Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors


 ,
, 

Abstract
:1. Introduction
2. Ligand-Dependent Triple-Membrane-Passing-Signal (TMPS) Mechanisms: Role of Membrane-Bound Matrix Metalloproteases (MMPs) and the A Disintegrin and Metalloproteases (ADAMs)
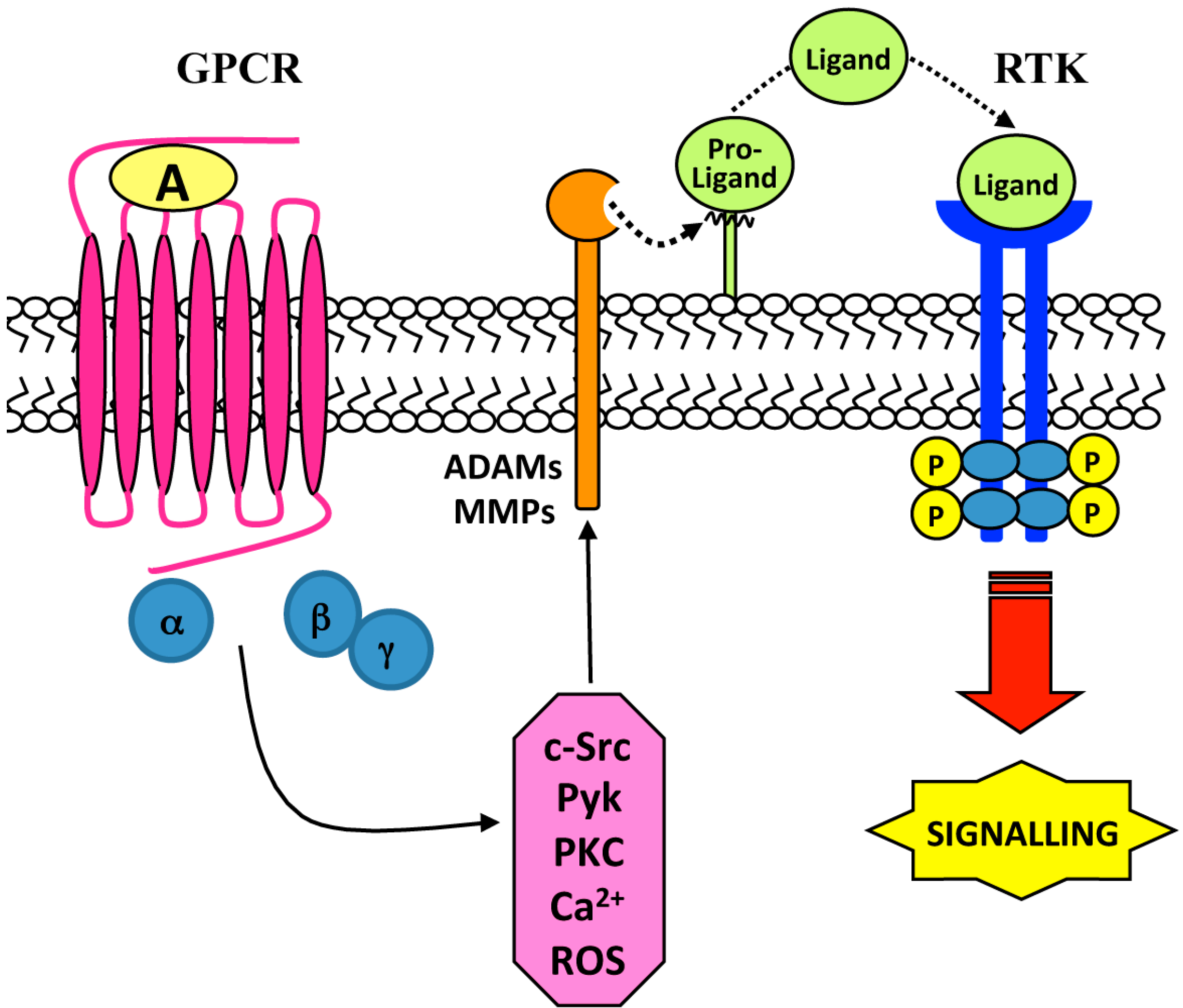

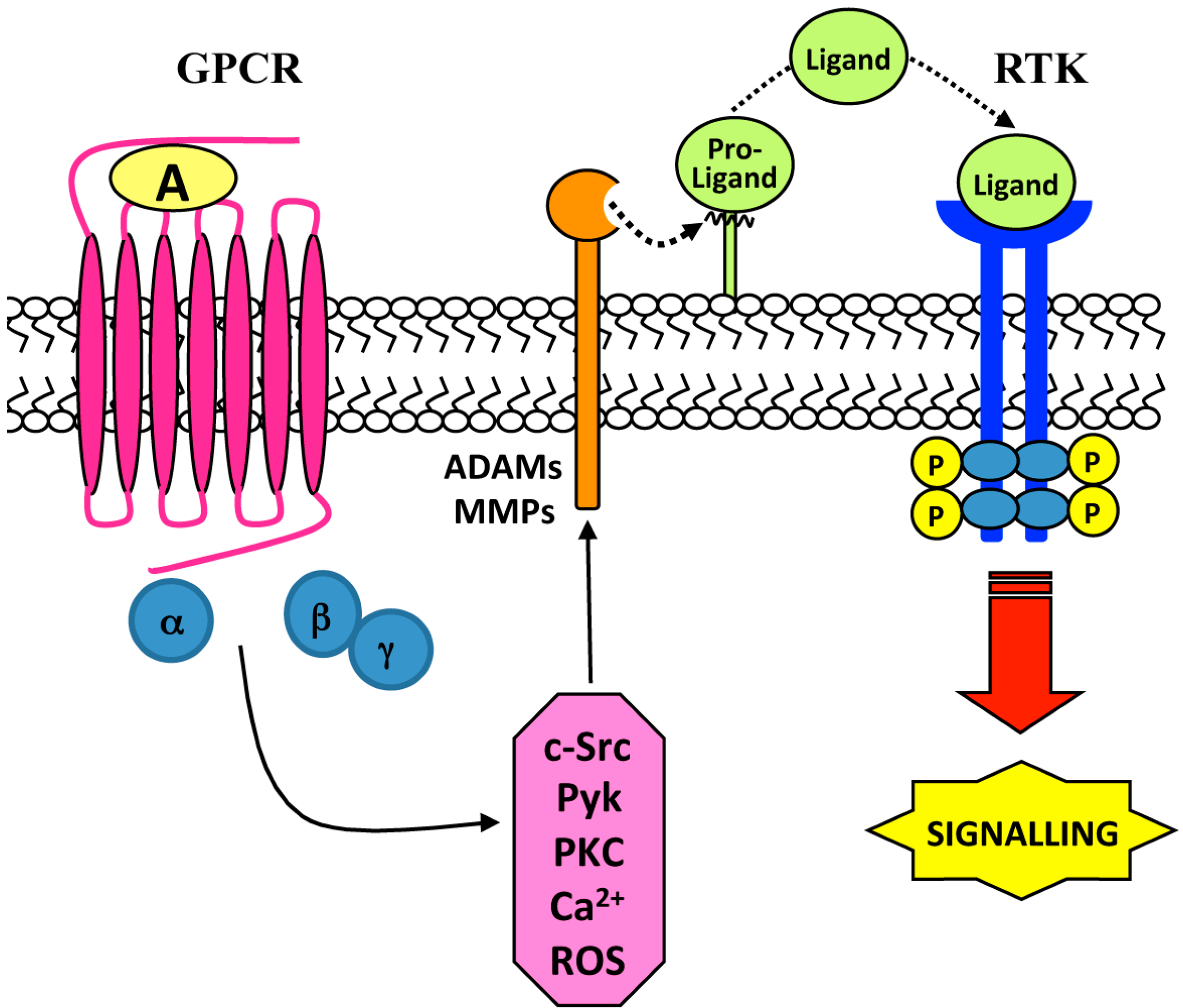{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Stimulus | GPRC | Metalloproteases | RTK | Biological Responses | Ref. |
|---|---|---|---|---|---|---|
| Pancreatic cancer cells | EGF | Neuromedin B | MMP-9 | EGFR | EGFR transactivation, cancer growth and metastatic spread | [24] |
| Isolated preovulatory ovarian follicles, Y1 adrenal cells | LH | LHRH | MMP-2–9 | EGFR | EGFR transactivation, steroidogenesis | [31] |
| Gonadrotropic cells | GnRH | GnRHR | MMP-2–9 | EGFR | EGFR transactivation, Src, Ras and ERKs activation | [32] |
| Mesenteric arteries | Phenylephrine | α1B-Adrenoreceptor | MMP-7 | EGFR | EGFR transactivation, vasoconstriction, growth | [33] |
| Gastrics epithelial cells | Histamine | H2R | MMP-1 | EGFR | EGFR transactivation, MAPK activation | [34] |
| Chondrocytes | Thrombin | PARs | MMP-13 | EGFR | EGFR transactivation, PI3K/Akt pathway and AP1 activation | [35] |
| 18Co | LPA, TNF-α | LPA1 | MMP | EGFR | EGFR transactivation, MAPK phosphorylation, COX2 expression | [36] |
| Corneal epithelial cells | LPA | LPA1 | MMP | EGFR | EGFR transactivation, ERK-Akt activation, wound healing, proliferation | [37] |
| Cardiomyocytes | Ang II | AT1 | ADAM17 | EGFR | EGFR transactivation, MAPK activation, angiogenesis | [42] |
| Kidney cancer cells, Bludder carcinoma cells, Caki2, A498, TccSup | LPA | LPA1 | ADAM10–15–17 | EGFR | EGFR transactivation, MAPK activation, tumor cell migration and invasion, TGF-β shedding | [43] |
| SCC-9 | LPA, Carbachol | LPA, AChR | ADAM17 | EGFR | EGFR transactivation, amphiregulin shedding, ERKs activation, PI3K/Akt activation, cell proliferation, migration | [44] |
| Neuroectodermal cells | Serotonin, Nor-epinephrine | 5-HT2B, α1D-Adrenoreceptor | ADAM17 | EGFR | EGFR transactivation, NADPH oxidase activation | [46] |
| Astrocytoma cells | UTP | P2Y2R | ADAM10–17 | EGFR | EGFR transactivation, amyloid precursor shedding | [47] |
| Colon cancer cells | Interleukin-8 | CXCR1, CXCR2 | ADAMs | EGFR | EGFR transactivation, MAPK activation, cell growth | [48] |
| CHO, EC-4 (TACE+/+) EC-2 (TACE ΔZn/ΔZn) | ATP | P2Y2R | ADAM17 | EGFR | EGFR transactivation | [49] |
3. Ligand-Independent Mechanisms: Role of Reactive Oxygen Species (ROS)
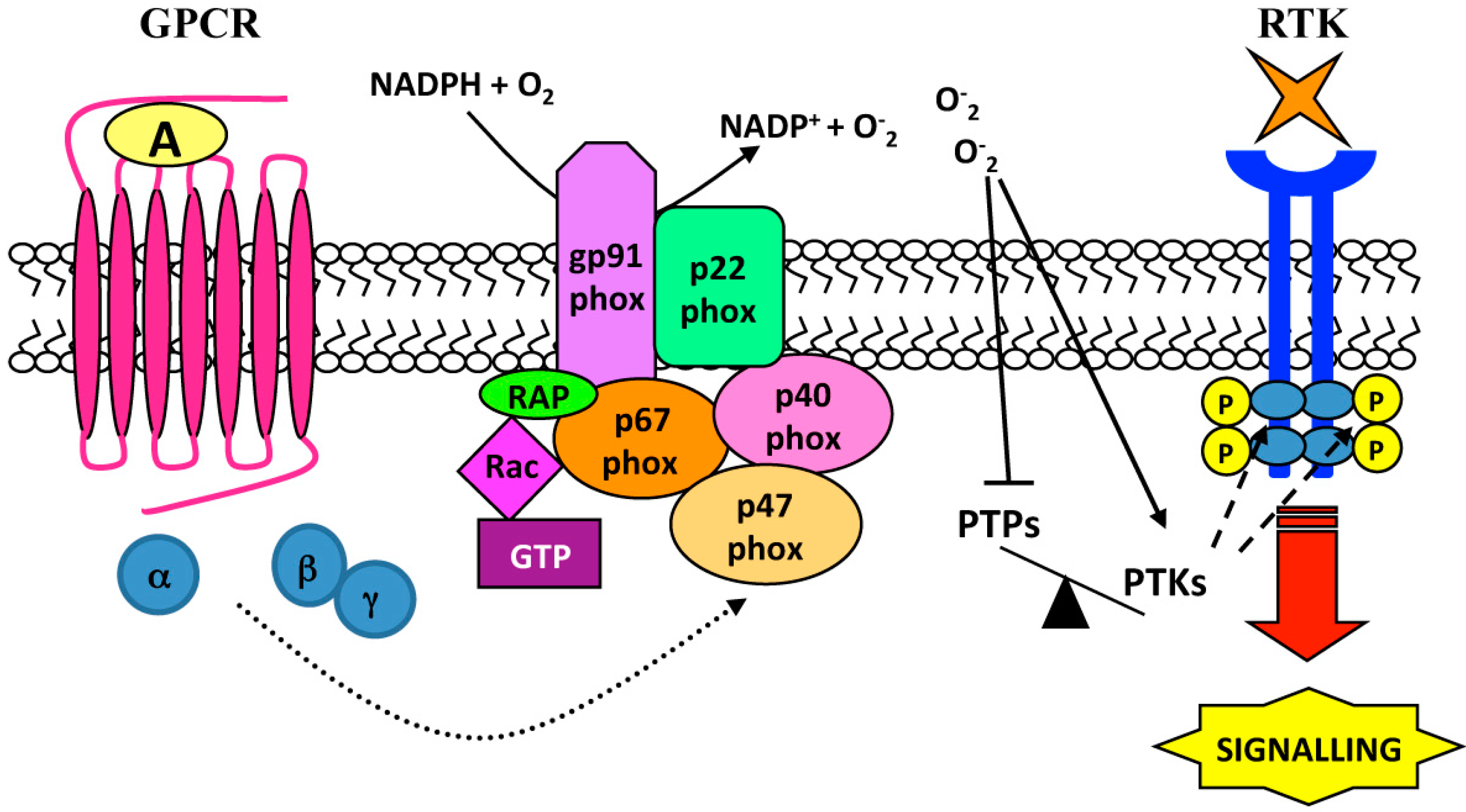
| Cell Lines | Stimulus | GPCR | Source of ROS | RTKs | Biological Responses | Ref. |
|---|---|---|---|---|---|---|
| VSMCs | Ang II | AT1 | NADPH oxidase | EGFR | EGFR transactivation, ERKs activation, growth | [62] |
| SMC | Thrombin | PARs | NOX1 | EGFR | EGFR transactivation, PI3K-Akt and ATF-1 activation, migration and proliferation, N-cadherin shedding mediated by MMP-9, ERKs activation | [64,65] |
| SH-SY5Y | 5-HT | 5-HTR | NADPH oxidase | PDGFR-β, TrkB | PDGFR-β transactivation, TrkB transactivation | [66] |
| Calu-6 | WKYMVm | FPR2 | NADPH oxidase | EGFR | EGFR transactivation, cell growth, STAT3 activation, PI3K/Akt activation | [67] |
| VSMCs | Ang II | AT1 | NADPH oxidase | EGFR | EGFR transactivation, increase of intracellular Ca2+ concentration, MAPK activation | [72] |
| DAN-G, HepG2, HuH7 | LPA, Bradykinin, Thrombin, Carbachol, Endothelin | LPA1, BDKRB1–2, PARs, mAChRs, EDNRs | NADPH oxidase | EGFR, c-Met | EGFR and c-Met transactivation, β-catenin nuclear traslocation, cell motility | [77] |
| PNT1A | WKYMVm | FPR2 | NADPH oxidase | c-Met | c-Met transactivation, cell proliferation, STAT3 activation, PI3K/Akt activation, PLCγ/PKCα activation | [79] |
| Monocytes | N-fMLP | FPR | NADPH oxidase | EGFR, TrkA | EGFR and TrkA transactivation, CD11b membrane up-regulation | [80] |
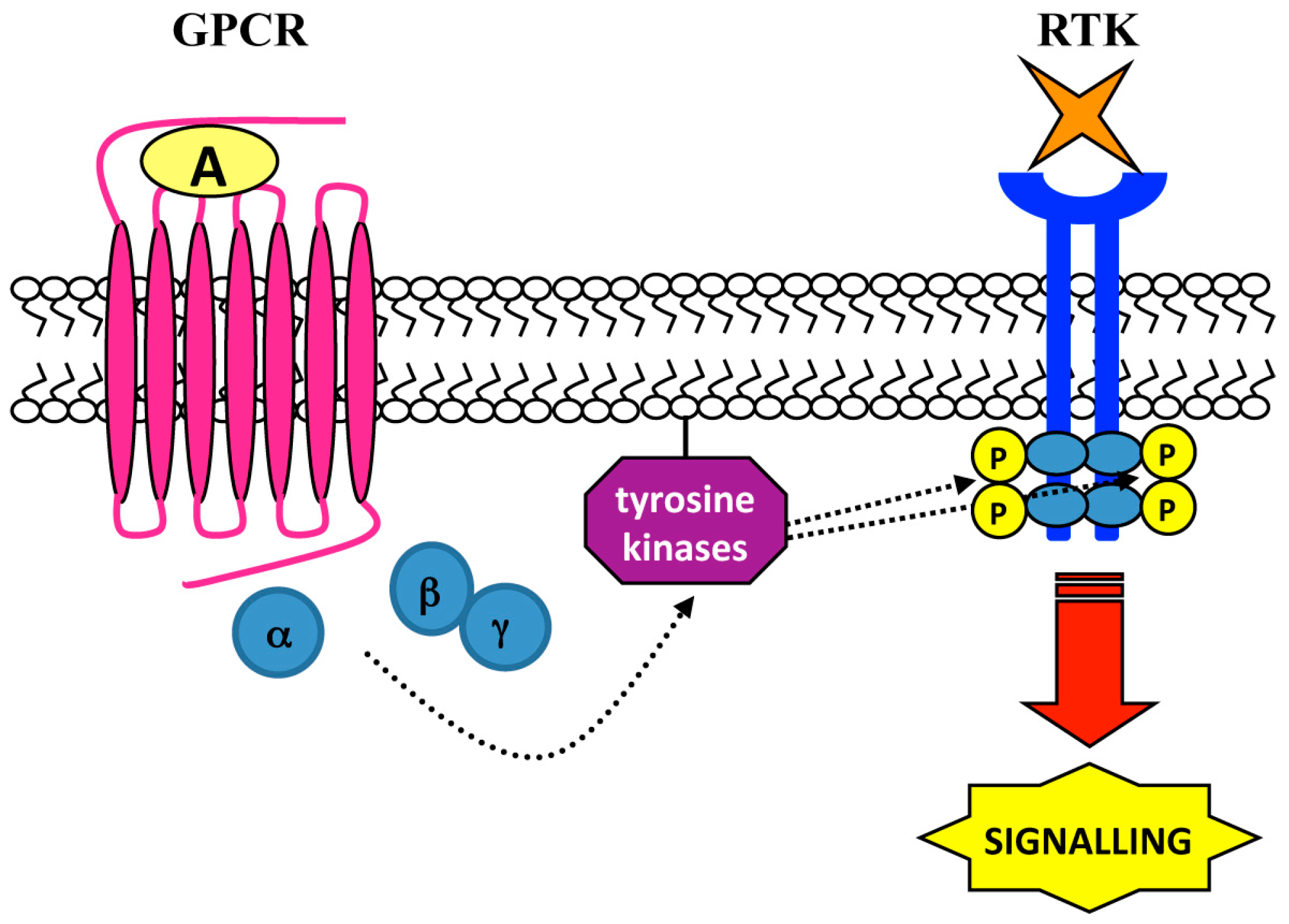
4. Ligand-Independent Mechanisms: Role of Intracellular Tyrosine Kinases
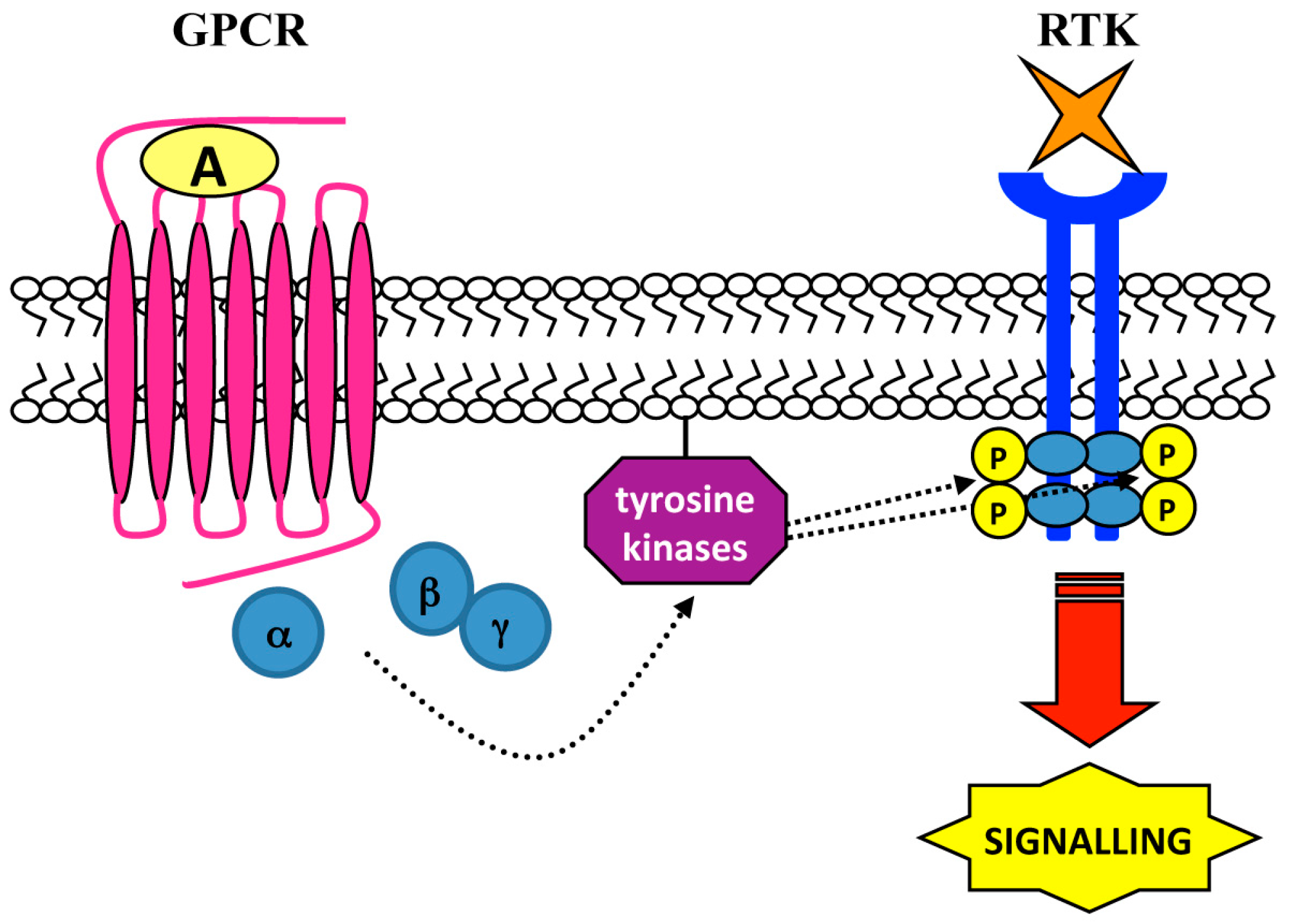
| Cell Lines | Stimulus | GPCR | Tyrosine Kinases | RTK | Biological Responses | Ref. |
|---|---|---|---|---|---|---|
| PC3 | Neurotensin | NTRs | c-Src | EGFR | EGFR transactivation, cell proliferation, DNA synthesis, STAT5-b activation | [83] |
| COS-7 | α2-AR agonists | α2-AR | c-Src | EGFR | EGFR transactivation, ERK activation | [85] |
| C9 | Ang II | AT1 | c-Src/Pyk2 | EGFR | EGFR transactivation, ERKs phosphorilation | [86] |
| HEK293 | AVP | V2R | c-Src | IGFR | IGFR transactivation, ERKs activation | [87] |
| PC12-615 | Adenosine, GCS21680 | Adenosine receptor | Fyn | TrkA | TrkA transactivation. | [91] |
| SH-SY5Y | 5-HT | 5-HT1A | c-Src | PDGFR-β | PDGFR-β transactivation | [92] |
| COS-7 | Isoproterenol | β2AR | c-Src | EGFR | EGFR transactivation, ERKs activation | [97] |
| Gastric mucosal cells | Isoproterenol | β2-AR | c-Src | EGFR | EGFR transactivation and regulation of gastric mucin secretion | [98] |
| Cardiomyocytes | Endothelin-1 | ET-1 | Pyk2 | EGFR | EGFR transactivation, MAPK activation | [103] |
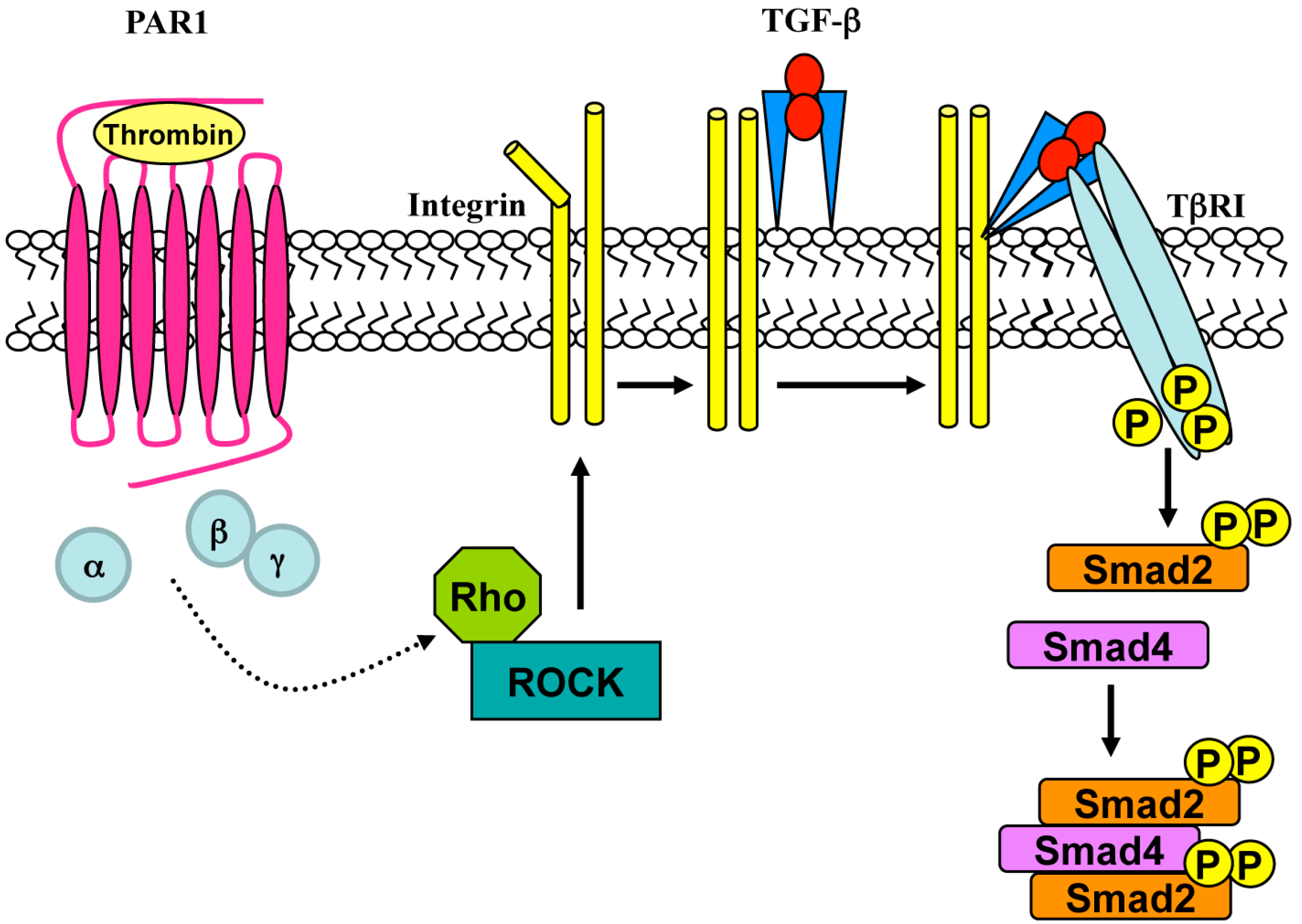
5. Role of G Proteins and β-Arrestins in Transactivation
6. GPCR-Mediated Transactivation of Serine/Threonine kinase (S/TK) Receptors and Toll-Like Receptors (TLRs)

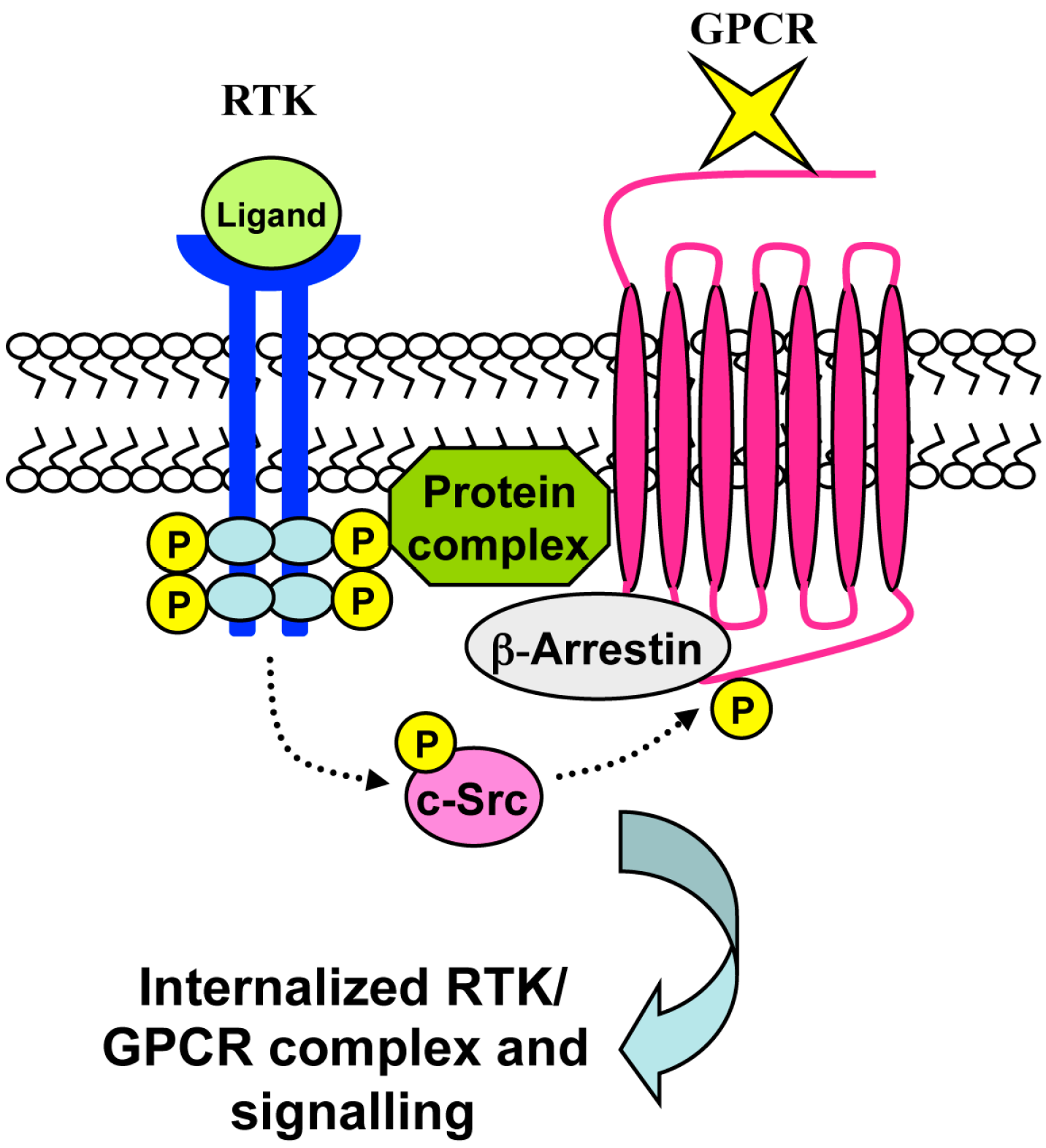
7. RTK-Mediated GPCR Transactivation
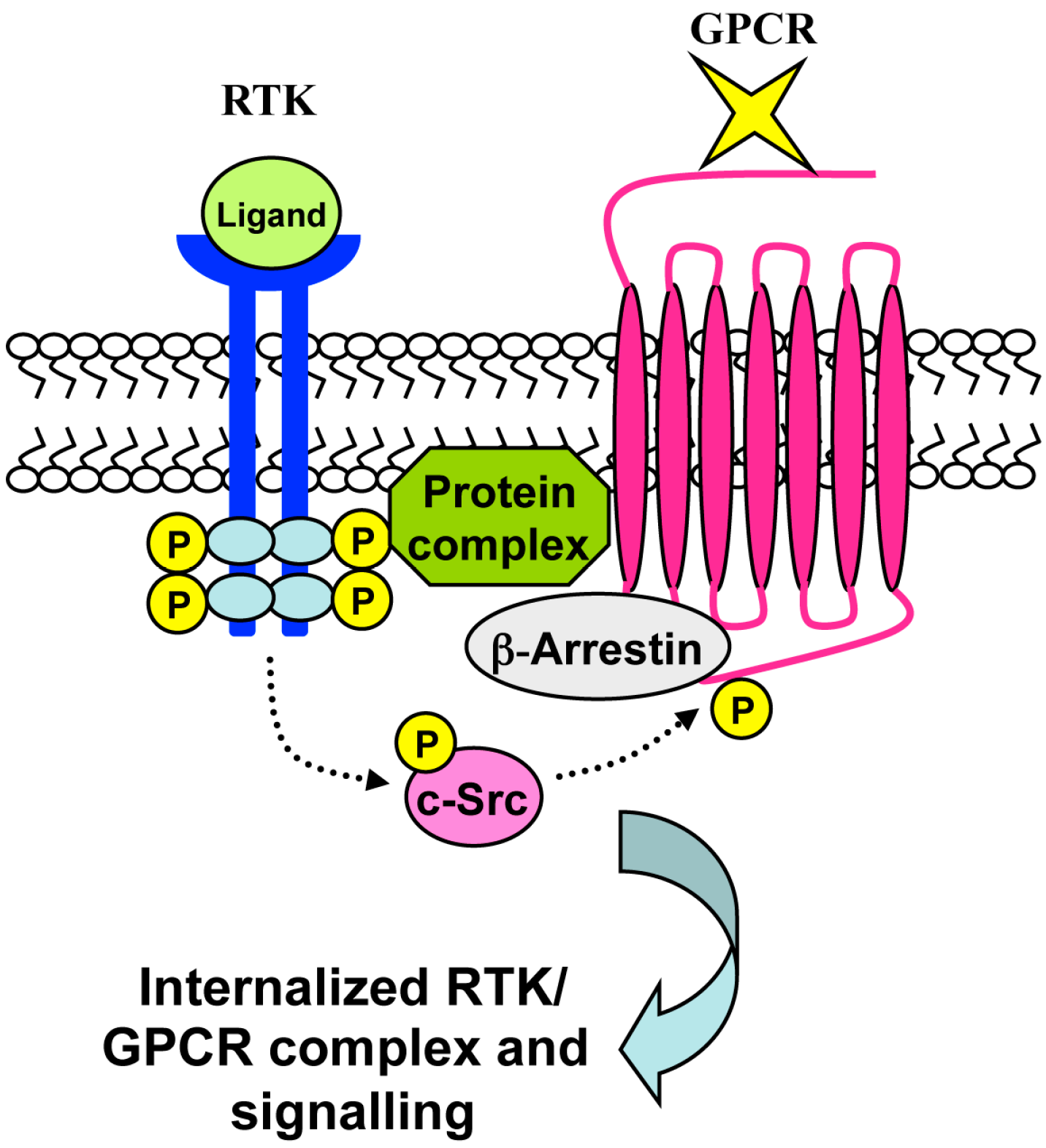
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamato, D.; Burch, M.L.; Osman, N.; Zheng, W.; Little, P.J. Therapeutic implications of endothelin and thrombin G-protein-coupled receptor transactivation of tyrosine and serine/threonine kinase cell surface receptors. J. Pharm. Pharmacol. 2013, 65, 465–473. [Google Scholar]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar]
- De Caestecker, M.P.; Parks, W.T.; Frank, C.J.; Castagnino, P.; Bottaro, D.P.; Roberts, A.B.; Lechleider, R.J. Smad2 transduces common signals from receptor serine-threonine and tyrosine kinases. Genes Dev. 1998, 12, 1587–1592. [Google Scholar]
- Neves, S.R.; Ram, P.T.; Lyengar, R. G protein pathways. Science 2002, 296, 1636–1639. [Google Scholar]
- George, A.J.; Hannan, R.D.; Thomas, W.G. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013, 280, 5258–5268. [Google Scholar]
- Luttrell, L.M.; Lefkowitz, R.J. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115, 455–465. [Google Scholar]
- García-Sáinz, J.A.; Romero-Ávila, M.T.; Medina, L.D.C. Dissecting how receptor tyrosine kinases modulate G protein-coupled receptor function. Eur. J. Pharmacol. 2010, 648, 1–5. [Google Scholar]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 6565, 557–560. [Google Scholar]
- Leserer, M.; Gschwind, A.; Ullrich, A. Epidermal growth factor receptor signal transactivation. IUBMB Life 2000, 49, 405–409. [Google Scholar]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 8, 443–450. [Google Scholar]
- Burch, M.L.; Osman, N.; Getachew, R.; Al-Aryahi, S.; Poronnik, P.; Zheng, W.; Hill, M.A.; Little, P.J. G protein coupled receptor transactivation: Extending the paradigm to include serine/threonine kinase receptors. Int. J. Biochem. Cell Biol. 2012, 44, 722–727. [Google Scholar]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptortransactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 6764, 884–888. [Google Scholar]
- Andreev, J.; Galisteo, M.L.; Kranenburg, O.; Logan, S.K.; Chiu, E.S.; Okigaki, M.; Cary, L.A.; Moolenaar, W.H.; Schlessinger, J. Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem. 2001, 276, 20130–20135. [Google Scholar]
- Esposito, G.; Perrino, C.; Cannavo, A.; Schiattarella, G.G.; Borgia, F.; Sannino, A.; Pironti, G.; Gargiulo, G.; di Serafino, L.; Franzone, A.; et al. EGFR trans-activation by urotensin II receptor is mediated by β-arrestin recruitment and confers cardioprotection in pressure overload-induced cardiac hypertrophy. Basic Res. Cardiol. 2011, 106, 577–589. [Google Scholar]
- Gesty-Palmer, D.; Yuan, L.; Martin, B.; Wood, W.H.; Lee, M.H.; Janech, M.G.; Tsoi, L.C.; Zheng, W.J.; Luttrell, L.M.; Maudsley, S. β-Arrestin-selective G protein-coupled receptor agonists engender unique biological efficacy in vivo. Mol. Endocrinol. 2013, 27, 296–314. [Google Scholar]
- El-Shewy, H.M.; Abdel-Samie, S.A.; Al Qalam, A.M.; Lee, M.H.; Kitatani, K.; Anelli, V.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Phospholipase C and protein kinase C-β2 mediate insulin-like growth factor II-dependent sphingosine kinase 1 activation. Mol. Endocrinol. 2011, 25, 2144–2156. [Google Scholar]
- Chen, J.; Chen, J.K.; Harris, R.C. Angiotensin II induces epithelial-to-mesenchymal transition in renal epithelial cells through reactive oxygen species/Src/caveolin-mediated activation of an epidermal growth factor receptor-extracellular signal-regulated kinase signaling pathway. Mol. Cell. Biol. 2012, 32, 981–991. [Google Scholar]
- Tilley, D.G.; Kim, I.M.; Patel, P.A.; Violin, J.D.; Rockman, H.A. Arrestin mediates β1-adrenergic receptor epidermal growth factor receptor interaction and downstream signaling. J. Biol. Chem. 2009, 284, 20375–20386. [Google Scholar]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar]
- Uchiyama-Tanaka, Y.; Matsubara, H.; Mori, Y.; Kosaki, A.; Kishimoto, N.; Amano, K.; Higashiyama, S.; Iwasaka, T. Involvement of HB-EGF and EGF receptor transactivation in TGF-β-mediated fibronectin expression in mesangial cells. Kidney Int. 2002, 62, 799–808. [Google Scholar]
- Kleine-Brueggeney, M.; Gradinaru, I.; Babaeva, E.; Schwinn, D.A.; Oganesian, A. Alpha1a-Adrenoceptor genetic variant induces cardiomyoblast-to-fibroblast-like cell transition via distinct signaling Pathways. Cell. Signal. 2014, 26, 1985–1997. [Google Scholar]
- Gilmour, A.M.; Abdulkhalek, S.; Cheng, T.S.; Alghamdi, F.; Jayanth, P.; O'Shea, L.K.; Geen, O.; Arvizu, L.A.; Szewczuk, M.R. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell. Signal. 2013, 25, 2587–2603. [Google Scholar]
- Nagareddy, P.R.; Chow, F.L.; Hao, L.; Wang, X.; Nishimura, T.; MacLeod, K.M.; McNeill, J.H.; Fernandez-Patron, C. Maintenance of adrenergic vascular tone by MMP transactivation of the EGFR requires PI3K and mitochondrial ATP synthesis. Cardiovasc. Res. 2009, 84, 368–377. [Google Scholar]
- Gschwind, A.; Prenzel, N.; Ullrich, A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002, 62, 6329–6336. [Google Scholar]
- Tanimoto, T.; Jin, Z.G.; Berk, B.C. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS). J. Biol. Chem. 2002, 277, 42997–43001. [Google Scholar]
- Thuringer, D.; Maulon, L.; Frelin, C. Rapid transactivation of the vascular endothelial growth factor receptor KDR/Flk-1 by the bradykinin B2 receptor contributes to endothelial nitric-oxide synthase activation in cardiac capillary endothelial cells. J. Biol. Chem. 2002, 277, 2028–2032. [Google Scholar]
- Tsai, C.L.; Chen, W.C.; Lee, I.T.; Chi, P.L.; Cheng, S.E.; Yang, C.M. c-Src-dependent transactivation of PDGFR contributes to TNF-α-induced MMP-9 expression and functional impairment in osteoblasts. Bone 2014, 60, 186–197. [Google Scholar]
- Yang, C.M.; Hsieh, H.L.; Yao, C.C.; Hsiao, L.D.; Tseng, C.P.; Wu, C.B. Protein kinase C-δ transactivates platelet-derived growth factor receptor-α in mechanical strain-induced collagenase 3 (matrix metalloproteinase-13) expression by osteoblast-like cells. J. Biol. Chem. 2009, 284, 26040–26050. [Google Scholar]
- Carbajal, L.; Biswas, A.; Niswander, L.M.; Prizant, H.; Hammes, S.R. GPCR/EGFR cross talk is conserved in gonadal and adrenal steroidogenesis but is uniquely regulated by matrix metalloproteinases 2 and 9 in the ovary. Mol. Endocrinol. 2011, 25, 1055–1065. [Google Scholar]
- Roelle, S.; Grosse, R.; Aigner, A.; Krell, H.W.; Czubayko, F.; Gudermann, T. Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J. Biol. Chem. 2003, 278, 47307–47318. [Google Scholar]
- Hao, L.; Du, M.; Lopez-Campistrous, A.; Fernandez-Patron, C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ. Res. 2004, 94, 68–76. [Google Scholar]
- Anchaa, H.R.; Kurella, R.R.; Stewart, C.A.; Damerac, G.; Ceresa, B.P.; Harty, R.F. Histamine stimulation of MMP-1 (collagenase-1) secretion and gene expression in gastric epithelial cells: Role of EGFR transactivation and the MAP kinase pathway. Int. J. Biochem. CellBiol. 2007, 39, 2143–2152. [Google Scholar]
- Huang, C.-Y.; Lin, H.-J.; Chen, H.-S.; Cheng, S.-Y.; Hsu, H.-C.; Tang, C.-H. Thrombin promotes matrix metalloproteinase-13 expression through the PKCδ/c-Src/EGFR/PI3K/Akt/AP-1 signaling pathway in human chondrocytes. Mediat. Inflamm. 2013, 2013, 326041. [Google Scholar]
- Yoo, J.; Rodriguez Perez, C.E.; Nie, W.; Sinnett-Smith, J.; Rozengurt, E. TNF-α and LPA promote synergistic expression of COX-2 in human colonic myofibroblasts: Role of LPA-mediated transactivation of upregulated EGFR. BMC Gastroenterol. 2013, 13, 90. [Google Scholar]
- Xu, K.P.; Yin, J.; Yu, F.S. Lysophosphatidic acid promoting corneal epithelial wound healing by transactivation of epidermal growth factor receptor. Investig. Ophthalmol. Vis. Sci. 2007, 48, 636–643. [Google Scholar]
- Klein, T.; Bischoff, R. Active metalloproteases of the a disintegrin and metalloprotease (ADAM) family: Biological function and structure. J. Proteome Res. 2011, 10, 17–33. [Google Scholar]
- Murphy, G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Semin. Cell Dev. Biol. 2009, 20, 138–145. [Google Scholar]
- Dang, M.; Armbruster, N.; Miller, M.A.; Cermeno, E.; Hartmann, M.; Bell, G.W.; Root, D.E.; Lauffenburger, D.A.; Lodish, H.F.; Herrlich, A. Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 9776–9781. [Google Scholar]
- Ohtsu, H.; Dempsey, P.J.; Educhi, S. ADAMs as mediators of EGF receptor transactivation by Gprotein-coupled receptors. Am. J. Physiol. Cell Physiol. 2006, 291, C1–C10. [Google Scholar]
- Thomas, W.G.; Brandenburger, Y.; Autelitano, D.J.; Pham, T.; Qian, H.; Hannan, R.D. Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ. Res. 2002, 90, 135–142. [Google Scholar]
- Schafer, B.; Gschwind, A.; Ullrich, A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene 2004, 23, 991–999. [Google Scholar]
- Gschwind, A.; Hart, S.; Fischer, O.M.; Ullrich, A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003, 22, 2411–2421. [Google Scholar]
- Chalaris, A.; Rabe, B.; Paliga, K.; Lange, H.; Laskay, T.; Fielding, C.A.; Jones, S.A.; Rose-John, S.; Scheller, J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 2007, 110, 1748–1755. [Google Scholar]
- Pietri, M.; Schneider, B.; Mouillet-Richard, S.; Ermonval, M.; Mutel, V.; Launay, J.M.; Kellermann, O. Reactive oxygen species-dependent TNF-α converting enzyme activation through stimulation of 5-HT2B and α1D autoreceptors in neuronal cells. FASEB J. 2005, 19, 1078–1087. [Google Scholar]
- Camden, J.M.; Schrader, A.M.; Camden, R.E.; Gonzalez, F.A.; Erb, L.; Seye, C.I.; Weisman, G.A. P2Y2 nucleotide receptors enhance α-secretase-dependent amyloid precursor protein processing. J. Biol. Chem. 2005, 280, 18696–18702. [Google Scholar]
- Itoh, Y.; Joh, T.; Tanida, S.; Sasaki, M.; Kataoka, H.; Itoh, K.; Oshima, T.; Ogasawara, N.; Togawa, S.; Wada, T.; et al. IL-8 promotes cell proliferation and migration through metalloproteinase-cleavage proHB-EGF in human colon carcinoma cells. Cytokine 2005, 29, 275–282. [Google Scholar]
- Myers, T.J.; Brennaman, L.H.; Stevenson, M.; Higashiyama, S.; Russell, W.E.; Lee, D.C.; Sunnarborg, S.W. Mitochondrial reactive oxygen species mediate GPCR-induced TACE/ADAM17-dependent transforming growth factor-α shedding. Mol. Biol. Cell 2009, 24, 5236–5249. [Google Scholar]
- Lee, S.J.; Seo, K.W.; Yun, M.R.; Bae, S.S.; Lee, W.S.; Hong, K.W.; Kim, C.D. 4-Hydroxynonenal enhances MMP-2 production in vascular smooth muscle cells via mitochondrial ROS-mediated activation of the Akt/NF-κB signaling pathways. Free Radic. Biol. Med. 2008, 45, 1487–1492. [Google Scholar]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Prenzel, N.; Ullrich, A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol. Cell. Biol. 2004, 24, 5172–5183. [Google Scholar]
- Frank, G.D.; Eguchi, S. Activation of tyrosine kinases by reactive oxygen species in vascular smooth muscle cells: Significance and involvement of EGF receptor transactivation by angiotensin II. Antioxid. Redox Signal. 2003, 5, 771–780. [Google Scholar]
- Adrain, C.; Freeman, M. Regulation of receptor tyrosine kinase ligand processing. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett. 2000, 476, 52–54. [Google Scholar]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Viña, J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: Role of p38 MAPKinase and E3 ubiquitin ligases. PLoS One 2012, 7, e46668. [Google Scholar]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. Signal. 2000. pe1. [Google Scholar]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar]
- Brandes, R.P.; Weissmann, N.; Schröder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar]
- Lambeth, J.D. Nox/Duox family of nicotinamide adenine dinucleotide (phosphate) oxidases. Curr. Opin. Hematol. 2002, 9, 11–17. [Google Scholar]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar]
- Frank, G.D.; Eguchi, S.; Inagami, T.; Motley, E.D. N-Acetylcysteine inhibits angiotensin II-mediated activation of extracellular signal-regulated kinase and epidermal growth factor receptor. Biochem. Biophys. Res. Commun. 2001, 280, 1116–1119. [Google Scholar]
- Frank, G.D.; Mifune, M.; Inagami, T.; Ohba, M.; Sasaki, T.; Higashiyama, S.; Dempsey, P.J.; Eguchi, S. Distinct mechanisms of receptor and nonreceptor tyrosine kinase activation by reactive oxygen species in vascular smooth muscle cells: Role of metalloprotease and protein kinase C-δ. Mol. Cell. Biol. 2003, 23, 1581–1589. [Google Scholar]
- Miller, F.J., Jr.; Chu, X.; Stanic, B.; Tian, X.; Sharma, R.V.; Davisson, R.L.; Lamb, F.S. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid. Redox Signal. 2010, 12, 583–593. [Google Scholar]
- Jagadeesha, D.K.; Takapoo, M.; Banfi, B.; Bhalla, R.C.; Miller, F.J., Jr. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc. Res. 2012, 93, 406–413. [Google Scholar]
- Kruk, J.S.; Vasefi, M.S.; Heikkila, J.J.; Beazely, M.A. Reactive oxygen species are required for 5-HT-induced transactivation of neuronal platelet-derived growth factor and TrkB receptors, but not for ERK1/2 activation. PLoS One 2013, 8, e77027. [Google Scholar]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar]
- Seshiah, P.N.; Weber, D.S.; Rocic, P.; Valppu, L.; Taniyama, Y.; Griendling, K.K. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ. Res. 2002, 91, 406–413. [Google Scholar]
- Chen, K.; Vita, J.A.; Berk, B.C.; Keaney, J.F., Jr. c-Jun N-terminal kinase activation by hydrogen peroxide in endothelial cells involves SRC-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 2001, 276, 16045–16050. [Google Scholar]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 2007, 213, 589–602. [Google Scholar]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar]
- Eguchi, S.; Numaguchi, K.; Iwasaki, H.; Matsumoto, T.; Yamakawa, T.; Utsunomiya, H.; Motley, E.D.; Kawakatsu, H.; Owada, K.M.; Hirata, Y.; et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J. Biol. Chem. 1998, 273, 8890–8896. [Google Scholar]
- Frank, G.D.; Motley, E.D.; Inagami, T.; Eguchi, S. PYK2/CAKβ represents a redox-sensitive tyrosine kinase in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2000, 270, 761–765. [Google Scholar]
- Lee, S.H.; Na, S.I.; Heo, J.S.; Kim, M.H.; Kim, Y.H.; Lee, M.Y.; Kim, S.H.; Lee, Y.J.; Han, H.J. Arachidonic acid release by H2O2 mediated proliferation of mouse embryonic stem cells: Involvement of Ca2+/PKC and MAPKs-induced EGFR transactivation. J. Cell. Biochem. 2009, 106, 787–797. [Google Scholar]
- Iaccio, A.; Collinet, C.; Gesualdi, N.M.; Ammendola, R. Protein kinase C-α and -δ are required for NADPH oxidase activation in WKYMVm-stimulated IMR90 human fibroblasts. Arch. Biochem. Biophys. 2007, 459, 288–294. [Google Scholar]
- Saito, S.; Frank, G.D.; Mifune, M.; Ohba, M.; Utsunomiya, H.; Motley, E.D.; Inagami, T.; Eguchi, S. Ligand-independent trans-activation of the platelet-derived growth factor receptor by reactive oxygen species requires protein kinase C-δ and c-Src. J. Biol. Chem. 2002, 277, 44695–44700. [Google Scholar]
- Fischer, O.M.; Giordano, S.; Comoglio, P.M.; Ullrich, A. Reactive oxygen species mediate Met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J. Biol. Chem. 2004, 279, 28970–28978. [Google Scholar]
- Pavone, L.M.; Cattaneo, F.; Rea, S.; de Pasquale, V.; Spina, A.; Sauchelli, E.; Mastellone, V.; Ammendola, R. Intracellular signaling cascades triggered by the NK1 fragment of hepatocyte growth factor in human prostate epithelial cell line PNT1A. Cell Signal. 2011, 23, 1961–1971. [Google Scholar]
- Cattaneo, F.; Parisi, M.; Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013, 587, 1536–1542. [Google Scholar]
- El Zein, N.; D’Hondt, S.; Sariban, E. Crosstalks between the receptors tyrosine kinase EGFR and TrkA and the GPCR, FPR, in human monocytes are essential for receptors-mediated cell activation. Cell Signal. 2010, 22, 1437–1447. [Google Scholar]
- Luttrell, D.K.; Luttrell, L.M. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene 2004, 23, 7969–7978. [Google Scholar]
- Fan, G.; Shumay, E.; Malbon, C.C.; Wang, H. c-Src tyrosine kinase binds the β2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2, and mediates agonist-induced receptor desensitization. J. Biol. Chem. 2001, 276, 13240–13247. [Google Scholar]
- Amorino, G.P.; Deeble, P.D.; Parsons, S.J. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene 2007, 26, 745–756. [Google Scholar]
- Rodrìguez-Fernàndez, J.L.; Rozengurt, E. Bombesin, bradykinin, vasopressin, and phorbol esters rapidly and transiently activate Src family tyrosine kinases in Swiss 3T3 cel. Dissociation from tyrosine phosphorylation of p125 focal adhesion kinase. J. Biol. Chem. 1996, 271, 27895–27901. [Google Scholar]
- Pierce, K.L.; Tohgo, A.; Ahn, S.; Field, M.E.; Luttrell, L.M.; Lefkowitz, R.J. Epidermal growth factor (EGF) receptor dependent ERK activation by G protein-coupled receptors: A co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J. Biol. Chem. 2001, 276, 23155–23160. [Google Scholar]
- Shah, B.H.; Catt, K.J. Calcium-independent activation of extracellularly regulated kinases 1 and 2 by angiotensin II in hepatic C9 cells: Roles of protein kinase Cδ, Src/proline-rich tyrosine kinase 2, and epidermal growth factor receptor trans-activation. Mol. Pharmacol. 2002, 61, 343–351. [Google Scholar]
- Oligny-Longpré, G.; Corbani, M.; Zhou, J.; Hogue, M.; Guillon, G.; Bouvier, M. Engagement of β-arrestin by transactivated insulin-like growth factor receptor is needed for V2 vasopressin receptor-stimulated ERK1/2 activation. Proc. Natl. Acad. Sci. USA 2012, 109, E1028–E1037. [Google Scholar]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar]
- Rajagopal, R.; Chao, M.V. A role for Fyn in Trk receptor transactivation by G-protein-coupled receptor signaling. Mol. Cell. Neurosci. 2006, 33, 36–46. [Google Scholar]
- Kruk, J.S.; Vasefi, M.S.; Liu, H.; Heikkila, J.J.; Beazely, M.A. 5-HT1A receptors transactivate the platelet-derived growth factor receptor type β in neuronal cells. Cell Signal. 2013, 25, 133–143. [Google Scholar]
- Waters, C.M.; Connell, M.C.; Pyne, S.; Pyne, N.J. c-Src is involved in regulating signal transmission from PDGFβ receptor-GPCR(s) complexes in mammalian cells. Cell Signal. 2005, 17, 263–277. [Google Scholar]
- Lei, H.; Kazlauskas, A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor α. Mol. Cell. Biol. 2014, 34, 110–122. [Google Scholar]
- Waters, C.; Sambi, B.; Kong, K.C.; Thompson, D.; Pitson, S.M.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate and platelet-derived growth factor (PDGF) act via PDGFβ receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J. Biol. Chem. 2003, 278, 6282–6290. [Google Scholar]
- Long, J.S.; Natarajan, V.; Tigyi, G.; Pyne, S.; Pyne, N.J. The functional PDGFβ receptor-S1P1 receptor signalling complex is involved in regulating migration of mouse embryonic fibroblasts in response to platelet derived growth factor. Prostaglandins Other Lipid Mediat. 2006, 80, 74–80. [Google Scholar]
- Maudsley, S.; Pierce, K.L.; Zamah, A.M.; Miller, W.E.; Ahn, S.; Daaka, Y.; Lefkowitz, R.J.; Luttrell, L.M. The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J. Biol. Chem. 2000, 275, 9572–9580. [Google Scholar]
- Slomiany, B.L.; Slomiany, A. Gastric mucin secretion in response to β-adrenergic G-protein-coupled receptor activation is mediated by Src kinase-dependent epidermal growth factor receptor transactivation. J. Physiol. Pharmacol. 2005, 56, 247–258. [Google Scholar]
- Delcourt, N.; Thouvenot, E.; Chanrion, B.; Galéotti, N.; Jouin, P.; Bockaert, J.; Marin, P. PACAP type 1 receptor transactivation is essential for IGF-1 receptor signalling and anti-apoptotic activity in neurones. EMBO J. 2007, 26, 1542–1551. [Google Scholar]
- Moughal, N.A.; Waters, C.; Sambi, B.; Pyne, S.; Pyne, N.J. Nerve growth factor signalling involves interaction between the TrkA receptor and LPA receptor 1 systems: Nuclear translocation of the LPA receptor 1 and TrkA receptors in pheochromocytoma 12 cells. Cell Signal. 2004, 16, 127–136. [Google Scholar]
- Bergelin, N.; Löf, C.; Balthasar, S.; Kalhori, V.; Törnquist, K. S1P1 and VEGFR-2 form a signalling complex with extracellularly regulated kinase 1/2 and protein kinase C-α regulating ML-1 thyroid carcinoma cell migration. Endocrinology 2010, 151, 2994–3005. [Google Scholar]
- Wetzker, R.; Böhmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 2003, 4, 651–657. [Google Scholar]
- Kodama, H.; Fukuda, K.; Takahashi, T.; Sano, M.; Kato, T.; Tahara, S.; Hakuno, D.; Sato, T.; Manabe, T.; Konishi, F.; et al. Role of EGF receptor and Pyk2 in endothelin-1-induced ERK activation in rat cardiomyocytes. J. Mol. Cell. Cardiol. 2002, 34, 139–150. [Google Scholar]
- Akekawatchai, C.; Holland, J.D.; Kochetkova, M.; Wallace, J.C.; McColl, S.R. Transactivation of CXCR4 by the insulin-like growth factor-1receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J. Biol. Chem. 2005, 280, 39701–39708. [Google Scholar]
- Shan, D.; Chen, L.; Wang, D.; Tan, Y.C.; Gu, J.L.; Huang, X.Y. The G protein Gα13 is required for growth factor-induced cell migration. Dev. Cell 2006, 10, 707–718. [Google Scholar]
- Rakhit, S.; Pyne, S.; Pyne, N.J. Nerve growth factor stimulation of p42/p44 mitogen activated protein kinase in PC12 cells: Role of Gi/o, G protein coupled receptor kinase 2, b-arrestin I, and endocytic processing. Mol. Pharmacol. 2001, 60, 63–70. [Google Scholar]
- Dalle, S.; Imamura, T.; Rose, D.W.; Worrall, D.S.; Ugi, S.; Hupfeld, C.J.; Olefsky, J.M. Insulin induces heterologous desensitization of G protein-coupled receptor and insulin-like growth factor I signalling by downregulating β-arrestin-1. Mol. Cell. Biol. 2002, 22, 6272–6285. [Google Scholar]
- Gschwind, A.; Zwick, E.; Prenzel, N.; Leserer, M.; Ullrich, A. Cell communication networks: Epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 2001, 20, 1594–1600. [Google Scholar]
- Noma, T.; Lemaire, A.; Naga Prasad, S.V.; Barki-Harrington, L.; Tilley, D.G.; Chen, J.; le Corvoisier, P.; Violin, J.D.; Wei, H.; Lefkowitz, R.J.; et al. β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig. 2007, 117, 2445–2458. [Google Scholar]
- Zajac, M.; Law, J.; Cvetkovic, D.D.; Pampillo, M.; McColl, L.; Pape, C.; di Guglielmo, G.M.; Postovit, L.M.; Babwah, A.V.; Bhattacharya, M. GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS One 2011. [Google Scholar] [CrossRef]
- Zheng, H.; Worrall, C.; Shen, H.; Issad, T.; Seregard, S.; Girnita, A.; Girnita, L. Selective recruitment of G protein-coupled receptor kinases (GRKs) controls signaling of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 7055–7060. [Google Scholar]
- Spartà, A.; Baiula, M.; Campbell, G.; Spampinato, S. β-Arrestin 2-mediated heterologous desensitization of IGF-IR by prolonged exposure of SH-SY5Y neuroblastoma cells to a mu opioid agonist. FEBS Lett. 2010, 584, 3580–3586. [Google Scholar]
- Ryu, J.M.; Baek, Y.B.; Shin, M.S.; Park, J.H.; Park, S.H.; Lee, J.H.; Han, H.J. Sphingosine-1-phosphate-induced Flk-1 transactivation stimulates mouse embryonic stem cell proliferation through S1P1/S1P3-dependent β-arrestin/c-Src pathways. Stem Cell Res. 2014, 12, 69–85. [Google Scholar]
- Little, P.J.; Burch, M.L.; Al-Aryahi, S.; Zheng, W. The paradigm of G protein receptor transactivation: A mechanistic definition and novel example. Sci. World J. 2011, 11, 709–714. [Google Scholar]
- Burch, M.L.; Ballinger, M.L.; Yang, S.N.; Getachew, R.; Itman, C.; Loveland, K.; Osman, N.; Little, P.J. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor β type I receptor. J. Biol. Chem. 2010, 285, 26798–26805. [Google Scholar]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces αvβ6 integrin-mediated TGF-β activation via the LPA2 receptor and the small G protein Gαq. Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances αvβ6 integrin-dependent TGF-β activation and promotes acute lung injury. J. Clin. Investig. 2006, 116, 1606–1614. [Google Scholar]
- Burch, M.L.; Getachew, R.; Osman, N.; Febbraio, M.A.; Little, P.J. Thrombin-mediated proteoglycan synthesis utilizes both protein-tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J. Biol. Chem. 2013, 288, 7410–7419. [Google Scholar]
- Belmadani, S.; Zerfaoui, M.; Boulares, H.A.; Palen, D.I.; Matrougui, K. Microvessel vascular smooth muscle cells contribute to collagen type I deposition through ERK1/2 MAP kinase, αvβ3-integrin, and TGF-β1 in response to ANG II and high glucose. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H69–H76. [Google Scholar]
- Liu, Y.; Ren, W.; Warburton, R.; Toksoz, D.; Fanburg, B.L. Serotonin induces Rho/ROCK-dependentactivation of Smads 1/5/8 in pulmonary artery smooth muscle cells. FASEB J. 2009, 23, 2299–2306. [Google Scholar]
- Shi, G.X.; Harrison, K.; Han, S.B.; Moratz, C.; Kehrl, J.H. Toll-like receptor signaling alters the expression of regulator of G protein signaling proteins in dendritic cells: Implications for G protein-coupled receptor signalling. J. Immunol. 2004, 172, 5175–5184. [Google Scholar]
- Abdulkhalek, S.; Guo, M.; Amith, S.R.; Jayanth, P.; Szewczuk, M.R. G-protein coupled receptor agonists mediate Neu1 sialidase and matrix metalloproteinase-9 cross-talk to induce transactivation of TOLL-like receptors and cellular signaling. Cell Signal. 2012, 24, 2035–2042. [Google Scholar]
- Amith, S.R.; Jayanth, P.; Finlay, T.; Franchuk, S.; Gilmour, A.; Abdulkhalek, S.; Szewczuk, M.R. Detection of Neu1 sialidase activity in regulating Toll-like receptor activation. J. Vis. Exp. 2010, 43, 2142. [Google Scholar]
- Abdulkhalek, S.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk regulates nucleic acid-induced endosomal TOLL-like receptor-7 and -9 activation, cellular signaling and pro-inflammatory responses. Cell Signal. 2013, 25, 2093–2105. [Google Scholar]
- Abdulkhalek, S.; Amith, S.R.; Franchuk, S.L.; Jayanth, P.; Guo, M.; Finlay, T.; Gilmour, A.; Guzzo, C.; Gee, K.; Beyaert, R.; et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for Toll-like receptor activation and cellular signaling. J. Biol. Chem. 2011, 286, 36532–36549. [Google Scholar]
- Hobson, J.P.; Rosenfeldt, H.M.; Barak, L.S.; Olivera, A.; Poulton, S.; Caron, M.G.; Milstien, S.; Spiegel, S. Role of the sphingosine-1-phosphate receptor EDG-1 in PDGF-induced cell motility. Science 2001, 291, 1800–1803. [Google Scholar]
- Mira, E.; Lacalle, R.A.; González, M.A.; Gómez-Moutón, C.; Abad, J.L.; Bernad, A.; Martínez, A.C.; Mañes, S. A role for chemokine receptor transactivation in growth factor signaling. EMBO Rep. 2001, 2, 151–156. [Google Scholar]
- Toman, R.E.; Payne, S.G.; Watterson, K.R.; Maceyka, M.; Lee, N.H.; Milstien, S.; Bigbee, J.W.; Spiegel, S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 2004, 166, 381–392. [Google Scholar]
- El-Shewy, H.M.; Johnson, K.R.; Lee, M.H.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J. Biol. Chem. 2006, 281, 31399–31407. [Google Scholar]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-jacking: From a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar]
- Valiquette, M.; Parent, S.; Loisel, T.P.; Bouvier, M. Mutation of tyrosine-141 inhibits insulin-promoted tyrosine phosphorylation and increased responsiveness of the human β2-adrenergic receptor. EMBO J. 1995, 14, 5542–5549. [Google Scholar]
- Baltensperger, K.; Karoor, V.; Paul, H.; Ruoho, A.; Czech, M.P.; Malbon, C.C. The β-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 1061–1064. [Google Scholar]
- Doronin, S.; Shumay, E.; Wang, H.Y.; Malbon, C.C. Akt mediates sequestration of the β2-adrenergic receptor in response to insulin. J. Biol. Chem. 2002, 277, 15124–15131. [Google Scholar]
- Gavi, S.; Yin, D.; Shumay, E.; Wang, H.Y.; Malbon, C.C. The 15-amino acid motif of the C terminus of the β2-adrenergic receptor is sufficient to confer insulin-stimulated counterregulation to the β1-adrenergic receptor. Endocrinology 2005, 146, 450–457. [Google Scholar]
- Karoor, V.; Malbon, C.C. Insulin-like growth factor receptor-1 stimulates phosphorylation of the β2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J. Biol. Chem. 1996, 271, 29347–29352. [Google Scholar]
- García-Sáinz, J.A.; Romero-Ávila, M.T.; Molina-Munoz, T.; García-Pasquel, M.-J. G-protein-coupledreceptor-receptor tyrosine kinase crosstalk. Regulation of receptor sensitivity and roles of autocrine feedback loops and signal integration. Curr. Signal. Transduct. Ther. 2008, 3, 174–182. [Google Scholar]
- Molina-Muñoz, T.; Romero-Ávila, M.T.; García-Sáinz, J.A. Insulin-like growth factor-I induces α1B-adrenergic receptor phosphorylation through Gβγ and epidermal growth factor receptor transactivation. Mol. Endocrinol. 2006, 20, 2773–2783. [Google Scholar]
- Pyne, N.J.; Waters, C.M.; Long, J.S.; Moughal, N.A.; Tigyi, G.; Pyne, S. Receptor tyrosine kinase-G-protein coupled receptor complex signaling in mammalian cells. Adv. Enzym. Regul. 2007, 47, 271–280. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 19700-19728. https://doi.org/10.3390/ijms151119700
Cattaneo F, Guerra G, Parisi M, De Marinis M, Tafuri D, Cinelli M, Ammendola R. Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2014; 15(11):19700-19728. https://doi.org/10.3390/ijms151119700
Chicago/Turabian StyleCattaneo, Fabio, Germano Guerra, Melania Parisi, Marta De Marinis, Domenico Tafuri, Mariapia Cinelli, and Rosario Ammendola. 2014. "Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors" International Journal of Molecular Sciences 15, no. 11: 19700-19728. https://doi.org/10.3390/ijms151119700
APA StyleCattaneo, F., Guerra, G., Parisi, M., De Marinis, M., Tafuri, D., Cinelli, M., & Ammendola, R. (2014). Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors. International Journal of Molecular Sciences, 15(11), 19700-19728. https://doi.org/10.3390/ijms151119700