Stem Cell Treatment for Alzheimer’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathophysiology of AD
3. General Treatment for AD
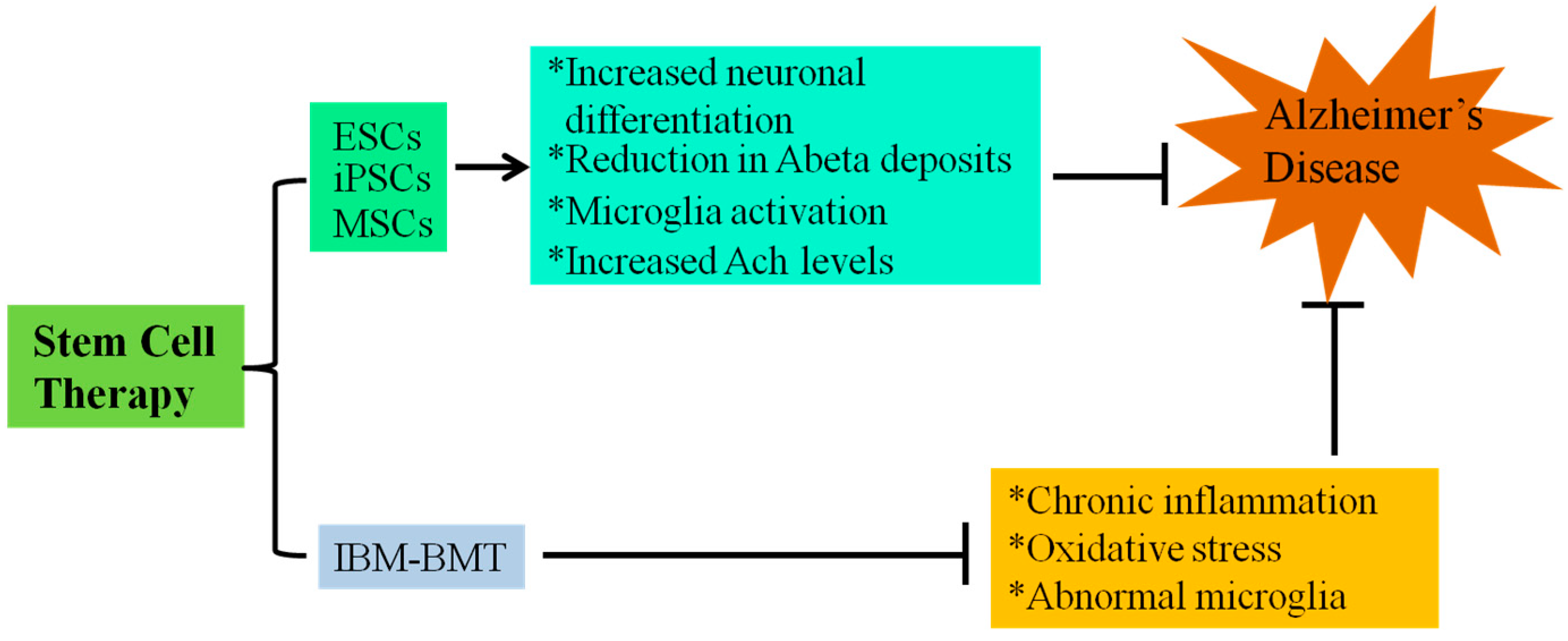
4. Stem Cell Treatment for AD
5. Intra-Bone Marrow-Bone Marrow Transplantation (IBM-BMT) and AD Mouse Model
5.1. IBM-BMT and Senescence-Accelerated Mouse Prone 8 (SAMP8)
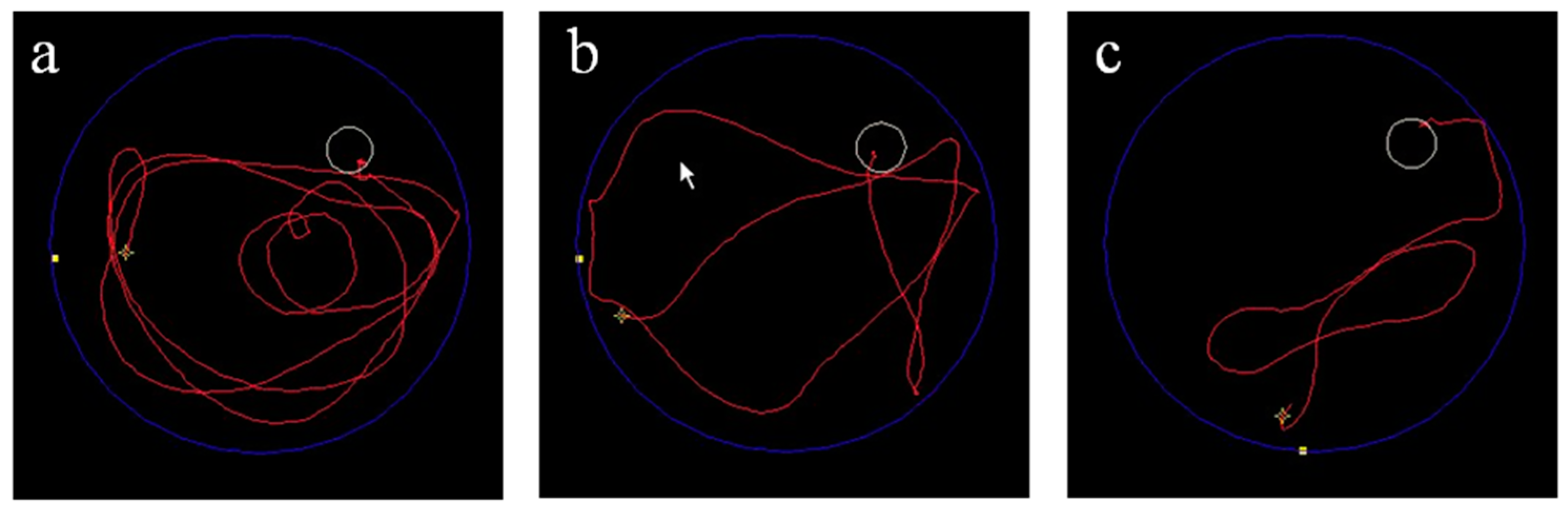
5.2. IBM-BMT and SAMP10
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [PubMed]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative stress in Alzheimer’s disease: Why did antioxidant therapy fail? Oxid. Med. Cell Longev. 2014, 2014, 427318. [Google Scholar]
- Demuro, A.; Smith, M.; Parker, I. Single-channel Ca2+ imaging implicates Aβ 1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [PubMed]
- Kuhla, B.; Haase, C.; Flach, K.; Luth, H.J.; Arendt, T.; Munch, G. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J. Biol. Chem. 2007, 282, 6984–6991. [Google Scholar] [CrossRef] [PubMed]
- Eikelenboom, P.; Bate, C.; van Gool, W.A.; Hoozemans, J.J.; Rozemuller, J.M.; Veerhuis, R.; Williams, A. Neuroinflammation in Alzheimer’s disease and prion disease. Glia 2002, 40, 232–239. [Google Scholar] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar] [PubMed]
- Patel, A.N.; Jhamandas, J.H. Neuronal receptors as targets for the action of amyloid-β protein (Aβ) in the brain. Expert Rev. Mol. Med. 2012, 14, e2. [Google Scholar]
- Iqbal, K.; Alonso, A.C.; Gong, C.X.; Khatoon, S.; Pei, J.J.; Wang, J.Z.; Grundke-Iqbal, I. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J. Neural Transm. Suppl. 1998, 53, 169–180. [Google Scholar] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [PubMed]
- Millington, C.; Sonego, S.; Karunaweera, N.; Rangel, A.; Aldrich-Wright, J.R.; Campbell, I.L.; Gyengesi, E.; Munch, G. Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. Biomed. Res. Int. 2014, 2014, 309129. [Google Scholar]
- Munch, G.; Schinzel, R.; Loske, C.; Wong, A.; Durany, N.; Li, J.J.; Vlassara, H.; Smith, M.A.; Perry, G.; Riederer, P. Alzheimer’s disease—Synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J. Neural Transm. 1998, 105, 439–461. [Google Scholar] [PubMed]
- Loane, D.J.; Byrnes, K.R. Role of microglia in neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F. Microglia: Phagocyte and glia cell. Int. J. Biochem. Cell Biol. 2005, 37, 17–21. [Google Scholar] [PubMed]
- Baune, B.T.; Ponath, G.; Rothermundt, M.; Roesler, A.; Berger, K. Association between cytokines and cerebral MRI changes in the aging brain. J. Geriatr. Psychiatry Neurol. 2009, 22, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Velayutham, M.; Hemann, C.; Zweier, J.L. Removal of H2O2 and generation of superoxide radical: Role of cytochrome c and NADH. Free Radic. Biol. Med. 2011, 51, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar]
- Federico, A.; Cardaioli, E.; da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [PubMed]
- Opazo, C.; Huang, X.; Cherny, R.A.; Moir, R.D.; Roher, A.E.; White, A.R.; Cappai, R.; Masters, C.L.; Tanzi, R.E.; Inestrosa, N.C.; et al. Metalloenzyme-like activity of Alzheimer’s disease β-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H2O2. J. Biol. Chem. 2002, 277, 40302–40308. [Google Scholar]
- Meraz-Rios, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernandez, J.; Campos-Pena, V. Inflammatory process in Alzheimer’s disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One 2012, 7, e34929. [Google Scholar]
- Chapman, H.A.; Riese, R.J.; Shi, G.P. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 1997, 59, 63–88. [Google Scholar] [PubMed]
- Hook, V.; Toneff, T.; Bogyo, M.; Greenbaum, D.; Medzihradszky, K.F.; Neveu, J.; Lane, W.; Hook, G.; Reisine, T. Inhibition of cathepsin B reduces β-amyloid production in regulated secretory vesicles of neuronal chromaffin cells: Evidence for cathepsin B as a candidate β-secretase of Alzheimer’s disease. Biol. Chem. 2005, 386, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; Gan, L. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Marr, R.A.; Rockenstein, E.; Mukherjee, A.; Kindy, M.S.; Hersh, L.B.; Gage, F.H.; Verma, I.M.; Masliah, E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. 2003, 23, 1992–1996. [Google Scholar] [PubMed]
- Massoud, F.; Leger, G.C. Pharmacological treatment of Alzheimer disease. Can. J. Psychiatry 2011, 56, 579–588. [Google Scholar] [PubMed]
- Mangialasche, F.; Kivipelto, M.; Mecocci, P.; Rizzuto, D.; Palmer, K.; Winblad, B.; Fratiglioni, L. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J. Alzheimers Dis. 2010, 20, 1029–1037. [Google Scholar] [PubMed]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.M.; Ayala, M.; Zhang, S.C. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Yang, Y.H.; Bae, D.K.; Lee, S.H.; Yang, G.; Kyung, J.; Kim, D.; Choi, E.K.; Lee, S.W.; Kim, G.H.; et al. Improvement of cognitive function and physical activity of aging mice by human neural stem cells over-expressing choline acetyltransferase. Neurobiol. Aging 2013, 34, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Yang, G.; Bae, D.K.; Lee, S.H.; Yang, Y.H.; Kyung, J.; Kim, D.; Choi, E.K.; Choi, K.C.; Kim, S.U.; et al. Human adipose tissue-derived mesenchymal stem cells improve cognitive function and physical activity in ageing mice. J. Neurosci. Res. 2013, 91, 660–670. [Google Scholar] [PubMed]
- Enciu, A.M.; Nicolescu, M.I.; Manole, C.G.; Muresanu, D.F.; Popescu, L.M.; Popescu, B.O. Neuroregeneration in neurodegenerative disorders. BMC Neurol. 2011, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xu, H.; Fan, X.; Li, D.; Rancourt, D.; Zhou, G.; Li, Z.; Yang, L. Embryonic stem cell-derived neural precursor cells improve memory dysfunction in Aβ(1–40) injured rats. Neurosci. Res. 2008, 62, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Campagnoli, C.; Roberts, I.A.; Kumar, S.; Bennett, P.R.; Bellantuono, I.; Fisk, N.M. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 2001, 98, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [PubMed]
- Erices, A.; Conget, P.; Minguell, J.J. Mesenchymal progenitor cells in human umbilical cord blood. Br. J. Haematol. 2000, 109, 235–242. [Google Scholar] [PubMed]
- Dominici, M.; le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar]
- Tang, D.Q.; Cao, L.Z.; Burkhardt, B.R.; Xia, C.Q.; Litherland, S.A.; Atkinson, M.A.; Yang, L.J. In vivo and in vitro characterization of insulin-producing cells obtained from murine bone marrow. Diabetes 2004, 53, 1721–1732. [Google Scholar]
- Beyth, S.; Borovsky, Z.; Mevorach, D.; Liebergall, M.; Gazit, Z.; Aslan, H.; Galun, E.; Rachmilewitz, J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood 2005, 105, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Fazekasova, H.; Lam, E.W.; Soeiro, I.; Lombardi, G.; Dazzi, F. Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation 2007, 83, 71–76. [Google Scholar] [PubMed]
- Aggarwal, S.M.; Pittenger, F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.M.; Barry, F.; Murphy, J.M.; Mahon, B.P. Interferon-γ does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin. Exp. Immunol. 2007, 149, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.M.; Ahmed, H.H.; Atta, H.M.; Ghazy, M.A.; Aglan, H.A. Potential of bone marrow mesenchymal stem cells in management of Alzheimer’s disease in female rats. Cell Biol. Int. 2014. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Bae, J.S. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 2009, 450, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [PubMed]
- Honmou, O.; Houkin, K.; Matsunaga, T.; Niitsu, Y.; Ishiai, S.; Onodera, R.; Waxman, S.G.; Kocsis, J.D. Intravenous administration of auto serum-expanded autologous mesenchymal stem cells in stroke. Brain 2011, 134, 1790–1807. [Google Scholar] [PubMed]
- Chen, J.; Tang, Y.X.; Liu, Y.M.; Hu, X.Q.; Liu, N.; Wang, S.X.; Zhang, Y.; Zeng, W.G.; Ni, H.J.; Zhao, B.; et al. Transplantation of adipose-derived stem cells is associated with neural differentiation and functional improvement in a rat model of intracerebral hemorrhage. CNS Neurosci. Ther. 2012, 18, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Ahn, S.; Kim, S.; Joo, Y.; Chong, Y.H.; Suh, Y.H.; Chang, K.A. In vivo imaging of human adipose-derived stem cells in Alzheimer’s disease animal model. J. Biomed. Opt. 2014, 19, 051206. [Google Scholar]
- Ma, T.; Gong, K.; Ao, Q.; Yan, Y.; Song, B.; Huang, H.; Zhang, X.; Gong, Y. Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer’s disease mice. Cell Transplant. 2013, 22, S113–S126. [Google Scholar] [CrossRef]
- Schira, J.; Gasis, M.; Estrada, V.; Hendricks, M.; Schmitz, C.; Trapp, T.; Kruse, F.; Kogler, G.; Wernet, P.; Hartung, H.P.; et al. Significant clinical, neuropathological and behavioural recovery from acute spinal cord trauma by transplantation of a well-defined somatic stem cell from human umbilical cord blood. Brain 2012, 135, 431–446. [Google Scholar] [PubMed]
- Darlington, D.; Deng, J.; Giunta, B.; Hou, H.; Sanberg, C.D.; Kuzmin-Nichols, N.; Zhou, H.D.; Mori, T.; Ehrhart, J.; Sanberg, P.R.; Tan, J. Multiple low-dose infusions of human umbilical cord blood cells improve cognitive impairments and reduce amyloid-β-associated neuropathology in Alzheimer mice. Stem Cells Dev. 2013, 22, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Z.; Wei, L.; Yang, S.; Zhu, Z.; Wang, P.; Zhao, C.; Bi, J. Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-β deposition in an AβPP/PS1 transgenic mouse model. Stem Cell Res. Ther. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, S.; Yasumizu, R.; Inaba, M.; Izui, S.; Hayakawa, K.; Sekita, K.; Toki, J.; Sugiura, K.; Iwai, H.; Nakamura, T.; et al. Long-term observations of autoimmune-prone mice treated for autoimmune disease by allogeneic bone marrow transplantation. Proc. Natl. Acad. Sci. USA 1989, 86, 3306–3310. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Inaba, M.; Hisha, H.; Sugiura, K.; Adachi, Y.; Nagata, N.; Ogawa, R.; Good, R.A.; Ikehara, S. Requirement of donor-derived stromal cells in the bone marrow for successful allogeneic bone marrow transplantation. Complete prevention of recurrence of autoimmune diseases in MRL/MP-Ipr/Ipr mice by transplantation of bone marrow plus bones (stromal cells) from the same donor. J. Immunol. 1994, 152, 3119–3127. [Google Scholar]
- Kushida, T.; Inaba, M.; Hisha, H.; Ichioka, N.; Esumi, T.; Ogawa, R.; Iida, H.; Ikehara, S. Intra-bone marrow injection of allogeneic bone marrow cells: A powerful new strategy for treatment of intractable autoimmune diseases in MRL/lpr mice. Blood 2001, 97, 3292–3299. [Google Scholar] [CrossRef] [PubMed]
- Kushida, T.; Ueda, Y.; Umeda, M.; Oe, K.; Okamoto, N.; Iida, H.; Abraham, N.G.; Gershwin, M.E.; Ikehara, S. Allogeneic intra-bone marrow transplantation prevents rheumatoid arthritis in SKG/Jcl mice. J. Autoimmun. 2009, 32, 216–222. [Google Scholar] [PubMed]
- Takada, K.; Inaba, M.; Ichioka, N.; Ueda, Y.; Taira, M.; Baba, S.; Mizokami, T.; Wang, X.; Hisha, H.; Iida, H.; et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells 2006, 24, 399–405. [Google Scholar] [PubMed]
- Li, M.; Inaba, M.; Guo, K.; Abraham, N.G.; Ikehara, S. Amelioration of cognitive ability in senescence-accelerated mouse prone 8 (SAMP8) by intra-bone marrow-bone marrow transplantation. Neurosci. Lett. 2009, 465, 36–40. [Google Scholar] [PubMed]
- Chiba, Y.; Shimada, A.; Kumagai, N.; Yoshikawa, K.; Ishii, S.; Furukawa, A.; Takei, S.; Sakura, M.; Kawamura, N.; Hosokawa, M. The senescence-accelerated mouse (SAM): A higher oxidative stress and age-dependent degenerative diseases model. Neurochem. Res. 2009, 34, 679–687. [Google Scholar] [PubMed]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Kiyota, Y.; Yamazaki, N.; Nagaoka, A.; Matsuo, T.; Nagawa, Y.; Takeda, T. Age-related changes in learning and memory in the senescence-accelerated mouse (SAM). Physiol. Behav. 1986, 38, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M.; Nakamura, S.; Akiguchi, I.; Ueno, M.; Oka, N.; Ishikawa, S.; Shimada, A.; Kimura, J.; Takeda, T. β/A4 proteinlike immunoreactive granular structures in the brain of senescence-accelerated mouse. Am. J. Pathol. 1993, 142, 1887–1897. [Google Scholar] [PubMed]
- Kumar, V.B.; Farr, S.A.; Flood, J.F.; Kamlesh, V.; Franko, M.; Banks, W.A.; Morley, J.E. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 2000, 21, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Flood, J.F.; Morley, J.E. Early onset of age-related impairment of aversive and appetitive learning in the SAMP8 mouse. J. Gerontol. 1992, 47, B52–B59. [Google Scholar] [CrossRef] [PubMed]
- Sjobeck, M.; Englund, E. Alzheimer’s disease and the cerebellum: A morphologic study on neuronal and glial changes. Dement. Geriatr. Cogn. Disord. 2001, 12, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, A.; Sultana, R.; Forster, S.; Perluigi, M.; Cenini, G.; Cini, C.; Cai, J.; Klein, J.B.; Farr, S.A.; Niehoff, M.L.; et al. Allan butterfield. Antisense directed against PS-1 gene decreases brain oxidative markers in aged senescence accelerated mice (SAMP8) and reverses learning and memory impairment: A proteomics study. Free Radic. Biol. Med. 2013, 65, 1–14. [Google Scholar]
- Lin, X.; Huang, R.; Zhang, S.; Wei, L.; Zhuo, L.; Wu, X.; Tang, A.; Huang, Q. Beneficial effects of asiaticoside on cognitive deficits in senescence-accelerated mice. Fitoterapia 2013, 87, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Skelton, R.W.; McNamara, R.K. Bilateral knife cuts to the perforant path disrupt spatial learning in the Morris water maze. Hippocampus 1992, 2, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.; Bauer, J.; Ganter, U.; Jonas, U.; Berger, M.; Volk, B. Detection of interleukin-6 and α 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer’s disease patients. Lab. Investig. 1992, 66, 223–230. [Google Scholar] [PubMed]
- Hosokawa, M. A higher oxidative status accelerates senescence and aggravates age-dependent disorders in SAMP strains of mice. Mech. Ageing Dev. 2002, 123, 1553–1561. [Google Scholar] [PubMed]
- Takeda, T.; Matsushita, T.; Kurozumi, M.; Takemura, K.; Higuchi, K.; Hosokawa, M. Pathobiology of the senescence-accelerated mouse (SAM). Exp. Gerontol. 1997, 32, 117–127. [Google Scholar] [PubMed]
- Shimada, A.; Hasegawa-Ishii, S. Senescence-accelerated mice (SAMs) as a model for brain aging and immunosenescence. Aging Dis. 2011, 2, 414–435. [Google Scholar] [PubMed]
- Hasegawa-Ishii, S.; Takei, S.; Chiba, Y.; Furukawa, A.; Umegaki, H.; Iguchi, A.; Kawamura, N.; Yoshikawa, K.; Hosokawa, M.; Shimada, A. Morphological impairments in microglia precede age-related neuronal degeneration in senescence-accelerated mice. Neuropathology 2011, 31, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [PubMed]
- Li, M.; Shi, M.; Abraham, N.G.; Ikehara, S. Improved expression of Sirt1 on thymic epithelial cells of SAMP10 after intra bone marrow-bone marrow transplantation. Cell Transplant. 2014, 23, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.H.; Seach, N.; Ueno, T.; Milton, M.K.; Liston, A.; Lew, A.M.; Goodnow, C.C.; Boyd, R.L. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood 2006, 108, 3777–3785. [Google Scholar] [PubMed]
- Gui, J.; Zhu, X.; Dohkan, J.; Cheng, L.; Barnes, P.F.; Su, D.M. The aged thymus shows normal recruitment of lymphohematopoietic progenitors but has defects in thymic epithelial cells. Int. Immunol. 2007, 19, 1201–1211. [Google Scholar] [PubMed]
- Calvanese, V.; Fraga, M.F. Sirt1 brings stemness closer to cancer and aging. Aging 2011, 3, 162–167. [Google Scholar] [PubMed]
- Zhang, J.; Lee, S.M.; Shannon, S.; Gao, B.; Chen, W.; Chen, A.; Divekar, R.; McBurney, M.W.; Braley-Mullen, H.; Zaghouani, H.; et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Investig. 2009, 119, 3048–3058. [Google Scholar] [PubMed]
- Takaki, T.; Hosaka, N.; Miyake, T.; Cui, W.; Nishida, T.; Inaba, M.; Ikehara, S. Presence of donor-derived thymic epithelial cells in B6→MRL/lpr mice after allogeneic intra-bone marrow-bone marrow transplantation (IBM-BMT). J. Autoimmun. 2008, 31, 408–415. [Google Scholar]
- Hasegawa-Ishii, S.; Shimada, A.; Inaba, M.; Li, M.; Shi, M.; Kawamura, N.; Takei, S.; Chiba, Y.; Hosokawa, M.; Ikehara, S. Selective localization of bone marrow-derived ramified cells in the brain adjacent to the attachments of choroid plexus. Brain Behav. Immun. 2013, 29, 82–97. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Guo, K.; Ikehara, S. Stem Cell Treatment for Alzheimer’s Disease. Int. J. Mol. Sci. 2014, 15, 19226-19238. https://doi.org/10.3390/ijms151019226
Li M, Guo K, Ikehara S. Stem Cell Treatment for Alzheimer’s Disease. International Journal of Molecular Sciences. 2014; 15(10):19226-19238. https://doi.org/10.3390/ijms151019226
Chicago/Turabian StyleLi, Ming, Kequan Guo, and Susumu Ikehara. 2014. "Stem Cell Treatment for Alzheimer’s Disease" International Journal of Molecular Sciences 15, no. 10: 19226-19238. https://doi.org/10.3390/ijms151019226
APA StyleLi, M., Guo, K., & Ikehara, S. (2014). Stem Cell Treatment for Alzheimer’s Disease. International Journal of Molecular Sciences, 15(10), 19226-19238. https://doi.org/10.3390/ijms151019226