From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The PLG Activation System
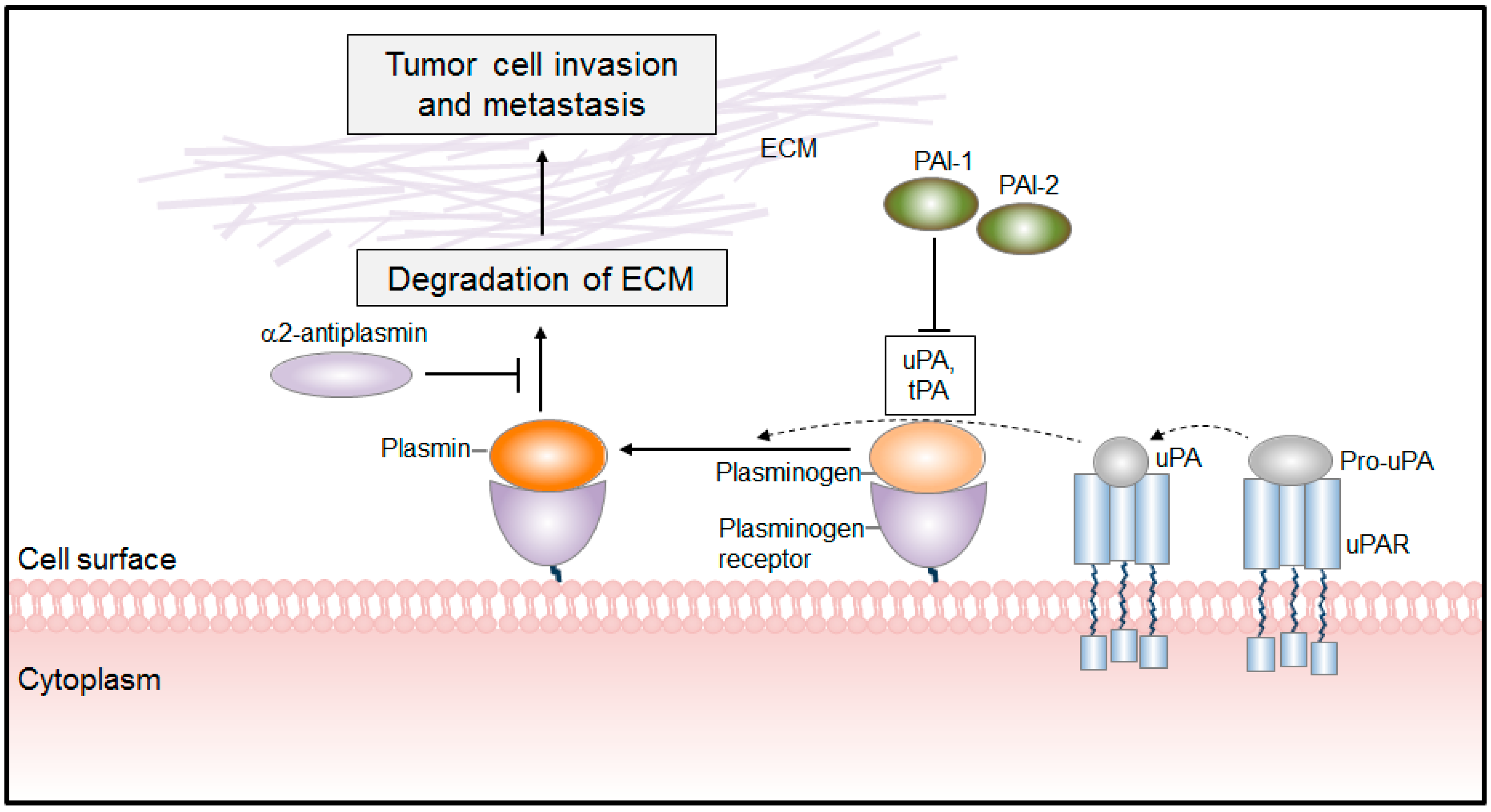
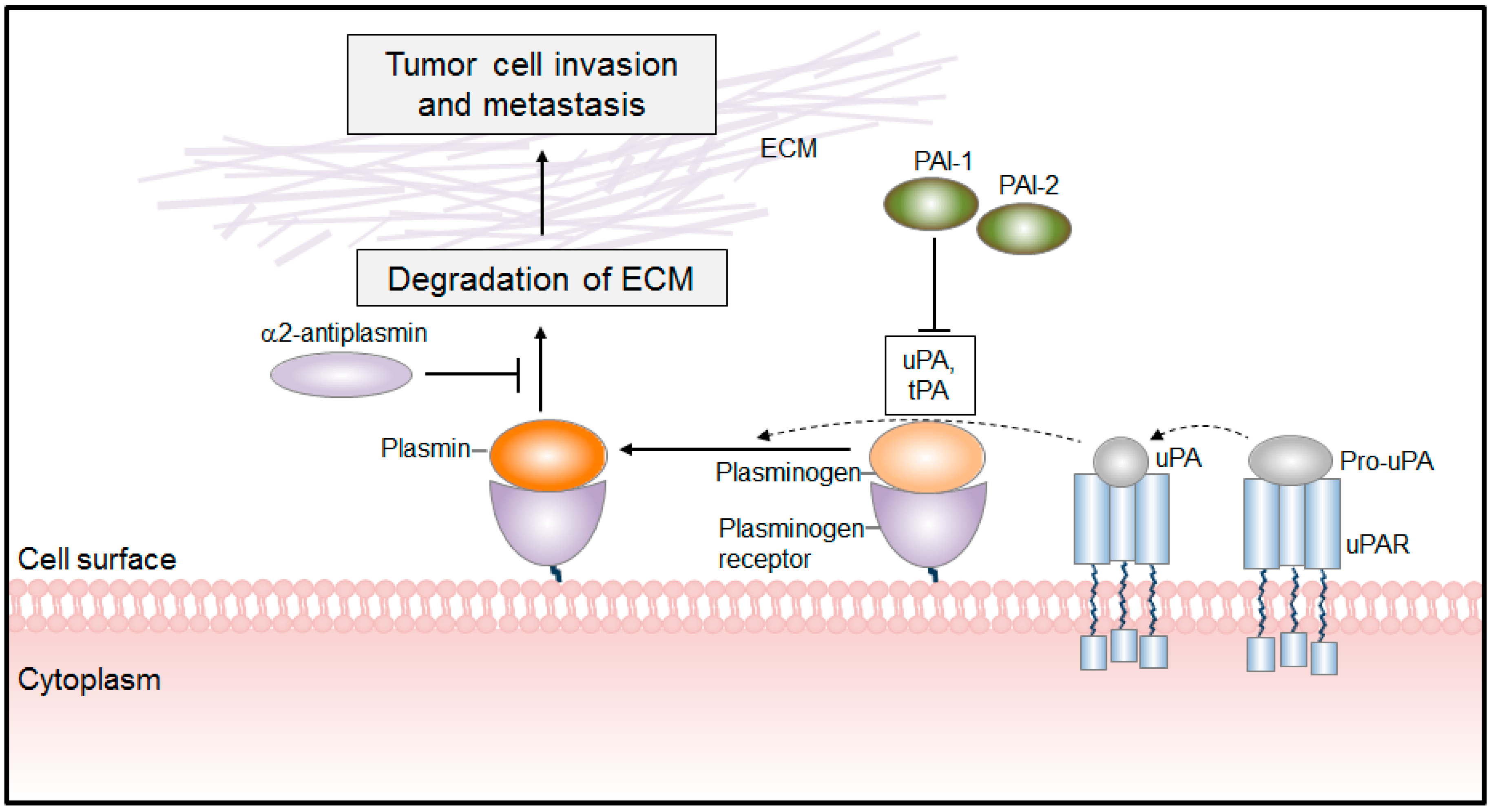
PLG/PLA System in Cancer
3. Plasminogen Receptors (PLG-R)
3.1. Classification of PLG-R
3.2. PLG-Rs in Cancer
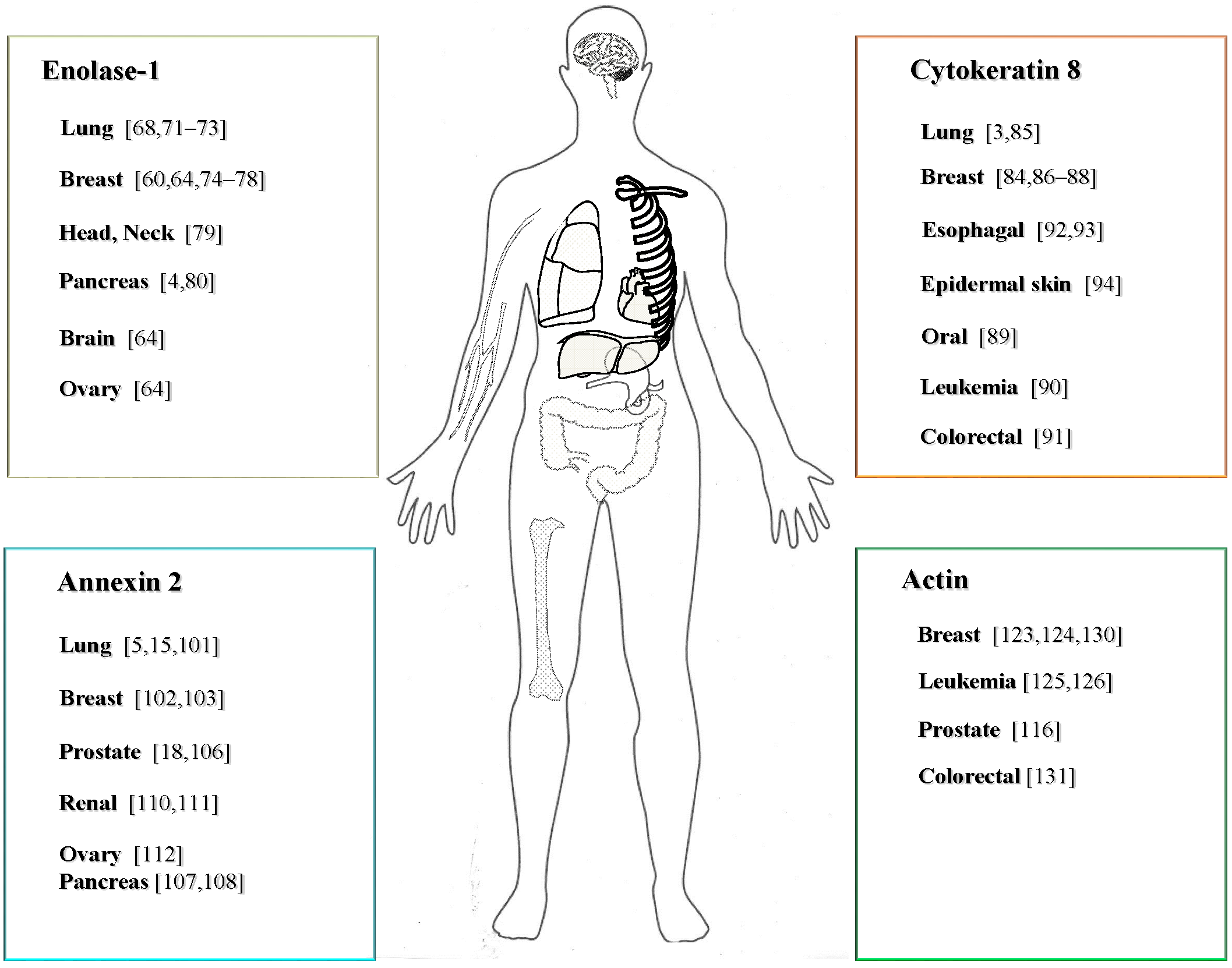
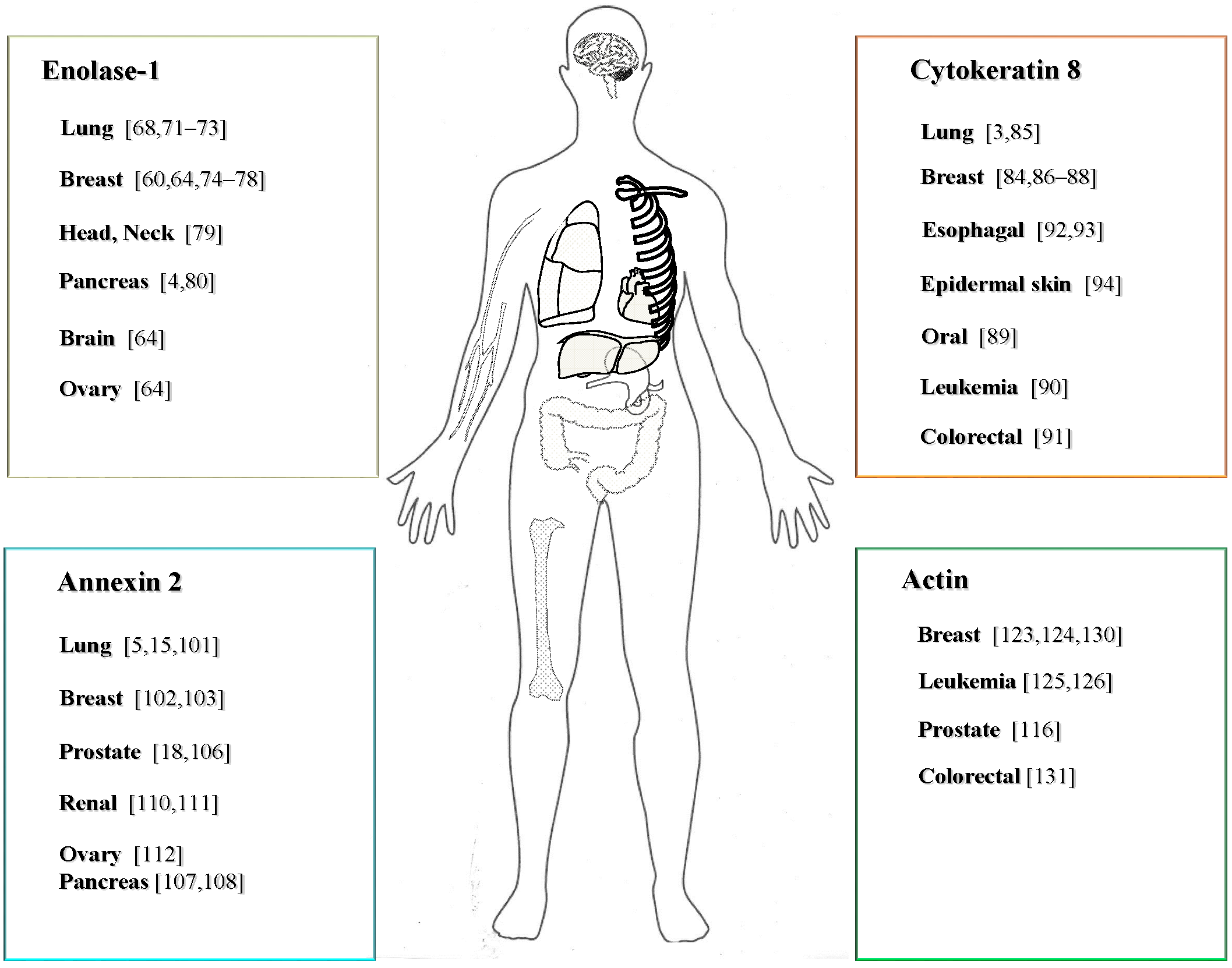
3.2.1. Expression and Function of ENO-1 in Cancer
3.2.2. The Role of CK8 in Cancer
3.2.3. The Expression and Function of ANX2 in Cancer
3.2.4. Expression and Function of ACT in Cancer
4. Mechanism of PLG-R Trafficking
5. Conclusions and Final Remarks
Acknowledgments
Conflicts of Interest
References
- Castellino, F.J.; Ploplis, V.A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [Google Scholar] [PubMed]
- Kwaan, H.C.; McMahon, B. The role of plasminogen-plasmin system in cancer. Cancer Treat. Res. 2009, 148, 43–66. [Google Scholar] [PubMed]
- Fukunaga, Y.; Bandoh, S.; Fujita, J.; Yang, Y.; Ueda, Y.; Hojo, S.; Dohmoto, K.; Tojo, Y.; Takahara, J.; Ishida, T. Expression of cytokeratin 8 in lung cancer cell lines and measurement of serum cytokeratin 8 in lung cancer patients. Lung Cancer 2002, 38, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Tomaino, B.; Chiarle, R.; Ceruti, P.; Novarino, A.; Castagnoli, C.; Migliorini, P.; Perconti, G.; Giallongo, A.; Milella, M.; et al. An integrated humoral and cellular response is elicited in pancreatic cancer by alpha-enolase, a novel pancreatic ductal adenocarcinoma-associated antigen. Int. J. Cancer 2009, 125, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Chen, C.L.; Tseng, Y.L.; Fang, Y.T.; Lin, Y.S.; Su, W.C.; Chen, C.C.; Chang, K.C.; Wang, Y.C.; Lin, C.F. Annexin A2 silencing induces G2 arrest of non-small cell lung cancer cells through p53-dependent and -independent mechanisms. J. Biol. Chem. 2012, 287, 32512–32524. [Google Scholar] [CrossRef] [PubMed]
- Plow, E.F.; Doeuvre, L.; Das, R. So many plasminogen receptors: Why? J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Syrovets, T.; Lunov, O.; Simmet, T. Plasmin as a proinflammatory cell activator. J. Leukoc. Biol. 2012, 92, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Janicke, F.; Moniwa, N.; Chucholowski, N.; Pache, L.; Graeff, H. Tumor-associated urokinase-type plasminogen activator: biological and clinical significance. Biol. Chem. Hoppe Seyler 1992, 373, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Syrovets, T.; Simmet, T. Novel aspects and new roles for the serine protease plasmin. Cell. Mol. Life Sci. 2004, 61, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Andronicos, N.M. Plasminogen binding and cancer: Promises and pitfalls. Front. Biosci. 2003, 8, S294–S304. [Google Scholar] [CrossRef] [PubMed]
- Lal, I.; Dittus, K.; Holmes, C.E. Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef]
- Kruithof, E.K.; Baker, M.S.; Bunn, C.L. Biological and clinical aspects of plasminogen activator inhibitor type 2. Blood 1995, 86, 4007–4024. [Google Scholar] [PubMed]
- Rifkin, D.B.; Mazzieri, R.; Munger, J.S.; Noguera, I.; Sung, J. Proteolytic control of growth factor availability. APMIS 1999, 107, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Laumonnier, Y.; Syrovets, T.; Simmet, T. Plasmin triggers cytokine induction in human monocyte-derived macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.H.; Liu, Q.Q.; Zhang, P.F.; Li, M.Y.; Chen, Z.C.; Liu, Y.F. Prognostic significance of annexin II expression in non-small cell lung cancer. Clin. Transl. Oncol. 2013, 15, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Syrovets, T.; Jendrach, M.; Rohwedder, A.; Schule, A.; Simmet, T. Plasmin-induced expression of cytokines and tissue factor in human monocytes involves AP-1 and IKKβ-mediated NF-κB activation. Blood 2001, 97, 3941–3950. [Google Scholar] [CrossRef] [PubMed]
- Burysek, L.; Syrovets, T.; Simmet, T. The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and janus kinase (JAK)/STAT signaling pathways. J. Biol. Chem. 2002, 277, 33509–33517. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.G.; Liu, J.; Yuan, Y.; Gopalakrishnan, V.K.; Johansson, S.L.; Dinda, A.K.; Gupta, N.P.; Trevino, L.; Vishwanatha, J.K. Expression of biomarkers modulating prostate cancer angiogenesis: Differential expression of annexin II in prostate carcinomas from India and USA. Mol. Cancer 2003, 2. [Google Scholar] [CrossRef]
- Henderson, M.C.; Azorsa, D.O. The genomic and proteomic content of cancer cell-derived exosomes. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef]
- Pluskota, E.; Soloviev, D.A.; Szpak, D.; Weber, C.; Plow, E.F. Neutrophil apoptosis: Selective regulation by different ligands of integrin αMβ2. J. Immunol. 2008, 181, 3609–3619. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Kombrinck, K.W.; Xiao, Q.; Holmback, K.; Daugherty, C.C.; Witte, D.P.; Degen, J.L. Growth and dissemination of Lewis lung carcinoma in plasminogen-deficient mice. Blood 1997, 90, 4522–4531. [Google Scholar] [PubMed]
- Perides, G.; Zhuge, Y.; Lin, T.; Stins, M.F.; Bronson, R.T.; Wu, J.K. The fibrinolytic system facilitates tumor cell migration across the blood-brain barrier in experimental melanoma brain metastasis. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Liu, H.; La Jeunesse, C.M.; Witte, D.P.; Degen, J.L. Plasminogen supports tumor growth through a fibrinogen-dependent mechanism linked to vascular patency. Blood 2003, 102, 2819–2827. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Gondi, C.; Chetty, C.; Chittivelu, S.; Joseph, P.A.; Lakka, S.S. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol. Cancer Ther. 2005, 4, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Gutova, M.; Najbauer, J.; Gevorgyan, A.; Metz, M.Z.; Weng, Y.; Shih, C.C.; Aboody, K.S. Identification of uPAR-positive chemoresistant cells in small cell lung cancer. PLoS One 2007, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Dass, K.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Sarkar, F.H. Evolving role of uPA/uPAR system in human cancers. Cancer Treat. Rev. 2008, 34, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, G.; Lund, I.K. Urokinase receptor variants in tissue and body fluids. Adv. Clin. Chem. 2007, 44, 65–102. [Google Scholar] [PubMed]
- Gondi, C.S.; Kandhukuri, N.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Down-regulation of uPAR and uPA activates caspase-mediated apoptosis and inhibits the PI3K/AKT pathway. Int. J. Oncol. 2007, 31, 19–27. [Google Scholar] [PubMed]
- Chaurasia, P.; Aguirre-Ghiso, J.A.; Liang, O.D.; Gardsvoll, H.; Ploug, M.; Ossowski, L. A region in urokinase plasminogen receptor domain III controlling a functional association with α5β1 integrin and tumor growth. J. Biol. Chem. 2006, 281, 14852–14863. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Peiro, S.; Roda, O.; Corominas, J.M.; Aguilar, S.; Ampurdanes, C.; Real, F.X.; Navarro, P. Tissue plasminogen activator induces pancreatic cancer cell proliferation by a non-catalytic mechanism that requires extracellular signal-regulated kinase 1/2 activation through epidermal growth factor receptor and annexin A2. Am. J. Pathol. 2007, 170, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- May, P.; Woldt, E.; Matz, R.L.; Boucher, P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007, 39, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Wilhelm, J.; Jablonska, E.; Zakrzewicz, D.; Preissner, K.T.; Seeger, W.; Guenther, A.; Markart, P. Shedding of low-density lipoprotein receptor-related protein-1 in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2011, 184, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Yang, J.; Tanaka, S.; Gonias, S.L.; Mars, W.M.; Liu, Y. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J. Biol. Chem. 2006, 281, 2120–2127. [Google Scholar] [CrossRef] [PubMed]
- Schmalfeldt, B.; Prechtel, D.; Harting, K.; Spathe, K.; Rutke, S.; Konik, E.; Fridman, R.; Berger, U.; Schmitt, M.; Kuhn, W.; et al. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clin. Cancer Res. 2001, 7, 2396–2404. [Google Scholar] [PubMed]
- Duffy, M.J. Urokinase-type plasminogen activator: A potent marker of metastatic potential in human cancers. Biochem. Soc. Trans. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.J.; Hawson, G.A.; Marsh, N.A. Impact of the fibrinolytic enzyme system on prognosis and survival associated with non-small cell lung carcinoma. Blood Coagul. Fibrinolysis 2001, 12, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Beyer, B.C.; Heiss, M.M.; Simon, E.H.; Gruetzner, K.U.; Babic, R.; Jauch, K.W.; Schildberg, F.W.; Allgayer, H. Urokinase system expression in gastric carcinoma: prognostic impact in an independent patient series and first evidence of predictive value in preoperative biopsy and intestinal metaplasia specimens. Cancer 2006, 106, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Bajou, K.; Noel, A.; Gerard, R.D.; Masson, V.; Brunner, N.; Holst-Hansen, C.; Skobe, M.; Fusenig, N.E.; Carmeliet, P.; Collen, D.; et al. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nat. Med. 1998, 4, 923–928. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G.A.; Petitclerc, E.; Stefansson, S.; Smith, E.; Wong, M.K.; Westrick, R.J.; Ginsburg, D.; Brooks, P.C.; Lawrence, D.A. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J. Biol. Chem. 2001, 276, 33964–33968. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Lee, J.W.; Juliano, R.L.; Church, F.C. Plasminogen activator inhibitor-1 and -3 increase cell adhesion and motility of MDA-MB-435 breast cancer cells. J. Biol. Chem. 2002, 277, 40950–40957. [Google Scholar] [CrossRef] [PubMed]
- Czekay, R.P.; Aertgeerts, K.; Curriden, S.A.; Loskutoff, D.J. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J. Cell Biol. 2003, 160, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ohba, K.; Miyata, Y.; Kanda, S.; Koga, S.; Hayashi, T.; Kanetake, H. Expression of urokinase-type plasminogen activator, urokinase-type plasminogen activator receptor and plasminogen activator inhibitors in patients with renal cell carcinoma: correlation with tumor associated macrophage and prognosis. J. Urol. 2005, 174, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Croucher, D.R.; Saunders, D.N.; Lobov, S.; Ranson, M. Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Rev. Cancer 2008, 8, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, M.; Sato, A.; Hayakawa, H.; Urano, T.; Takada, Y.; Takada, A. Plasminogen activators and their inhibitors in non-small cell lung cancer. Low content of type 2 plasminogen activator inhibitor associated with tumor dissemination. Cancer 1994, 73, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.M.; Yu, Y.B.; Laug, W.E. Overexpression of plasminogen activator inhibitor 2 in human melanoma cells inhibits spontaneous metastasis in scid/scid mice. Proc. Natl. Acad. Sci. USA 1995, 92, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Plow, E.F. Binding and activation of plasminogen on the platelet surface. J. Biol. Chem. 1985, 260, 4303–4311. [Google Scholar] [PubMed]
- Diaz-Ramos, A.; Roig-Borrellas, A.; Garcia-Melero, A.; Lopez-Alemany, R. α-Enolase, a multifunctional protein: its role on pathophysiological situations. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Andronicos, N.M.; O’Mullane, M.J.; Baker, M.S. Increased plasminogen binding is associated with metastatic breast cancer cells: Differential expression of plasminogen binding proteins. Br. J. Cancer 1998, 77, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Parmer, R.J. Plasminogen receptors: The first quarter century. Semin. Thromb. Hemost. 2013, 39, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Cell surface remodeling by plasmin: A new function for an old enzyme. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Fulde, M.; Steinert, M.; Bergmann, S. Interaction of streptococcal plasminogen binding proteins with the host fibrinolytic system. Front. Cell. Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Vasudevan, J.; Allietta, M.M.; Glass, W.F., 2nd; Gonias, S.L. A cytokeratin 8-like protein with plasminogen-binding activity is present on the external surfaces of hepatocytes, HepG2 cells and breast carcinoma cell lines. J. Cell Sci. 1995, 108, 1071–1082. [Google Scholar] [PubMed]
- Kassam, G.; Choi, K.S.; Ghuman, J.; Kang, H.M.; Fitzpatrick, S.L.; Zackson, T.; Zackson, S.; Toba, M.; Shinomiya, A.; Waisman, D.M. The role of annexin II tetramer in the activation of plasminogen. J. Biol. Chem. 1998, 273, 4790–4799. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Soloviev, D.A.; Bdeir, K.; Cines, D.B.; Plow, E.F. Integrin αMβ2 orchestrates and accelerates plasminogen activation and fibrinolysis by neutrophils. J. Biol. Chem. 2004, 279, 18063–18072. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, V. Multifunctional α-enolase: Its role in diseases. Cell. Mol. Life Sci. 2001, 58, 902–920. [Google Scholar] [CrossRef] [PubMed]
- Capello, M.; Ferri-Borgogno, S.; Cappello, P.; Novelli, F. α-Enolase: A promising therapeutic and diagnostic tumor target. FEBS J. 2011, 278, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Glynn, S.A.; Gammell, P.; Heenan, M.; O’Connor, R.; Liang, Y.; Keenan, J.; Clynes, M. A new superinvasive in vitro phenotype induced by selection of human breast carcinoma cells with the chemotherapeutic drugs paclitaxel and doxorubicin. Br. J. Cancer 2004, 91, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Stillfried, G.E.; Saunders, D.N.; Ranson, M. Plasminogen binding and activation at the breast cancer cell surface: the integral role of urokinase activity. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef]
- Miles, L.A.; Dahlberg, C.M.; Plescia, J.; Felez, J.; Kato, K.; Plow, E.F. Role of cell-surface lysines in plasminogen binding to cells: Identification of α-enolase as a candidate plasminogen receptor. Biochemistry 1991, 30, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar]
- Feo, S.; Arcuri, D.; Piddini, E.; Passantino, R.; Giallongo, A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 2000, 473, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Miller, D.M. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. J. Biol. Chem. 2000, 275, 5958–5965. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.C.; Shih, N.Y.; Fang, H.L.; Huang, T.S.; Kuo, C.C.; Chu, P.Y.; Hung, Y.M.; Chou, S.W.; Yang, Y.Y.; Chang, G.C.; et al. Surface α-enolase promotes extracellular matrix degradation and tumor metastasis and represents a new therapeutic target. PLoS One 2013, 8, e69354. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer statistics, 2006. CA Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Kim, H.; Rhee, H.; Kim, S.H.; Shin, D.H.; Chung, K.Y.; Park, K.S.; Paik, Y.K.; Chang, J.; Kim, H. Proteomic analysis distinguishes basaloid carcinoma as a distinct subtype of nonsmall cell lung carcinoma. Proteomics 2004, 4, 3394–3400. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.C.; Liu, K.J.; Hsieh, C.L.; Hu, T.S.; Charoenfuprasert, S.; Liu, H.K.; Luh, K.T.; Hsu, L.H.; Wu, C.W.; Ting, C.C.; et al. Identification of α-enolase as an autoantigen in lung cancer: Its overexpression is associated with clinical outcomes. Clin. Cancer Res. 2006, 12, 5746–5754. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Naka, T.; Serada, S.; Fujimoto, M.; Tanaka, T.; Hashimoto, S.; Shima, Y.; Yamadori, T.; Suzuki, H.; Hirashima, T.; et al. Proteomics-based identification of alpha-enolase as a tumor antigen in non-small lung cancer. Cancer Sci. 2007, 98, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.H.; Chang, C.C.; Chen, C.S.; Tam, K.W.; Wang, Y.J.; Lee, C.H.; Lin, H.W.; Cheng, T.C.; Huang, C.S.; Chu, J.S.; et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res. Treat. 2010, 121, 539–553. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Coussens, L.M. The inflammatory tumor microenvironment and its impact on cancer development. Contrib. Microbiol. 2006, 13, 118–137. [Google Scholar]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar]
- Ejma, M.; Misiuk-Hojlo, M.; Gorczyca, W.A.; Podemski, R.; Szymaniec, S.; Kuropatwa, M.; Rogozinska-Szczepka, J.; Bartnik, W. Antibodies to 46-kDa retinal antigen in a patient with breast carcinoma and cancer-associated retinopathy. Breast Cancer Res. Treat. 2008, 110, 269–271. [Google Scholar] [PubMed]
- Dowling, P.; Meleady, P.; Dowd, A.; Henry, M.; Glynn, S.; Clynes, M. Proteomic analysis of isolated membrane fractions from superinvasive cancer cells. Biochim. Biophys. Acta 2007, 1774, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.T.; Chien, I.H.; Shen, W.H.; Kuo, Y.Z.; Jin, Y.T.; Wong, T.Y.; Hsiao, J.R.; Wang, H.P.; Shih, N.Y.; Wu, L.W. ENO1, a potential prognostic head and neck cancer marker, promotes transformation partly via chemokine CCL20 induction. Eur. J. Cancer 2010, 46, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Mikuriya, K.; Kuramitsu, Y.; Ryozawa, S.; Fujimoto, M.; Mori, S.; Oka, M.; Hamano, K.; Okita, K.; Sakaida, I.; Nakamura, K. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int. J. Oncol. 2007, 30, 849–855. [Google Scholar] [PubMed]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, D.; Didiasova, M.; Zakrzewicz, A.; Hocke, A.C.; Uhle, F.; Markart, P.; Preissner, K.T.; Wygrecka, M. The interaction of enolase-1 with caveolae-associated proteins regulates its subcellular localization. Biochem. J. 2014, 460, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Andratschke, M.; Schmitt, B.; Mack, B.; Schaffrik, M. Cytokeratin 8 associates with the external leaflet of plasma membranes in tumour cells. Biochem. Biophys. Res. Commun. 2005, 328, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Godfroid, E.; Geuskens, M.; Dupressoir, T.; Parent, I.; Szpirer, C. Cytokeratins are exposed on the outer surface of established human mammary carcinoma cells. J. Cell Sci. 1991, 99, 595–607. [Google Scholar] [PubMed]
- Fujita, J.; Dobashi, N.; Ohtsuki, Y.; Ueda, Y.; Bandoh, S.; Yamadori, I.; Takahara, J. Detection of large molecular weight cytokeratin 8 as carrier protein of CA19-9 in non-small-cell lung cancer cell lines. Br. J. Cancer 1999, 81, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Hembrough, T.A.; Li, L.; Gonias, S.L. Cell-surface cytokeratin 8 is the major plasminogen receptor on breast cancer cells and is required for the accelerated activation of cell-associated plasminogen by tissue-type plasminogen activator. J. Biol. Chem. 1996, 271, 25684–25691. [Google Scholar] [CrossRef] [PubMed]
- Lehr, H.A.; Folpe, A.; Yaziji, H.; Kommoss, F.; Gown, A.M. Cytokeratin 8 immunostaining pattern and E-cadherin expression distinguish lobular from ductal breast carcinoma. Am. J. Clin. Pathol. 2000, 114, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Cimpean, A.M.; Suciu, C.; Ceausu, R.; Tatucu, D.; Muresan, A.M.; Raica, M. Relevance of the immunohistochemical expression of cytokeratin 8/18 for the diagnosis and classification of breast cancer. Rom. J. Morphol. Embryol. 2008, 49, 479–483. [Google Scholar] [PubMed]
- Fillies, T.; Werkmeister, R.; Packeisen, J.; Brandt, B.; Morin, P.; Weingart, D.; Joos, U.; Buerger, H. Cytokeratin 8/18 expression indicates a poor prognosis in squamous cell carcinomas of the oral cavity. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, M.; Andersson, L.C.; Virtanen, I. K562 erythroleukemia cells express cytokeratins 8, 18, and 19 and epithelial membrane antigen that disappear after induced differentiation. J. Cell Physiol. 1990, 143, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, B.; Stigbrand, T. A two-site enzyme-linked immunosorbent assay for cytokeratin 8. Int. J. Cancer 1990, 46, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Brattstrom, D.; Wagenius, G.; Sandstrom, P.; Dreilich, M.; Bergstrom, S.; Goike, H.; Hesselius, P.; Bergqvist, M. Newly developed assay measuring cytokeratins 8, 18 and 19 in serum is correlated to survival and tumor volume in patients with esophageal carcinoma. Dis. Esophagus 2005, 18, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kapur, S.; Chattopadhyay, I.; Purkayastha, J.; Sharma, J.; Mishra, A.; Hewitt, S.M.; Saxena, S. Cytokeratin immunoexpression in esophageal squamous cell carcinoma of high-risk population in Northeast India. Appl. Immunohistochem. Mol. Morphol. 2009, 17, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.L.; Bravo, A.; Martinez-Palacio, J.; Fernandez-Acenero, M.J.; Villanueva, C.; Larcher, F.; Conti, C.J.; Jorcano, J.L. Epidermal abnormalities and increased malignancy of skin tumors in human epidermal keratin 8-expressing transgenic mice. FASEB J. 2004, 18, 1556–1558. [Google Scholar] [PubMed]
- Lokman, N.A.; Ween, M.P.; Oehler, M.K.; Ricciardelli, C. The role of annexin A2 in tumorigenesis and cancer progression. Cancer Microenviron. 2011, 4, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Guevara, C.A.; Lev, E.; Dowling, K.; Chacko, J. Interaction of the fibrinolytic receptor, annexin II, with the endothelial cell surface. Essential role of endonexin repeat 2. J. Biol. Chem. 1996, 271, 21652–21659. [Google Scholar] [PubMed]
- Mai, J.; Finley, R.L., Jr.; Waisman, D.M.; Sloane, B.F. Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J. Biol. Chem. 2000, 275, 12806–12812. [Google Scholar] [PubMed]
- Hajjar, K.A.; Jacovina, A.T.; Chacko, J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J. Biol. Chem. 1994, 269, 21191–21197. [Google Scholar] [PubMed]
- Kassam, G.; Manro, A.; Braat, C.E.; Louie, P.; Fitzpatrick, S.L.; Waisman, D.M. Characterization of the heparin binding properties of annexin II tetramer. J. Biol. Chem. 1997, 272, 15093–15100. [Google Scholar] [CrossRef] [PubMed]
- Madureira, P.A.; Surette, A.P.; Phipps, K.D.; Taboski, M.A.; Miller, V.A.; Waisman, D.M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood 2011, 118, 4789–4797. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.W.; Wang, Y.L. Expression and function of Annexin II in lung cancer tissue. Asian Pac. J. Trop. Med. 2013, 6, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Hastie, C. Interferon gamma, a possible therapeutic approach for late-stage prostate cancer? Anticancer Res. 2008, 28, 2843–2849. [Google Scholar] [PubMed]
- Sharma, M.R.; Koltowski, L.; Ownbey, R.T.; Tuszynski, G.P.; Sharma, M.C. Angiogenesis-associated protein annexin II in breast cancer: Selective expression in invasive breast cancer and contribution to tumor invasion and progression. Exp. Mol. Pathol. 2006, 81, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Ownbey, R.T.; Sharma, M.C. Breast cancer cell surface annexin II induces cell migration and neoangiogenesis via tPA dependent plasmin generation. Exp. Mol. Pathol. 2010, 88, 278–286. [Google Scholar] [CrossRef] [PubMed]
- McColl, B.K.; Baldwin, M.E.; Roufail, S.; Freeman, C.; Moritz, R.L.; Simpson, R.J.; Alitalo, K.; Stacker, S.A.; Achen, M.G. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J. Exp. Med. 2003, 198, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Shen, J.J.; Tanzillo-Swarts, A.; Bhatia, B.; Maldonado, C.M.; Person, M.D.; Lau, S.S.; Tang, D.G. Annexin II expression is reduced or lost in prostate cancer cells and its re-expression inhibits prostate cancer cell migration. Oncogene 2003, 22, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Vishwanatha, J.K.; Chiang, Y.; Kumble, K.D.; Hollingsworth, M.A.; Pour, P.M. Enhanced expression of annexin II in human pancreatic carcinoma cells and primary pancreatic cancers. Carcinogenesis 1993, 14, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Nedjadi, T.; Kitteringham, N.; Campbell, F.; Jenkins, R.E.; Park, B.K.; Navarro, P.; Ashcroft, F.; Tepikin, A.; Neoptolemos, J.P.; Costello, E. S100A6 binds to annexin 2 in pancreatic cancer cells and promotes pancreatic cancer cell motility. Br. J. Cancer 2009, 101, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS One 2011, 6, e19390. [Google Scholar] [CrossRef] [PubMed]
- Domoto, T.; Miyama, Y.; Suzuki, H.; Teratani, T.; Arai, K.; Sugiyama, T.; Takayama, T.; Mugiya, S.; Ozono, S.; Nozawa, R. Evaluation of S100A10, annexin II and B-FABP expression as markers for renal cell carcinoma. Cancer Sci. 2007, 98, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Izumi, M.; Kawamura, T.; Nishimura, T.; Mukai, K.; Tachibana, M. Annexin II represents metastatic potential in clear-cell renal cell carcinoma. Br. J. Cancer 2009, 101, 287–294. [Google Scholar] [PubMed]
- Lokman, N.A.; Elder, A.S.; Ween, M.P.; Pyragius, C.E.; Hoffmann, P.; Oehler, M.K.; Ricciardelli, C. Annexin A2 is regulated by ovarian cancer-peritoneal cell interactions and promotes metastasis. Oncotarget 2013, 4, 1199–1211. [Google Scholar] [PubMed]
- Sharma, M.; Blackman, M.R.; Sharma, M.C. Antibody-directed neutralization of annexin II (ANX II) inhibits neoangiogenesis and human breast tumor growth in a xenograft model. Exp. Mol. Pathol. 2012, 92, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Herman, I.M. Actin isoforms. Curr. Opin. Cell Biol. 1993, 5, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Andronicos, N.M.; Baik, N.; Parmer, R.J. Cell-surface actin binds plasminogen and modulates neurotransmitter release from catecholaminergic cells. J. Neurosci. 2006, 26, 13017–13024. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Doll, J.A.; Jiang, K.; Cundiff, D.L.; Czarnecki, J.S.; Wilson, M.; Ridge, K.M.; Soff, G.A. Differential binding of plasminogen, plasmin, and angiostatin4.5 to cell surface β-actin: Implications for cancer-mediated angiogenesis. Cancer Res. 2006, 66, 7211–7215. [Google Scholar] [CrossRef] [PubMed]
- Le, P.U.; Nguyen, T.N.; Drolet-Savoie, P.; Leclerc, N.; Nabi, I.R. Increased beta-actin expression in an invasive moloney sarcoma virus-transformed MDCK cell variant concentrates to the tips of multiple pseudopodia. Cancer Res. 1998, 58, 1631–1635. [Google Scholar] [PubMed]
- Nowak, D.; Kochman, A.; Malicka-Blaszkiewicz, M. Identification of actin from hepatoma Morris 5123 cells. Acta Biochim. Pol. 1999, 46, 949–959. [Google Scholar] [PubMed]
- Nguyen, T.N.; Wang, H.J.; Zalzal, S.; Nanci, A.; Nabi, I.R. Purification and characterization of beta-actin-rich tumor cell pseudopodia: Role of glycolysis. Exp. Cell Res. 2000, 258, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Krawczenko, A.; Dus, D.; Malicka-Blaszkiewicz, M. Actin in human colon adenocarcinoma cells with different metastatic potential. Acta Biochim. Pol. 2002, 49, 823–828. [Google Scholar] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.L.; Kiaer, H.; Andersen, J.; Jensen, V.; Melsen, F. Prognostic comparison of three classifications for medullary carcinomas of the breast. Histopathology 1997, 30, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.H.; Nielsen, H.V.; Ditzel, H.J. Translocation of an intracellular antigen to the surface of medullary breast cancer cells early in apoptosis allows for an antigen-driven antibody response elicited by tumor-infiltrating B cells. J. Immunol. 2002, 169, 2701–2711. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, M.; Toyoshima, S.; Yao, T.; Tanaka, M.; Tsuneyoshi, M. Apoptosis and cell proliferation in medullary carcinoma of the breast: a comparative study between medullary and non-medullary carcinoma using the TUNEL method and immunohistochemistry. J. Surg. Oncol. 1999, 70, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Liaw, T.Y.; Chang, M.H.; Kavallaris, M. The cytoskeleton as a therapeutic target in childhood acute leukemia: Obstacles and opportunities. Curr. Drug Targets 2007, 8, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Huber, F.; Schnauss, J.; Ronicke, S.; Rauch, P.; Muller, K.; Futterer, C.; Kas, J. Emergent complexity of the cytoskeleton: from single filaments to tissue. Adv. Phys. 2013, 62, 1–112. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.R.; Castellino, F.J.; Chang, Y.; Deford, M.E.; Gray, H.; Villarreal, X.; Kondri, M.E.; Marti, D.N.; Llinas, M.; Schaller, J.; et al. Characterization of kringle domains of angiostatin as antagonists of endothelial cell migration, an important process in angiogenesis. FASEB J. 1998, 12, 1731–1738. [Google Scholar]
- Small, J.V.; Stradal, T.; Vignal, E.; Rottner, K. The lamellipodium: Where motility begins. Trends Cell Biol. 2002, 12, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Azios, N.G.; Dharmawardhane, S.F. Resveratrol and estradiol exert disparate effects on cell migration, cell surface actin structures, and focal adhesion assembly in MDA-MB-231 human breast cancer cells. Neoplasia 2005, 7, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Skwarek-Maruszewska, A.; Zemanek-Zboch, M.; Malicka-Blaszkiewicz, M. β-actin in human colon adenocarcinoma cell lines with different metastatic potential. Acta Biochim. Pol. 2005, 52, 461–468. [Google Scholar] [PubMed]
- Lopez-Alemany, R.; Correc, P.; Camoin, L.; Burtin, P. Purification of the plasmin receptor from human carcinoma cells and comparison to α-enolase. Thromb. Res. 1994, 75, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Burke, T.; Plow, E.F. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood 2007, 110, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, E.M.; van Deurs, B.; Hansen, G.H. “Nonclassical” secretion of annexin A2 to the lumenal side of the enterocyte brush border membrane. Biochemistry 2003, 42, 14670–14676. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Kirino, A.; Endo, S.; Morisaka, H.; Kuroda, K.; Takagi, M.; Ueda, M. Tracing putative trafficking of the glycolytic enzyme enolase via SNARE-driven unconventional secretion. Eukaryot. Cell. 2012, 11, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Marsh, L.M.; Morty, R.E.; Henneke, I.; Guenther, A.; Lohmeyer, J.; Markart, P.; Preissner, K.T. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 2009, 113, 5588–5598. [Google Scholar] [CrossRef] [PubMed]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem. 2004, 279, 43411–43418. [Google Scholar] [CrossRef] [PubMed]
- O’Mullane, M.J.; Baker, M.S. Elevated plasminogen receptor expression occurs as a degradative phase event in cellular apoptosis. Immunol. Cell Biol. 1999, 77, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ucker, D.S.; Jain, M.R.; Pattabiraman, G.; Palasiewicz, K.; Birge, R.B.; Li, H. Externalized glycolytic enzymes are novel, conserved, and early biomarkers of apoptosis. J. Biol. Chem. 2012, 287, 10325–10343. [Google Scholar] [CrossRef] [PubMed]
- Menke, M.; Gerke, V.; Steinem, C. Phosphatidylserine membrane domain clustering induced by annexin A2/S100A10 heterotetramer. Biochemistry 2005, 44, 15296–15303. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Plow, E.F. Phosphatidylserine as an anchor for plasminogen and its plasminogen receptor, histone H2B, to the macrophage surface. J. Thromb. Haemost. 2011, 9, 339–349. [Google Scholar] [CrossRef]
- Zhou, W.; Capello, M.; Fredolini, C.; Piemonti, L.; Liotta, L.A.; Novelli, F.; Petricoin, E.F. Mass spectrometry analysis of the post-translational modifications of alpha-enolase from pancreatic ductal adenocarcinoma cells. J. Proteome Res. 2010, 9, 2929–2936. [Google Scholar] [CrossRef] [PubMed]
- Tomaino, B.; Cappello, P.; Capello, M.; Fredolini, C.; Sperduti, I.; Migliorini, P.; Salacone, P.; Novarino, A.; Giacobino, A.; Ciuffreda, L.; et al. Circulating autoantibodies to phosphorylated α-enolase are a hallmark of pancreatic cancer. J. Proteome Res. 2011, 10, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Valapala, M.; Vishwanatha, J.K. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J. Biol. Chem. 2011, 286, 30911–30925. [Google Scholar] [CrossRef] [PubMed]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [Google Scholar] [CrossRef] [PubMed]
- Wendler, F.; Bota-Rabassedas, N.; Franch-Marro, X. Cancer becomes wasteful: Emerging roles of exosomes in cell-fate determination. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didiasova, M.; Wujak, L.; Wygrecka, M.; Zakrzewicz, D. From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer. Int. J. Mol. Sci. 2014, 15, 21229-21252. https://doi.org/10.3390/ijms151121229
Didiasova M, Wujak L, Wygrecka M, Zakrzewicz D. From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer. International Journal of Molecular Sciences. 2014; 15(11):21229-21252. https://doi.org/10.3390/ijms151121229
Chicago/Turabian StyleDidiasova, Miroslava, Lukasz Wujak, Malgorzata Wygrecka, and Dariusz Zakrzewicz. 2014. "From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer" International Journal of Molecular Sciences 15, no. 11: 21229-21252. https://doi.org/10.3390/ijms151121229
APA StyleDidiasova, M., Wujak, L., Wygrecka, M., & Zakrzewicz, D. (2014). From Plasminogen to Plasmin: Role of Plasminogen Receptors in Human Cancer. International Journal of Molecular Sciences, 15(11), 21229-21252. https://doi.org/10.3390/ijms151121229