Biomarkers of Treatment Toxicity in Combined-Modality Cancer Therapies with Radiation and Systemic Drugs: Study Design, Multiplex Methods, Molecular Networks

Abstract
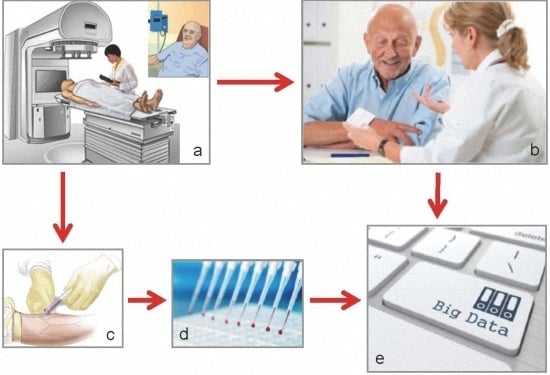
:
1. Introduction
1.1. Combined-Modality Radiotherapy
1.2. Treatment Toxicity
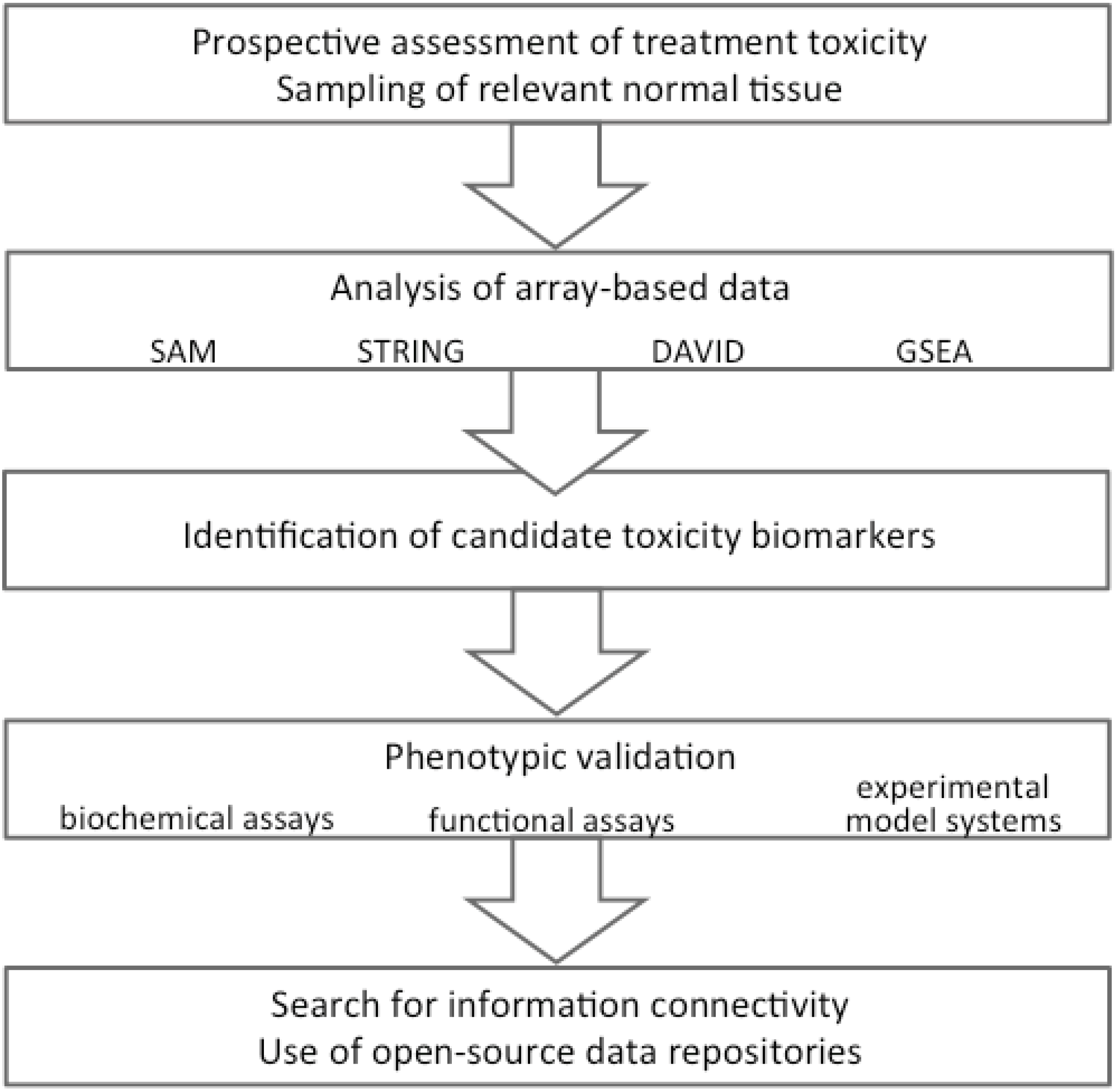
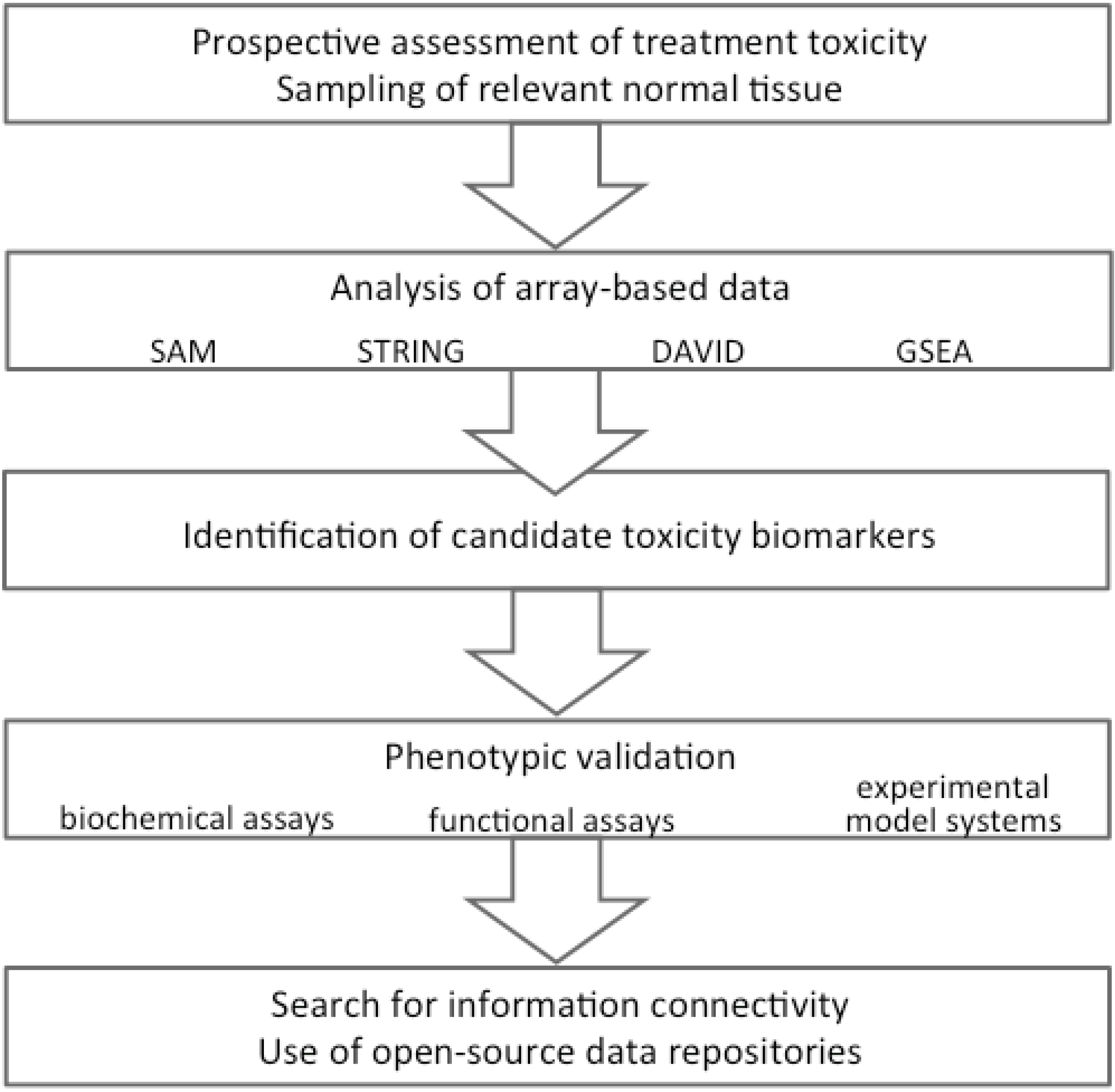
1.3. Exploring Toxicity Mechanisms—A “Top-Down” Strategy
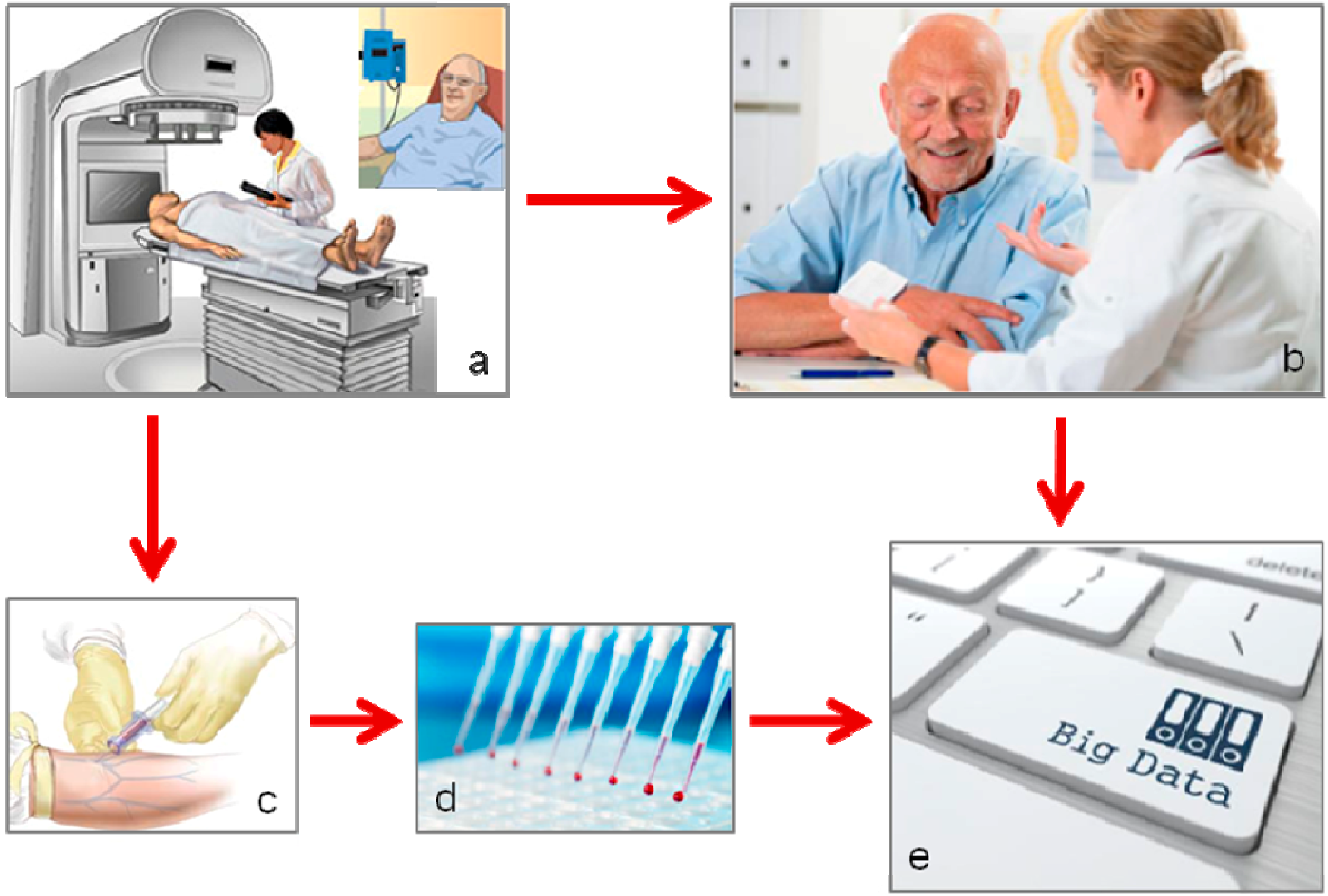
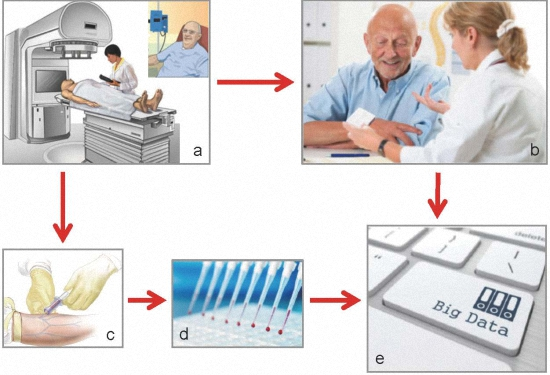
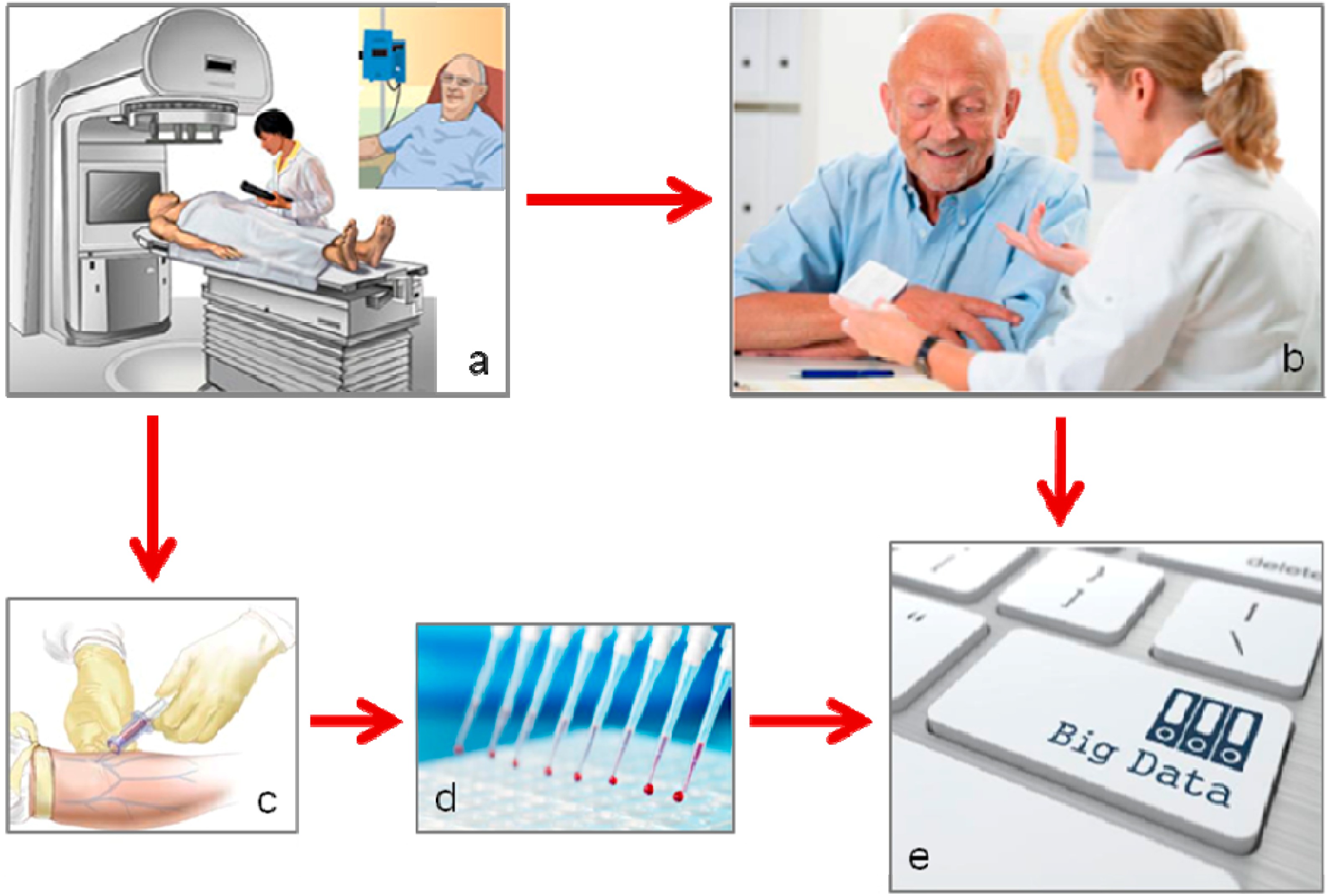
2. Biomarkers of Treatment Toxicity—Study Conduct
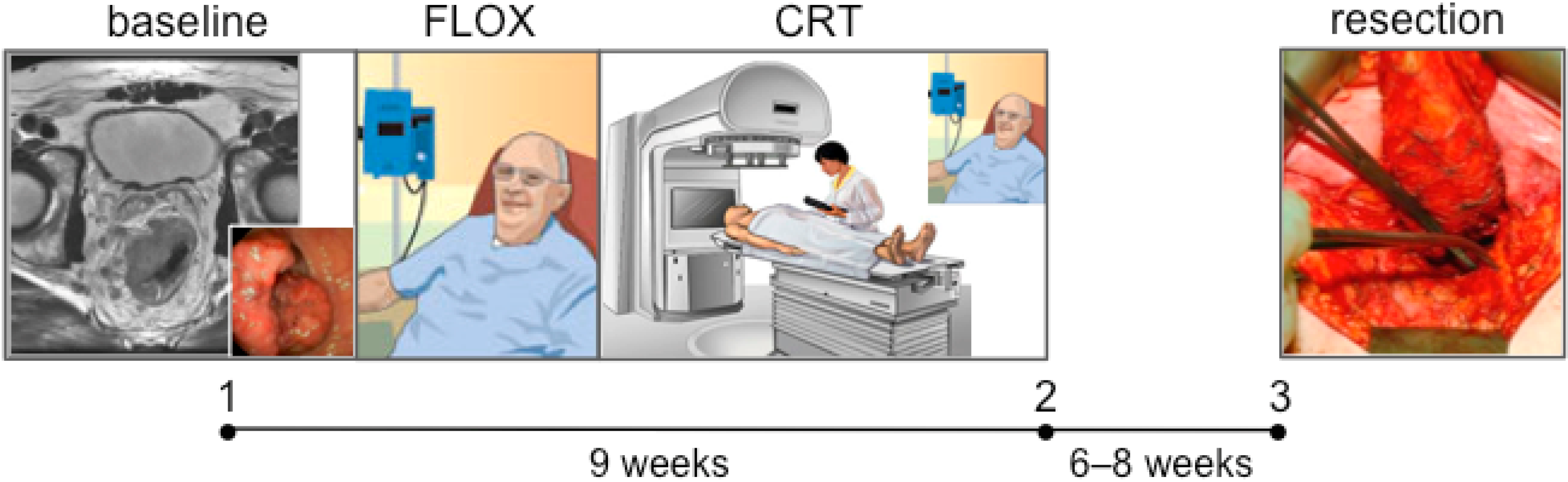
2.1. The Locally Advanced Rectal Cancer—Radiation Response Prediction (LARC-RRP) Study
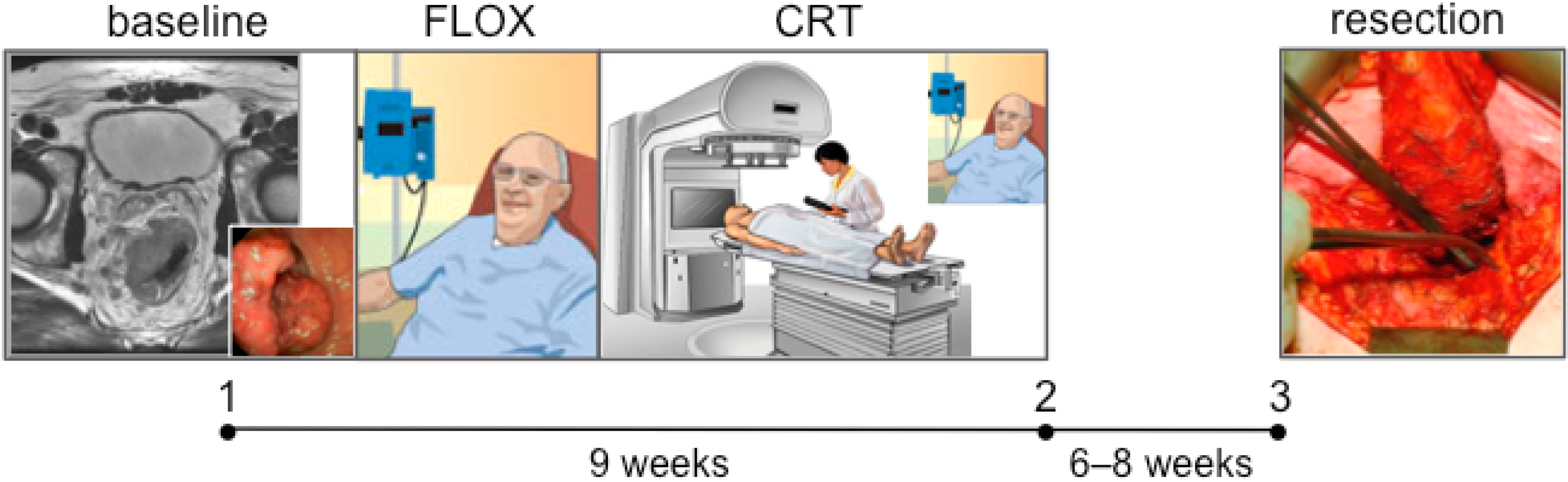
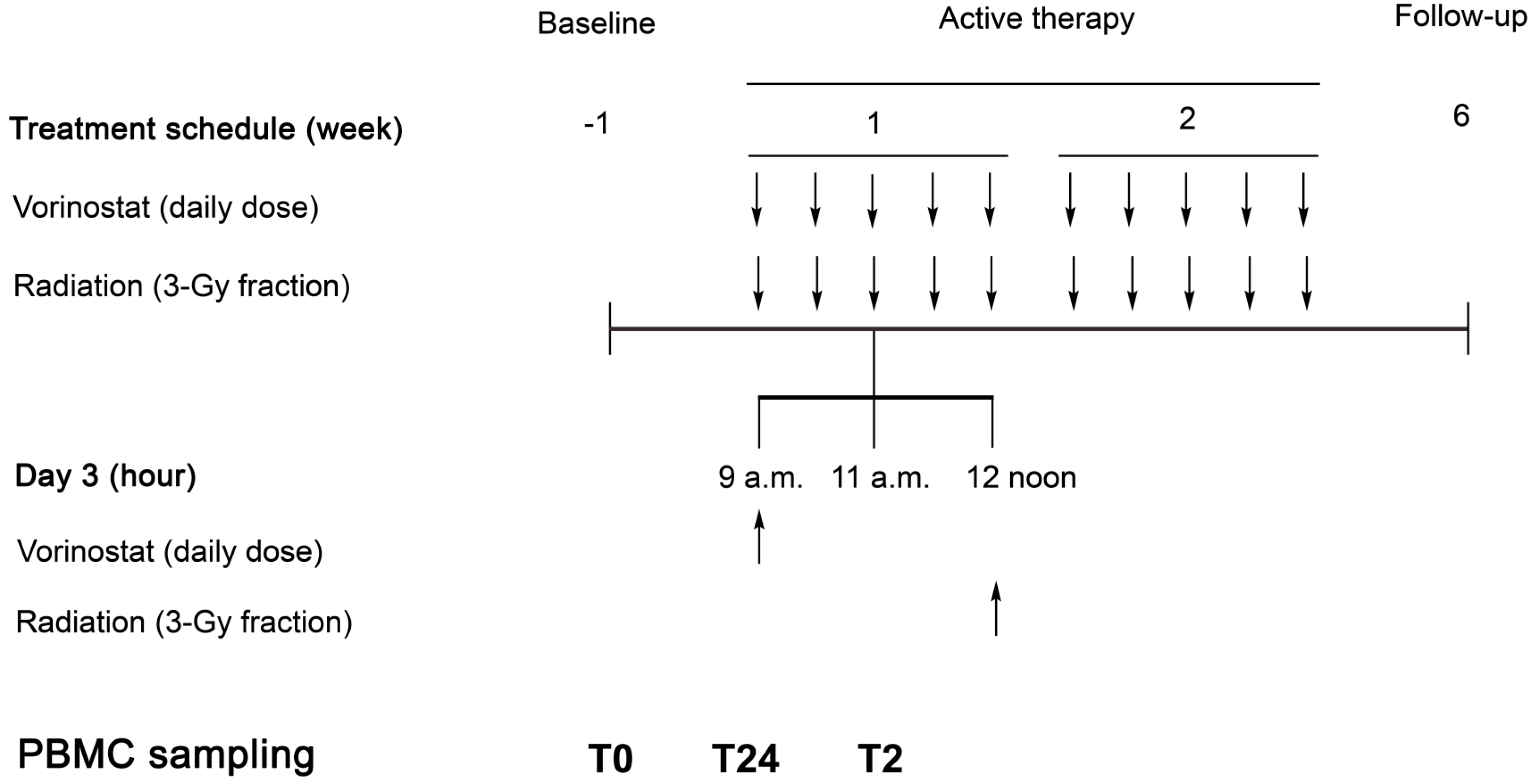
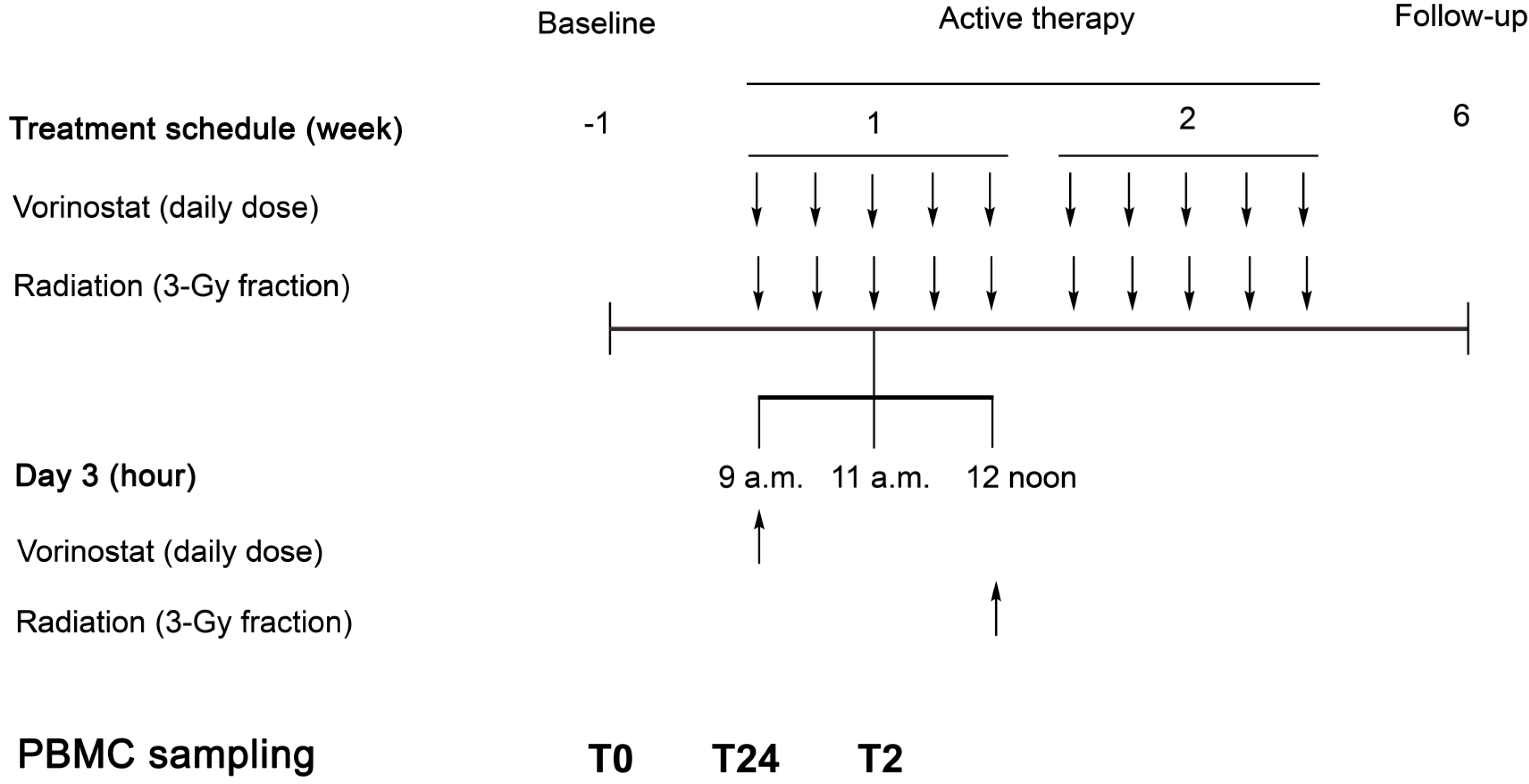
2.2. The Pelvic Radiation and Vorinostat (PRAVO) Study
3. Evaluation of Treatment Toxicity—Clinical Assessment
3.1. Common Terminology Criteria for Adverse Events (CTCAE)
3.2. Intestinal Toxicity in Pelvic Radiotherapy
3.3. LARC-RRP and PRAVO—Clinical Toxicity Profiles
3.3.1. Curative Combined-Modality Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTCAE Scoring | At Baseline | At Completion of FLOX + CRT | At Evaluation |
|---|---|---|---|
| CTCAE grade 0 diarrhea | 66 | 36 | 53 |
| CTCAE grade 1 diarrhea | 10 | 22 | 17 |
| CTCAE grade 2 diarrhea | 4 | 14 | 0 |
| CTCAE grade 3 diarrhea | 0 | 8 | 2 |
| CTCAE grade 4 diarrhea | 0 | 0 | 0 |
| not determined | 0 | 0 | 8 |
3.3.2. Non-Curative Combined-Modality Therapy
| Age of Study Patient (years) | Vorinostat Dose Level | SBV 30 (%) | CTCAE Grade 3 Adverse Events |
|---|---|---|---|
| 77 | 1 | 0 | |
| 49 | 2 | 11 | |
| 66 | 2 | 3 | |
| 87 | 3 | 40 | anorexia, fatigue |
| 47 | 3 | 14 | |
| 77 | 3 | 1 | |
| 82 | 3 | 0 | |
| 81 | 3 | 0 | |
| 55 | 4 | 6 | |
| 83 | 4 | 3 | diarrhea, anorexia, hyponatremia |
| 85 | 4 | 0 | |
| 75 | 4 | 0 | diarrhea, fatigue, hypokalemia |
| 62 | 4 | 0 | |
| 45 | 4 | 0 |
4. Biomarkers of Treatment Toxicity—Array-Based Analyses
4.1. Serial Serum Samples—Cytokine Profiling
4.2. Serial PBMC Samples—Gene Expression Profiling
5. Biomarkers of Treatment Toxicity—Computational Analyses
5.1. Integrated Systems Biology-Based Prediction of Treatment Safety
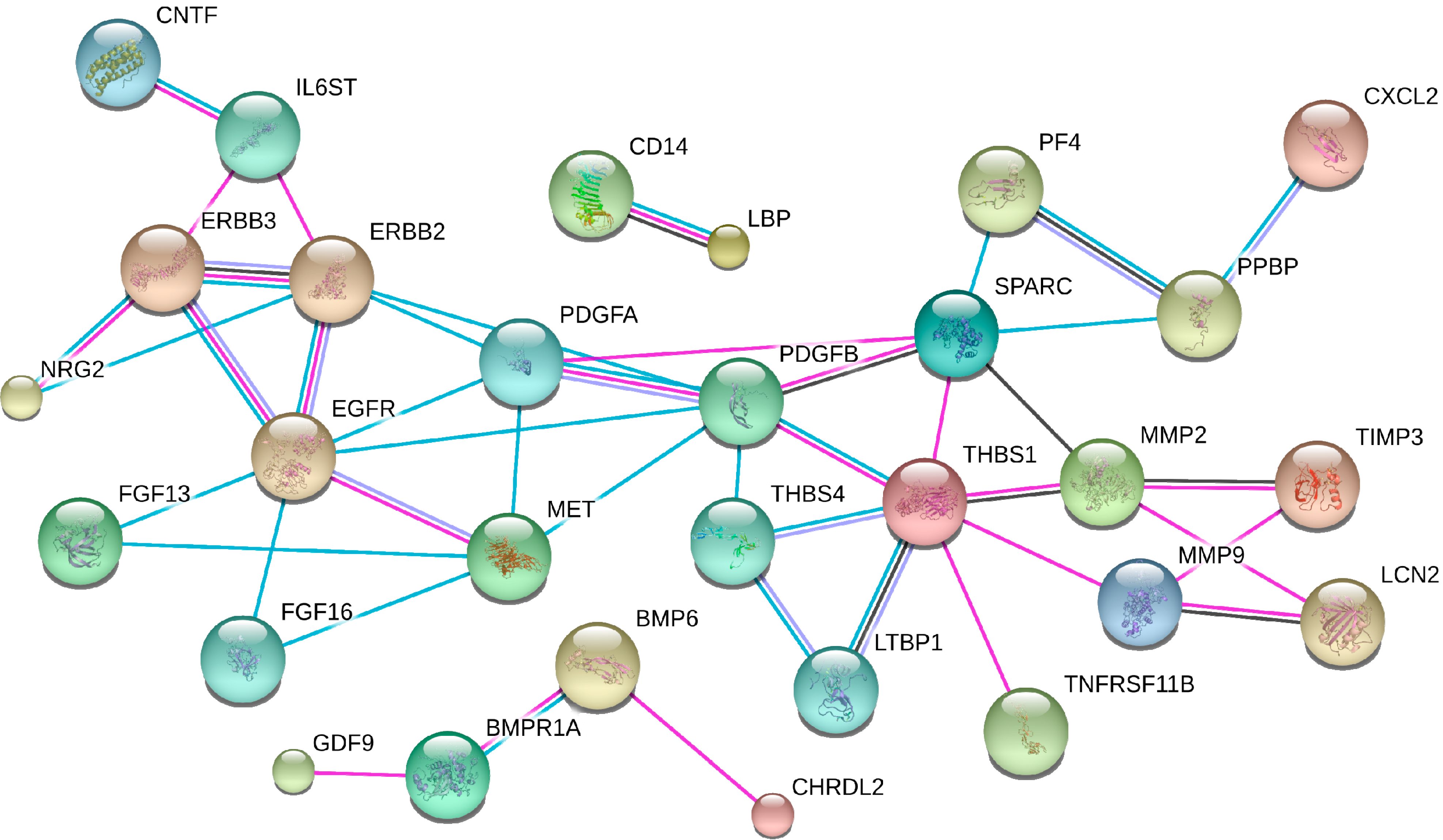
5.2. Exploring Proteomic Signatures

5.3. Exploring Transcriptomic Signatures
6. Prediction of Toxicity in Cancer Therapy—Integration of Multilevel Information
6.1. Defining Clinical Treatment Toxicity
6.2. Understanding Biological Mechanisms of Toxicity
6.2.1. Biochemical and Functional Markers
6.2.2. Pharmacogenetics
6.2.3. PBMC as Drug-Exposed Normal Tissue
6.2.4. Phenotypic Validation
6.3. Revealing Information Connectivity through Open Data Accessibility
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Good, J.S.; Harrington, K.J. The hallmarks of cancer and the radiation oncologist: Updating the 5Rs of radiobiology. Clin. Oncol. (R. Coll. Radiol.) 2013, 25, 569–577. [Google Scholar] [CrossRef]
- Valentini, V.; Beets-Tan, R.; Borras, J.M.; Krivokapic, Z.; Leer, J.W.; Påhlman, L.; Rödel, C.; Schmoll, H.J.; Scott, N.; Velde, C.V.; et al. Evidence and research in rectal cancer. Radiother. Oncol. 2008, 87, 449–474. [Google Scholar] [CrossRef] [PubMed]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rödel, C.; Kuo, L.J.; Calvo, F.A.; Garcia-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-term outcome in patients with a pathological complete response after chemoradiation for rectal cancer: A pooled analysis of individual patient data. Lancet Oncol. 2010, 11, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H.; Hollywood, D. Design and conduct of early-phase radiotherapy trials with targeted therapeutics: Lessons from the PRAVO experience. Radiother. Oncol. 2013, 108, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, B.D.; Pan, C.C.; Dawson, L.A.; Das, S.K.; Li, X.A.; Ten Haken, R.K.; Miften, M. Radiation dose-volume effects in the stomach and small bowel. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, S101–S107. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M.; Trotti, A. Evaluation of early and late toxicities in chemoradiation trials. J. Clin. Oncol. 2007, 25, 4096–4103. [Google Scholar] [CrossRef] [PubMed]
- Depositphotos. Available online: http://depositphotos.com (purchased on 12 June 2014).
- National Cancer Institute at the National Institutes of Health. Available online: http://www.cancer.gov (accessed on 12 June 2014).
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/show/NCT00278694 (accessed on 1 August 2014).
- Sørbye, H.; Glimelius, B.; Berglund, A.; Fokstuen, T.; Tveit, K.M.; Braendengen, M.; Øgreid, D.; Dahl, O. Multicenter phase II study of Nordic fluorouracil and folinic acid bolus schedule combined with oxaliplatin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Folkvord, S.; Flatmark, K.; Dueland, S.; de Wijn, R.; Grøholt, K.K.; Hole, K.H.; Nesland, J.M.; Ruijtenbeek, R.; Boender, P.J.; Johansen, M.; et al. Prediction of response to preoperative chemoradiotherapy in rectal cancer by multiplex kinase activity profiling. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H. A complex case of rectal neuroendocrine carcinoma with terminal delirium. Nat. Clin. Gastroenterol. Hepatol. 2006, 3, 408–413. [Google Scholar] [CrossRef]
- Flatmark, K.; Nome, R.V.; Folkvord, S.; Bratland, A.; Rasmussen, H.; Ellefsen, M.S.; Fodstad, Ø.; Ree, A.H. Radiosensitization of colorectal carcinoma cell lines by histone deacetylase inhibition. Radiat. Oncol. 2006, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H.; Folkvord, S.; Flatmark, K. HDAC2 deficiency and histone acetylation. Nat. Genet. 2008, 40, 812–813. [Google Scholar] [CrossRef] [PubMed]
- Folkvord, S.; Ree, A.H.; Furre, T.; Halvorsen, T.; Flatmark, K. Radiosensitization by SAHA in experimental colorectal carcinoma models—In vivo effects and relevance of histone acetylation status. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Saelen, M.G.; Ree, A.H.; Kristian, A.; Fleten, K.G.; Furre, T.; Hektoen, H.H.; Flatmark, K. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat. Oncol. 2012, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/show/NCT00455351 (accessed on 1 August 2014).
- Ree, A.H.; Dueland, S.; Folkvord, S.; Hole, K.H.; Seierstad, T.; Johansen, M.; Abrahamsen, T.W.; Flatmark, K. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: The Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010, 11, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Bratland, A.; Dueland, S.; Hollywood, D.; Flatmark, K.; Ree, A.H. Gastrointestinal toxicity of vorinostat: Reanalysis of phase 1 study results with emphasis on dose-volume effects of pelvic radiotherapy. Radiat. Oncol. 2011, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H.; Saelen, M.G.; Kalanxhi, E.; Østensen, I.H.; Schee, K.; Røe, K.; Abrahamsen, T.W.; Dueland, S.; Flatmark, K. Biomarkers of histone deacetylase inhibitor activity in a phase 1 combined-modality study with radiotherapy. PLoS One 2014, 9, e89750. [Google Scholar] [CrossRef] [PubMed]
- Common Terminology Criteria for Adverse Events v3.0 (CTCAE). Available online: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf (accessed on 1 August 2014).
- Trotti, A.; Colevas, A.D.; Setser, A.; Rusch, V.; Jaques, D.; Budach, V.; Langer, C.; Murphy, B.; Cumberlin, R.; Coleman, C.N.; et al. CTCAE v3.0: Development of a comprehensive grading system for the adverse effects of cancer treatment. Semin. Radiat. Oncol. 2003, 13, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; McLeod, H.L. Use of pharmacogenetics for predicting cancer prognosis and treatment exposure, response and toxicity. J. Hum. Genet. 2013, 58, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, S.M.; Harari, P.M.; Bernier, J. Exploitable mechanisms for combining drugs with radiation: Concepts, achievements and future directions. Nat. Clin. Pract. Oncol. 2007, 4, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Lee, J.J.; Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009, 101, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Baglan, K.L.; Frazier, R.C.; Yan, D.; Huang, R.R.; Martinez, A.A.; Robertson, J.M. The dose-volume relationship of acute small bowel toxicity from concurrent 5-FU-based chemotherapy and radiation therapy for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2002, 52, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.M.; Lockman, D.; Yan, D.; Wallace, M. The dose-volume relationship of small bowel irradiation and acute grade 3 diarrhea during chemoradiotherapy for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Sonis, S.T. The pathobiology of mucositis. Nat. Rev. Cancer 2004, 4, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Al-Dasooqi, N.; Sonis, S.T.; Bowen, J.M.; Bateman, E.; Blijlevens, N.; Gibson, R.J.; Logan, R.M.; Nair, R.G.; Stringer, A.M.; Yazbeck, R.; et al. Mucositis Study Group of Multinational Association of Supportive Cancer in Cancer/International Society of Oral Oncology (MASCC/ISOO). Emerging evidence on the pathobiology of mucositis. Support. Care Cancer 2013, 21, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Linard, C.; Busson, E.; Holler, V.; Strup-Perrot, C.; Lacave-Lapalun, J.V.; Lhomme, B.; Prat, M.; Devauchelle, P.; Sabourin, J.C.; Simon, J.M.; et al. Repeated autologous bone marrow-derived mesenchymal stem cell injections improve radiation-induced proctitis in pigs. Stem Cells Transl. Med. 2013, 2, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Jiang, W.; Yang, J.; Mao, Y.Q.; Zhang, Y.; Yang, W.; Yang, D.; Burkholder, B.; Huang, R.F.; Huang, R.P. A biotin label-based antibody array for high-content profiling of protein expression. Cancer Genomics Proteomics 2010, 7, 129–141. [Google Scholar] [PubMed]
- Gene Expression Omnibus. Available online: http://ww.ncbi.nlm.nih.gov/geo/ (accessed on 1 August 2014).
- Bai, J.P.; Abernethy, D.R. Systems pharmacology to predict drug toxicity: Integration across levels of biological organization. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 451–473. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, D.S.; Marina, O.; Ray, S.; Zhang, G.L.; Wu, C.J.; Brusic, V. Data processing and analysis for protein microarrays. Methods Mol. Biol. 2011, 723, 337–347. [Google Scholar] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Ramachandran, N. Protein microarrays as tools for functional proteomics. Curr. Opin. Chem. Biol. 2005, 9, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Roelofs, E.; Reyman, B.; Velazquez, E.R.; Buijsen, J.; Zegers, C.M.; Carvalho, S.; Leijenaar, R.T.; Nalbantov, G.; Oberije, C.; et al. ‘Rapid Learning health care in oncology’—An approach towards decision support systems enabling customised radiotherapy. Radiother. Oncol. 2013, 109, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L. Cancer pharmacogenomics: Early promise, but concerted effort needed. Science 2013, 339, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Rosmarin, D.; Palles, C.; Church, D.; Domingo, E.; Jones, A.; Johnstone, E.; Wang, H.; Love, S.; Julier, P.; Scudder, C.; et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: Investigation in the QUASAR2 study, systematic review, and meta-analysis. J. Clin. Oncol. 2014, 32, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- De Mello, V.D.; Kolehmanien, M.; Schwab, U.; Pulkkinen, L.; Uusitupa, M. Gene expression of peripheral blood mononuclear cells as a tool in dietary intervention studies: What do we know so far? Mol. Nutr. Food Res. 2012, 56, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- Connolly, P.H.; Caiozzo, V.J.; Zaldivar, F.; Nemet, D.; Larson, J.; Hung, S.P.; Heck, J.D.; Hatfield, J.D.; Cooper, D.M. Effects of exercise on gene expression in human peripheral blood mononuclear cells. J. Appl. Physiol. 2004, 97, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Rokutan, K.; Morita, K.; Masuda, K.; Tominaga, K.; Shikishima, M.; Teshima-Kondo, S.; Omori, T.; Sekiyama, A. Gene expression profiling in peripheral blood leukocytes as a new approach for assessment of human stress response. J. Med. Investig. 2005, 52, 137–144. [Google Scholar] [CrossRef]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar] [PubMed]
- Fensgård, Ø.; Kassahun, H.; Bombik, I.; Rognes, T.; Lindvall, J.M.; Nilsen, H. A two-tiered compensatory response to loss of DNA repair modulates aging and stress response pathways. Aging (Albany N. Y.) 2010, 31, 133–159. [Google Scholar]
- Arczewska, K.D.; Tomazella, G.G.; Lindvall, J.M.; Kassahun, H.; Maglioni, S.; Torgovnick, A.; Henriksson, J.; Matilainen, O.; Marquis, B.J.; Nelson, B.C.; et al. Active transcriptomic and proteomic reprogramming in the C. elegans nucleotide excision repair mutant xpa-1. Nucleic Acids Res. 2013, 41, 5368–5381. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Lindvall, J.M.; Thijssen, K.; Karambelas, A.E.; Cupac, D.; Fensgård, O.; Jansen, G.; Hoeijmakers, J.H.; Nilsen, H.; Vermaulen, W. DNA damage leads to progressive replicative decline but extends the life span of long-lived mutant animals. Cell Death Differ. 2013, 20, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Skjeldam, H.K.; Kassahun, H.; Fensgård, O.; SenGupta, T.; Babaie, E.; Lindvall, J.M.; Arczewska, K.; Nilsen, H. Loss of Caenorhabditis elegans UNG-1 uracil-DNA glycosylase affects apoptosis in response to DNA damaging agents. DNA Repair (Amst.) 2010, 5, 861–870. [Google Scholar] [CrossRef]
- Forthun, R.B.; Sengupta, T.; Skjeldam, H.K.; Lindvall, J.M.; McCormack, E.; Gjertsen, B.T.; Nilsen, H. Cross-species functional genomic analysis identifies resistance genes of the histone deacetylase inhibitor valproic acid. PLoS One 2012, 7, e48992. [Google Scholar] [CrossRef]
- SenGupta, T.; Torgersen, M.L.; Kassahun, H.; Vellai, T.; Simonsen, A.; Nilsen, H. Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat. Commun. 2013, 4, 2674. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.L.; Fabre, K.M.; Tagle, D.A. The National Institutes of Health Microphysiological Systems Program focuses on a critical challenge in the drug discovery pipeline. Stem Cell Res. Ther. 2013, 4, I1. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Long, C.J.; Berry, B.J.; McAleer, C.; Stancescu, M.; Molnar, P.; Miller, P.G.; Esch, M.B.; Prot, J.M.; Hickman, J.J.; et al. Microphysiological systems and low-cost microfluidic platform with analytics. Stem Cell Res. Ther. 2013, 4, S9. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Srinivasan, B.; Esch, M.B.; McLamb, W.T.; Bernabini, C.; Shuler, M.L.; Hickman, J.J. Using physiologically-based pharmacokinetic-guided “body-on-a-chip” systems to predict mammalian response to drug and chemical exposure. Exp. Biol. Med. (Maywood) 2014, 239, 1225–1239. [Google Scholar] [CrossRef]
- Kalanxhi, E.; Akershus University Hospital, Lørenskog, Norway. Unpublished work. 2014.
- Friend, S.H. The need for precompetitive integrative bionetwork disease model building. Clin. Pharmacol. Ther. 2010, 87, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Norman, T.C. Metcalfe’s law and the biology information commons. Nat. Biotechnol. 2013, 31, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J. Profile: Stephen Friend. The visionary. Science 2012, 335, 651–653. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ree, A.H.; Meltzer, S.; Flatmark, K.; Dueland, S.; Kalanxhi, E. Biomarkers of Treatment Toxicity in Combined-Modality Cancer Therapies with Radiation and Systemic Drugs: Study Design, Multiplex Methods, Molecular Networks. Int. J. Mol. Sci. 2014, 15, 22835-22856. https://doi.org/10.3390/ijms151222835
Ree AH, Meltzer S, Flatmark K, Dueland S, Kalanxhi E. Biomarkers of Treatment Toxicity in Combined-Modality Cancer Therapies with Radiation and Systemic Drugs: Study Design, Multiplex Methods, Molecular Networks. International Journal of Molecular Sciences. 2014; 15(12):22835-22856. https://doi.org/10.3390/ijms151222835
Chicago/Turabian StyleRee, Anne Hansen, Sebastian Meltzer, Kjersti Flatmark, Svein Dueland, and Erta Kalanxhi. 2014. "Biomarkers of Treatment Toxicity in Combined-Modality Cancer Therapies with Radiation and Systemic Drugs: Study Design, Multiplex Methods, Molecular Networks" International Journal of Molecular Sciences 15, no. 12: 22835-22856. https://doi.org/10.3390/ijms151222835
APA StyleRee, A. H., Meltzer, S., Flatmark, K., Dueland, S., & Kalanxhi, E. (2014). Biomarkers of Treatment Toxicity in Combined-Modality Cancer Therapies with Radiation and Systemic Drugs: Study Design, Multiplex Methods, Molecular Networks. International Journal of Molecular Sciences, 15(12), 22835-22856. https://doi.org/10.3390/ijms151222835