | [6] | |
| PhosTag | Pig/Heart | [87] | ||
| 32P | Pig/Heart | [90] | ||
| MS | T145 | Pig/Heart | [89] | |
| MS | S155 | Human/Muscle | [88] | |
| Glycosylation | Leptin resin | Bovine/Heart | [7] | |
| Ubiquitination | Ub Ab | Human colon cancer cells | [92] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antoniel, M.; Giorgio, V.; Fogolari, F.; Glick, G.D.; Bernardi, P.; Lippe, G. The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology. Int. J. Mol. Sci. 2014, 15, 7513-7536. https://doi.org/10.3390/ijms15057513
Antoniel M, Giorgio V, Fogolari F, Glick GD, Bernardi P, Lippe G. The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology. International Journal of Molecular Sciences. 2014; 15(5):7513-7536. https://doi.org/10.3390/ijms15057513
Chicago/Turabian StyleAntoniel, Manuela, Valentina Giorgio, Federico Fogolari, Gary D. Glick, Paolo Bernardi, and Giovanna Lippe. 2014. "The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology" International Journal of Molecular Sciences 15, no. 5: 7513-7536. https://doi.org/10.3390/ijms15057513
APA StyleAntoniel, M., Giorgio, V., Fogolari, F., Glick, G. D., Bernardi, P., & Lippe, G. (2014). The Oligomycin-Sensitivity Conferring Protein of Mitochondrial ATP Synthase: Emerging New Roles in Mitochondrial Pathophysiology. International Journal of Molecular Sciences, 15(5), 7513-7536. https://doi.org/10.3390/ijms15057513