TCDD Induces the Hypoxia-Inducible Factor (HIF)-1α Regulatory Pathway in Human Trophoblastic JAR Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
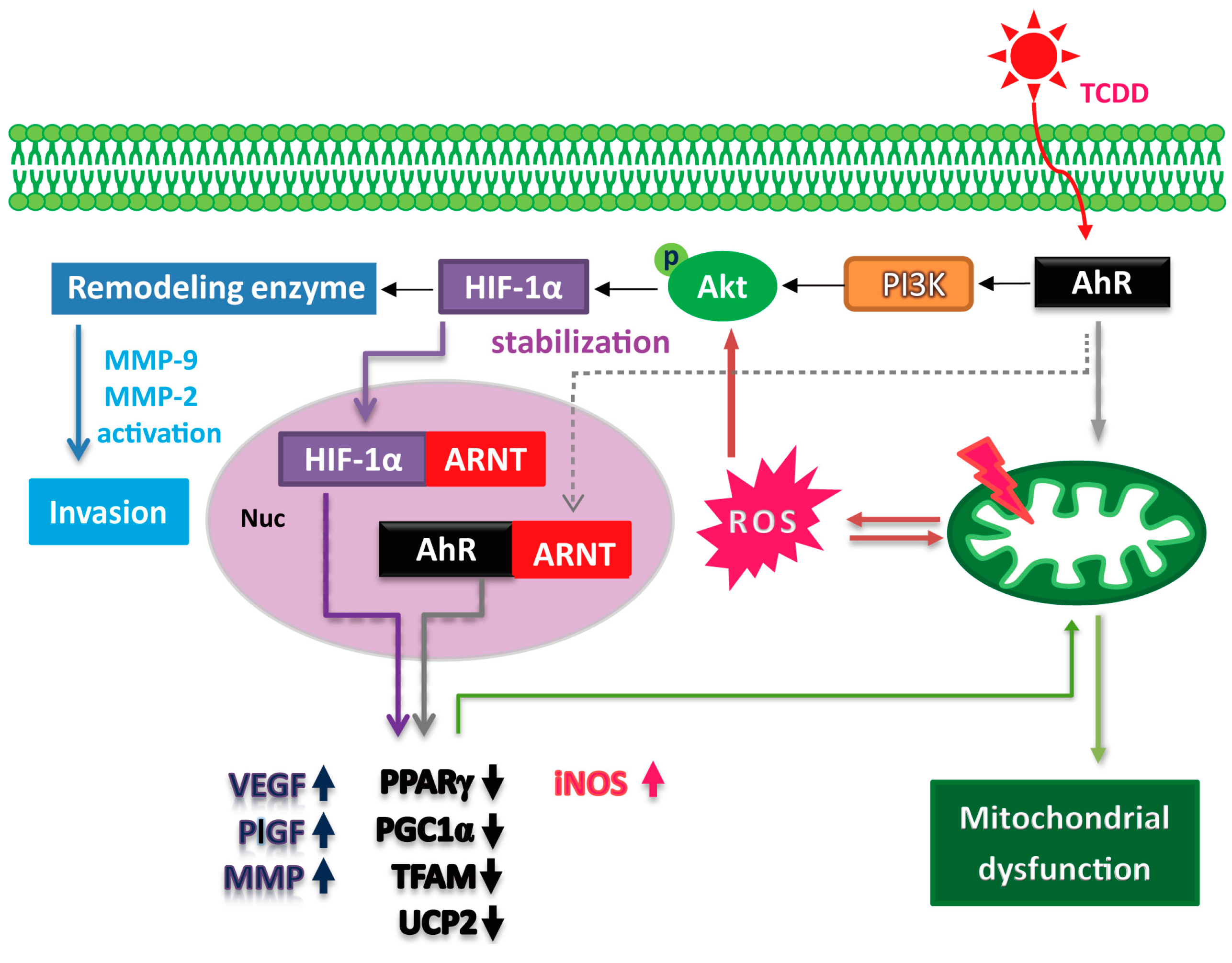{kind=link}
Abstract
:1. Introduction
2. Results
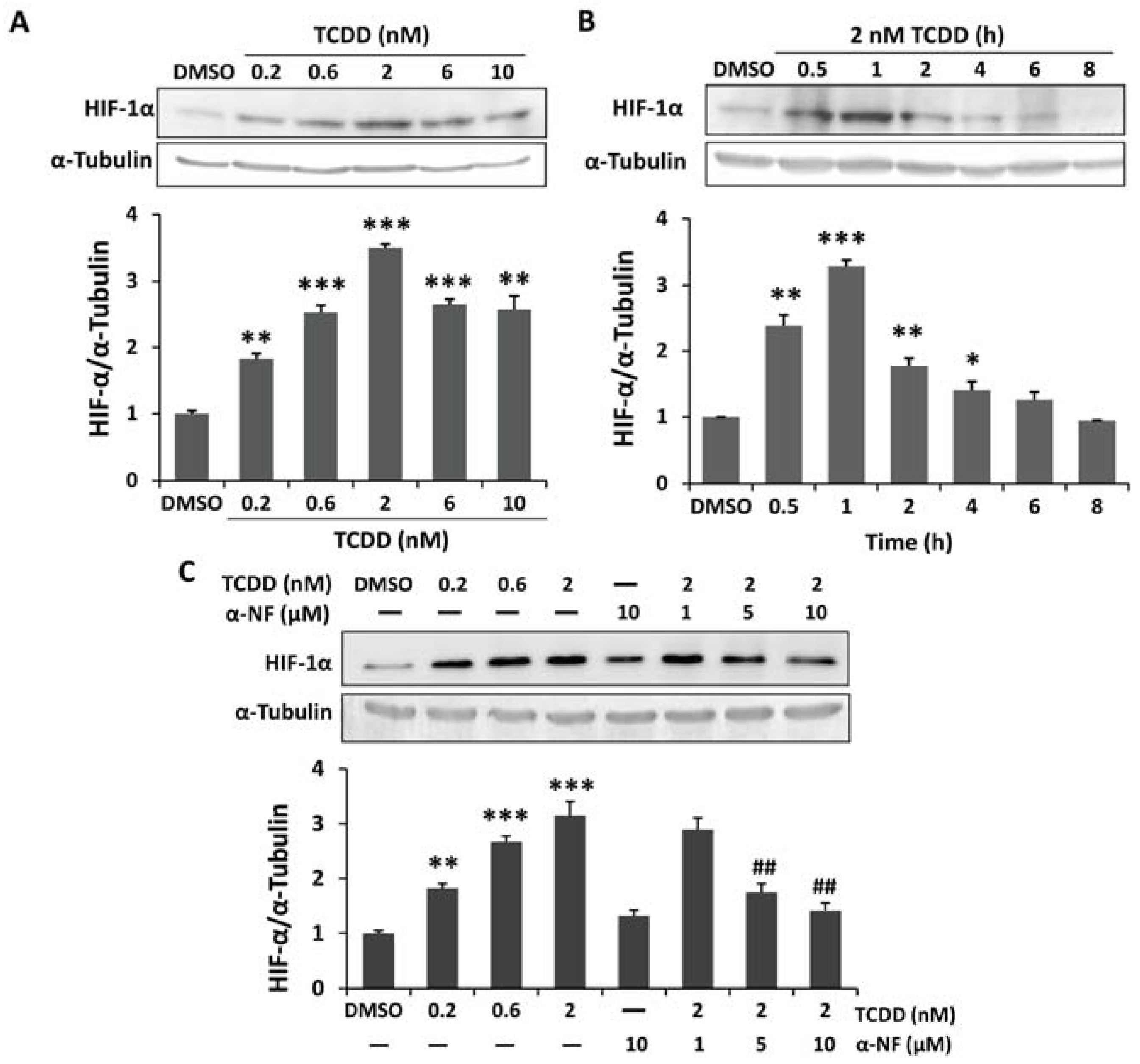
2.1. Induced Hypoxia-Inducible Factor-1 Alpha (HIF-1α) Stabilization in 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD)-Treated Human Trophoblastic Cells
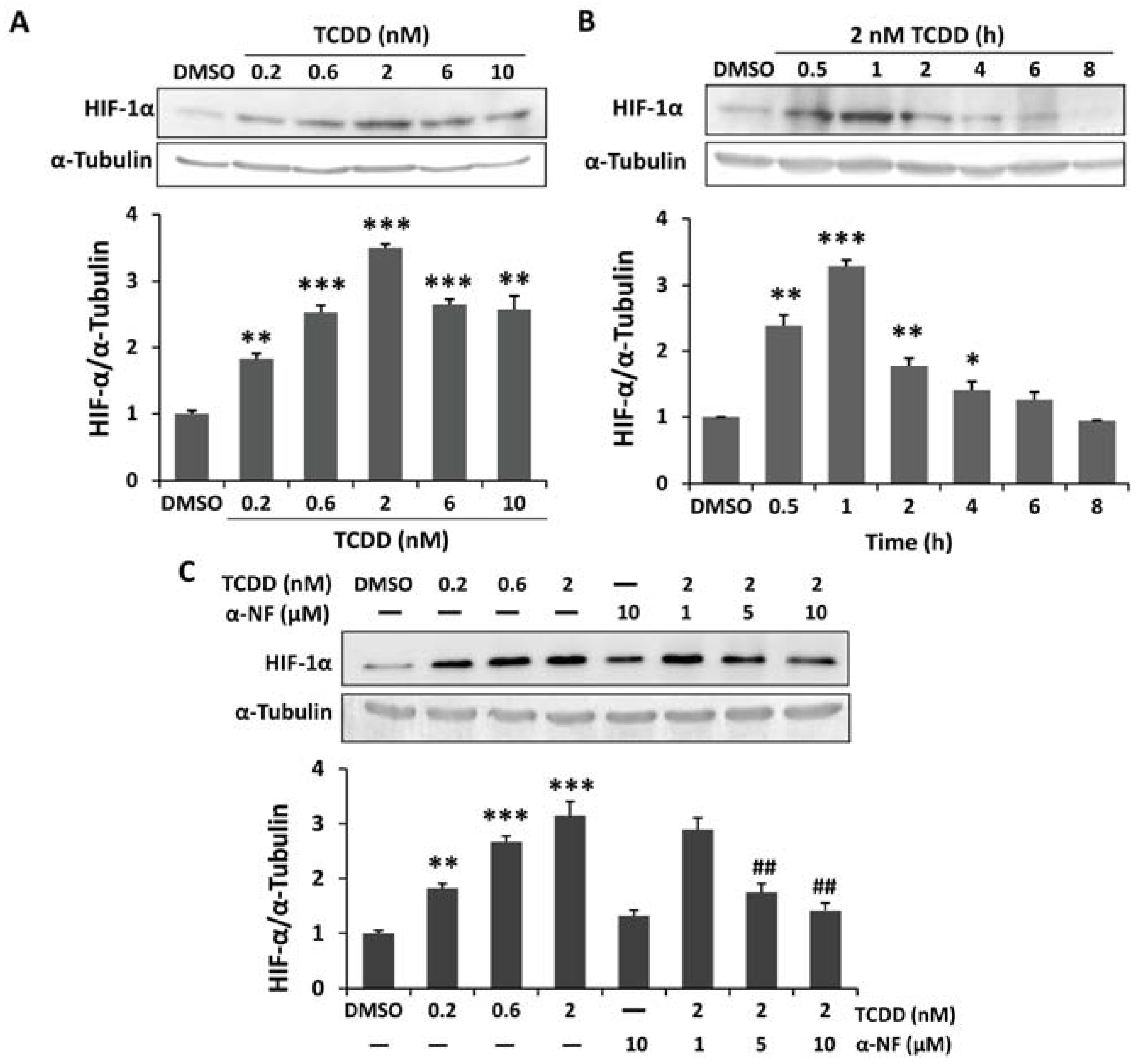
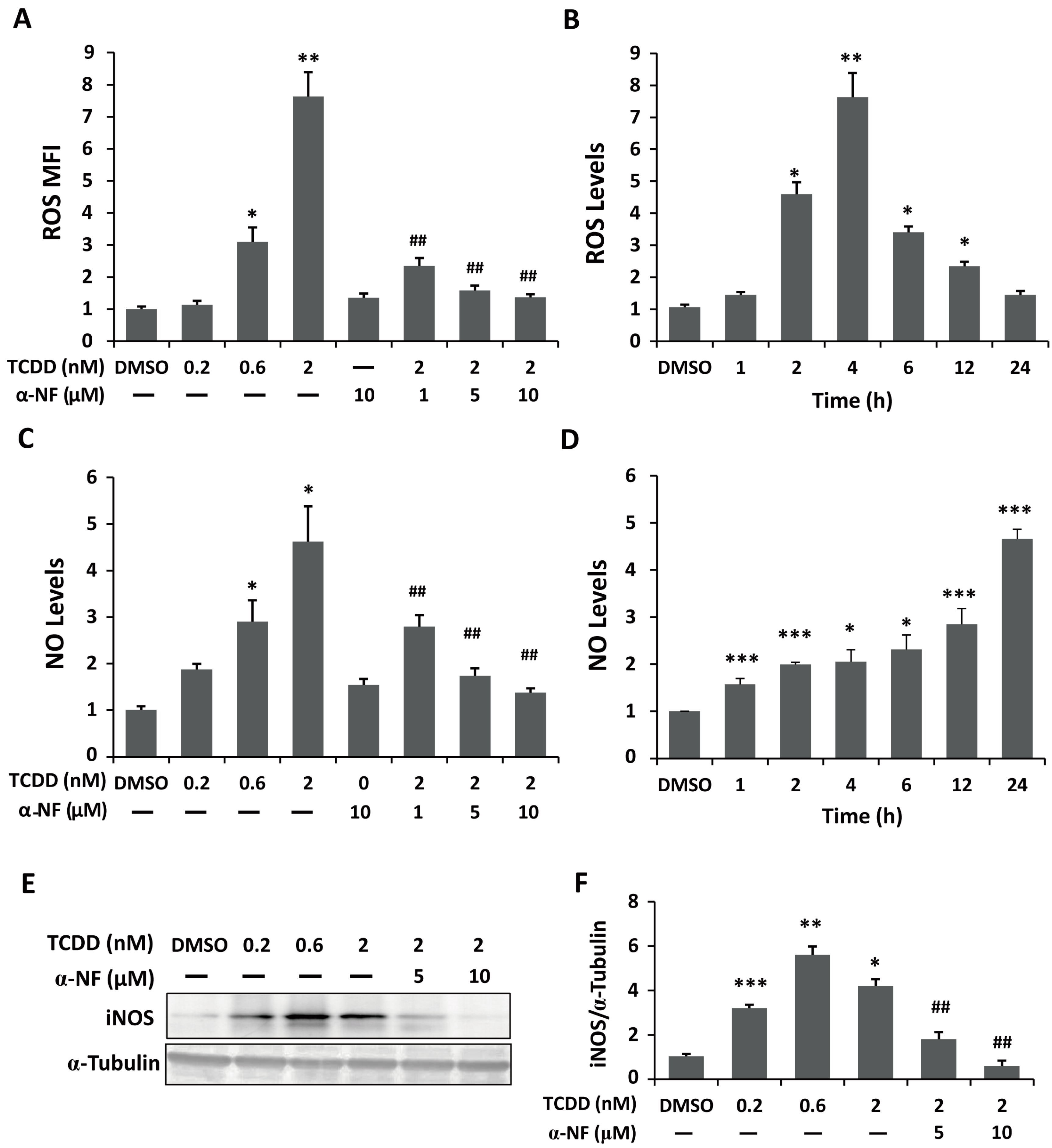
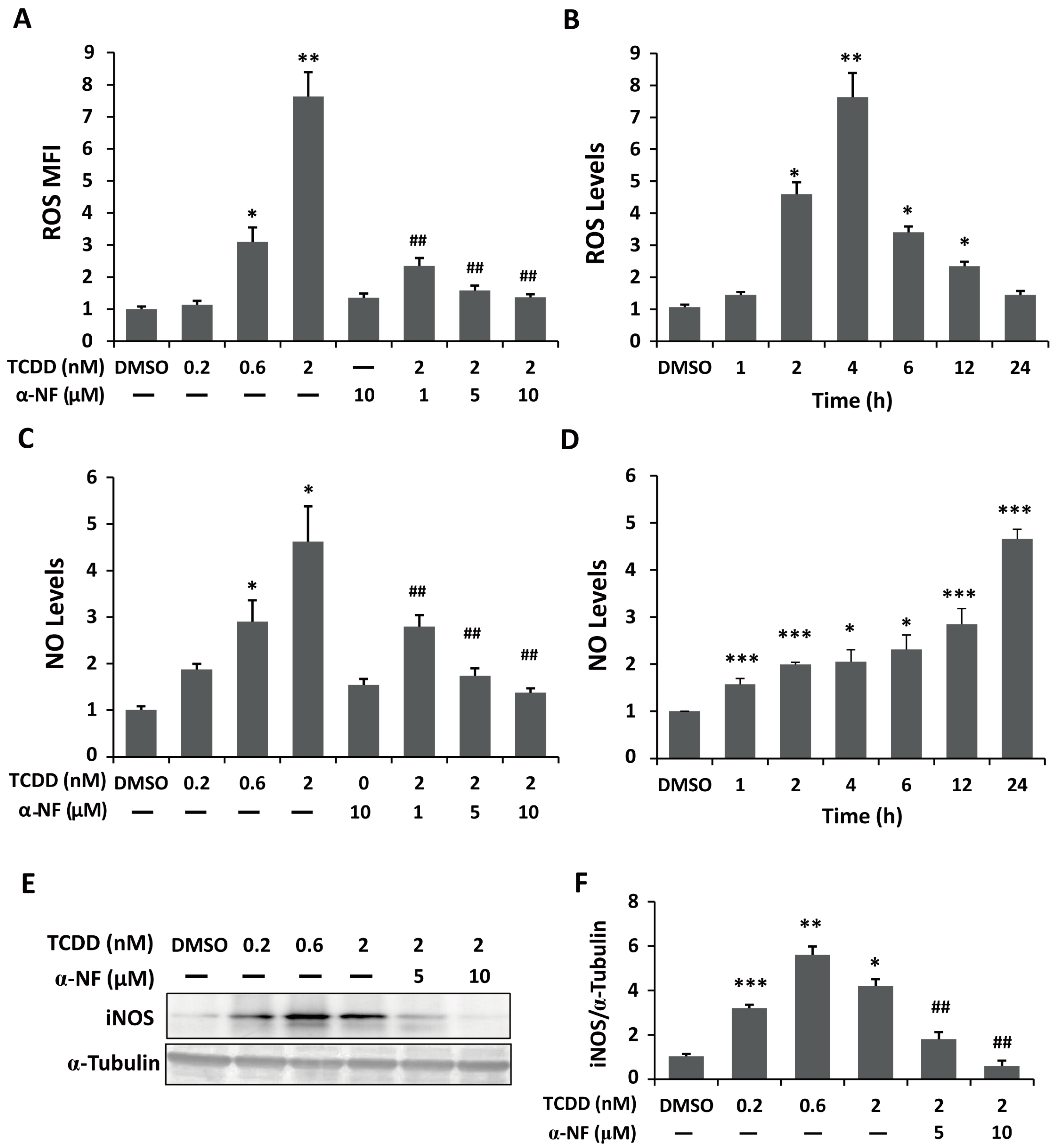
2.2. Enhanced Reactive Oxygen Species (ROS) and Nitric Oxide (NO) Generation in TCDD-Treated Human Trophoblastic Cells
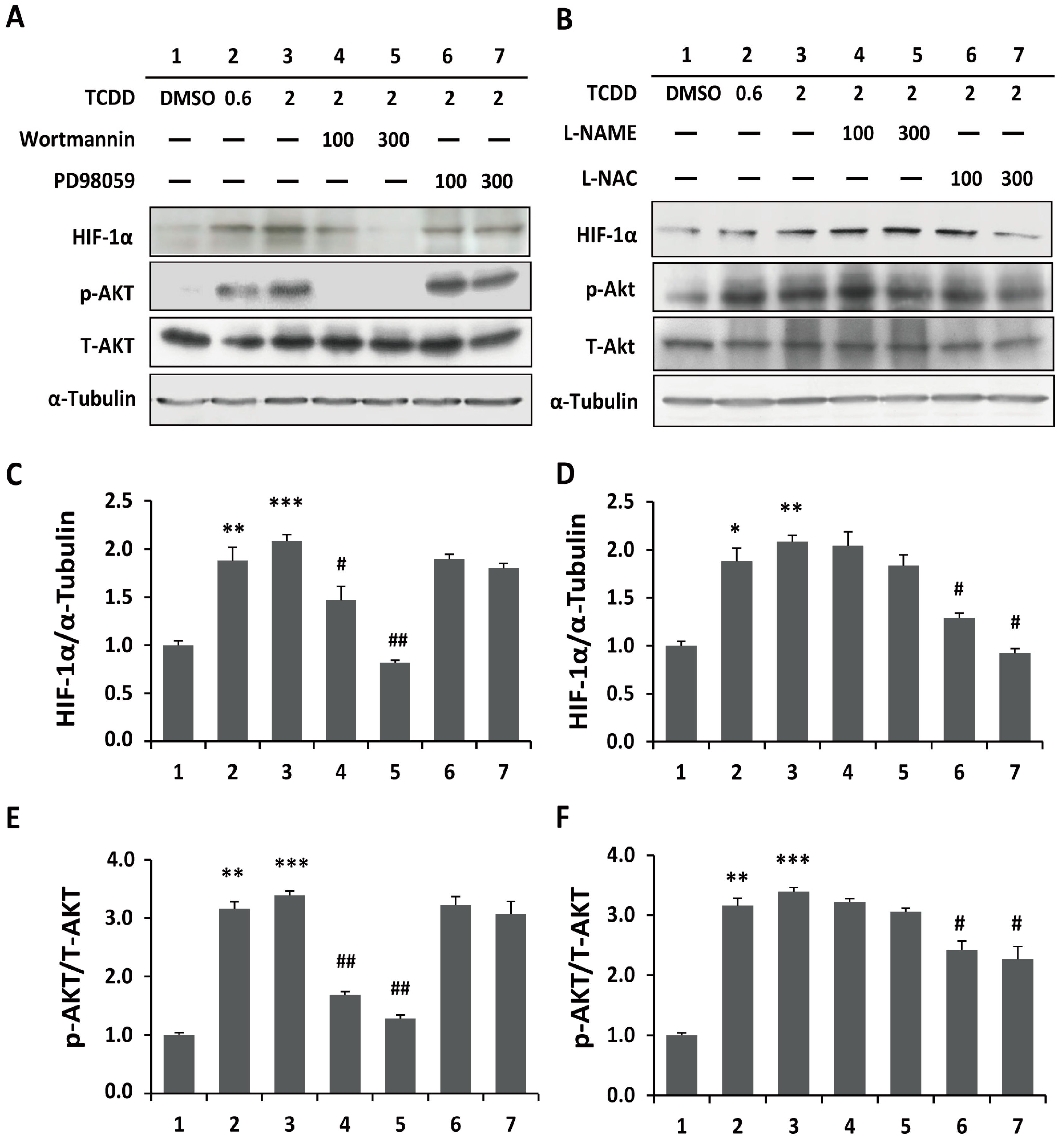
2.3. Phosphoinositide-3-Kinase and Akt Signaling Are Involved in TCDD-Induced Hypoxia and ROS Generation in Trophoblastic Cells
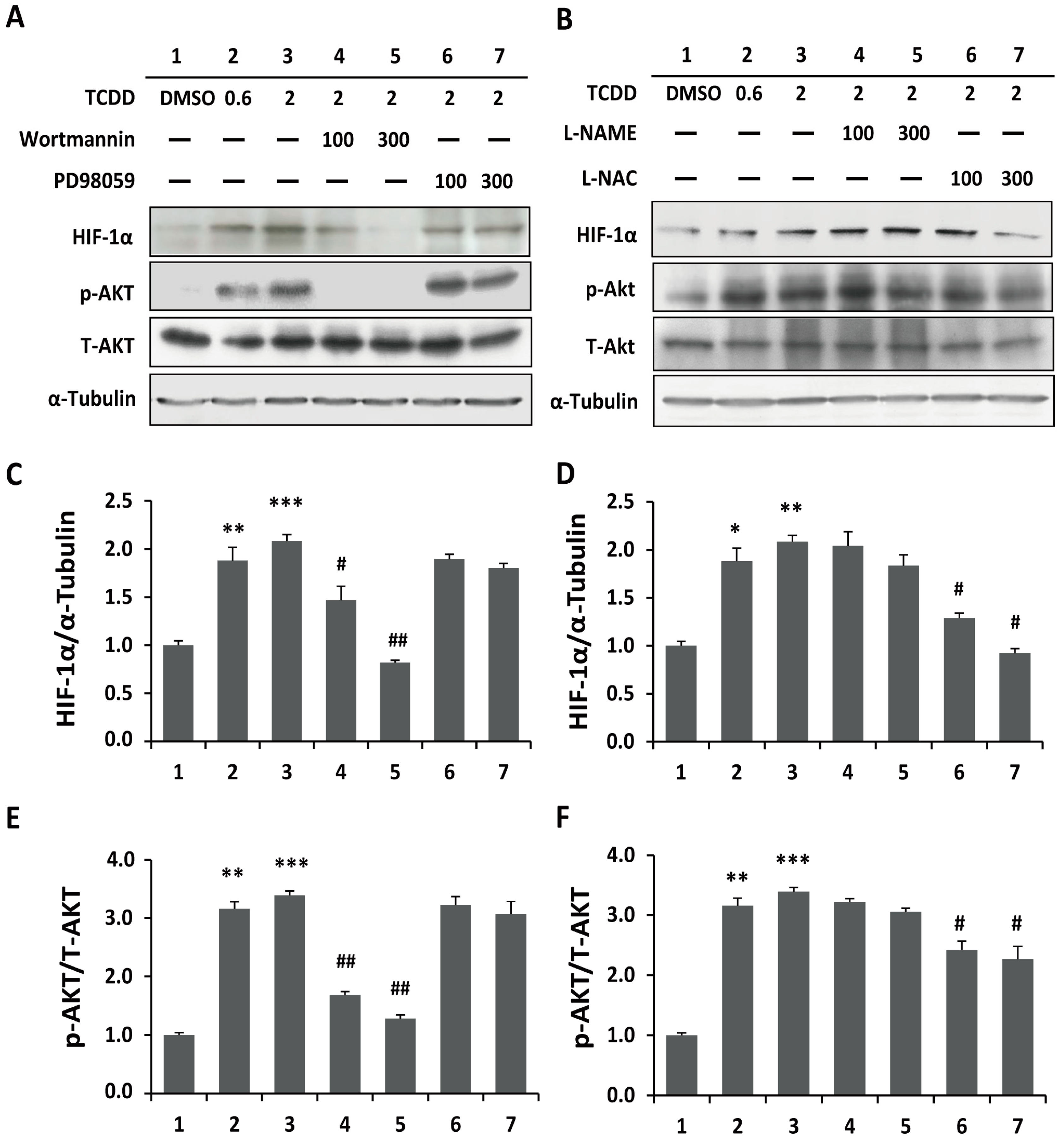
2.4. Induced Cell Invasion and Activation of Matrix Metalloproteinase in TCDD-Treated Trophoblastic Cells

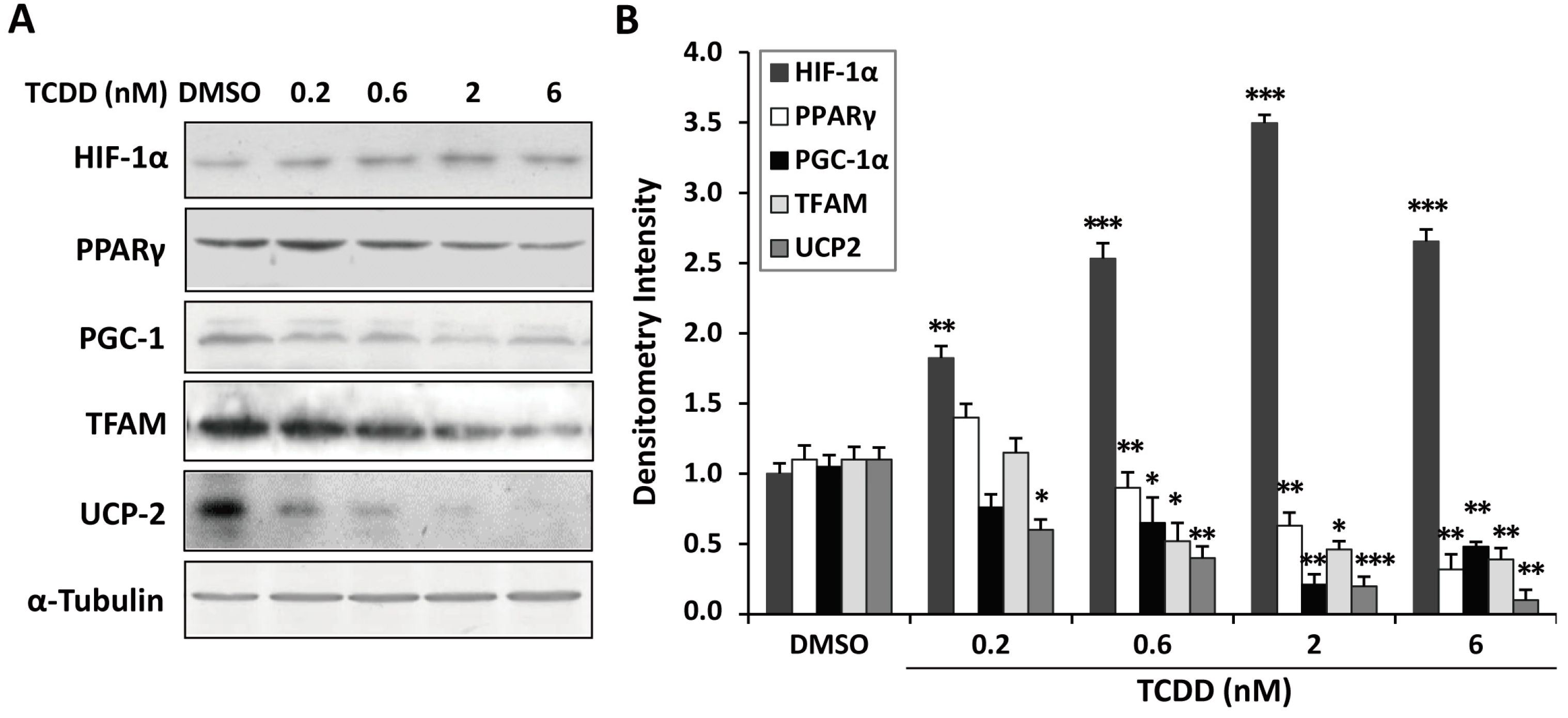
2.5. Inhibition of Peroxisome Proliferator-Activated Receptor γ (PPARγ) and PPARγ Coactivator-1α (PGC-1α) Expression in TCDD-Treated Trophoblastic Cells
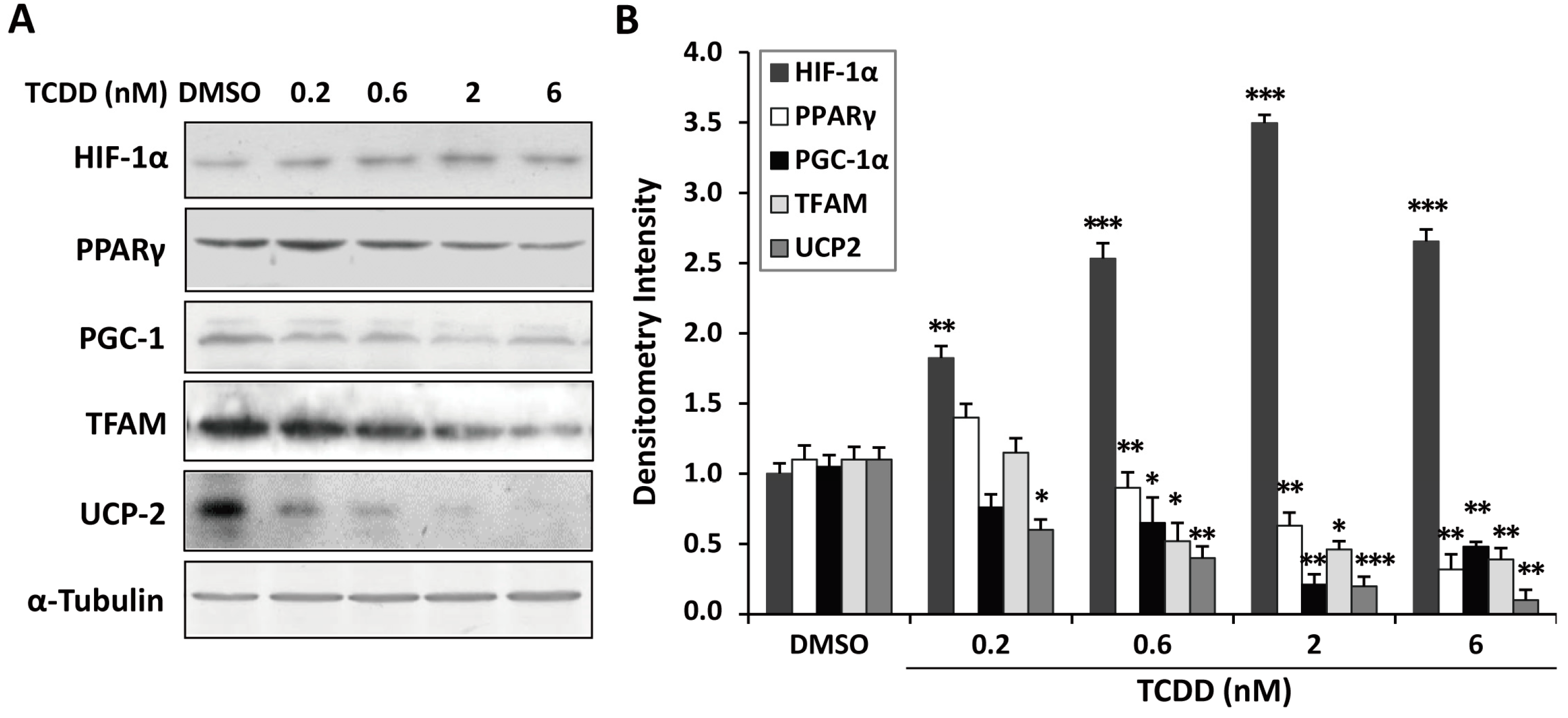
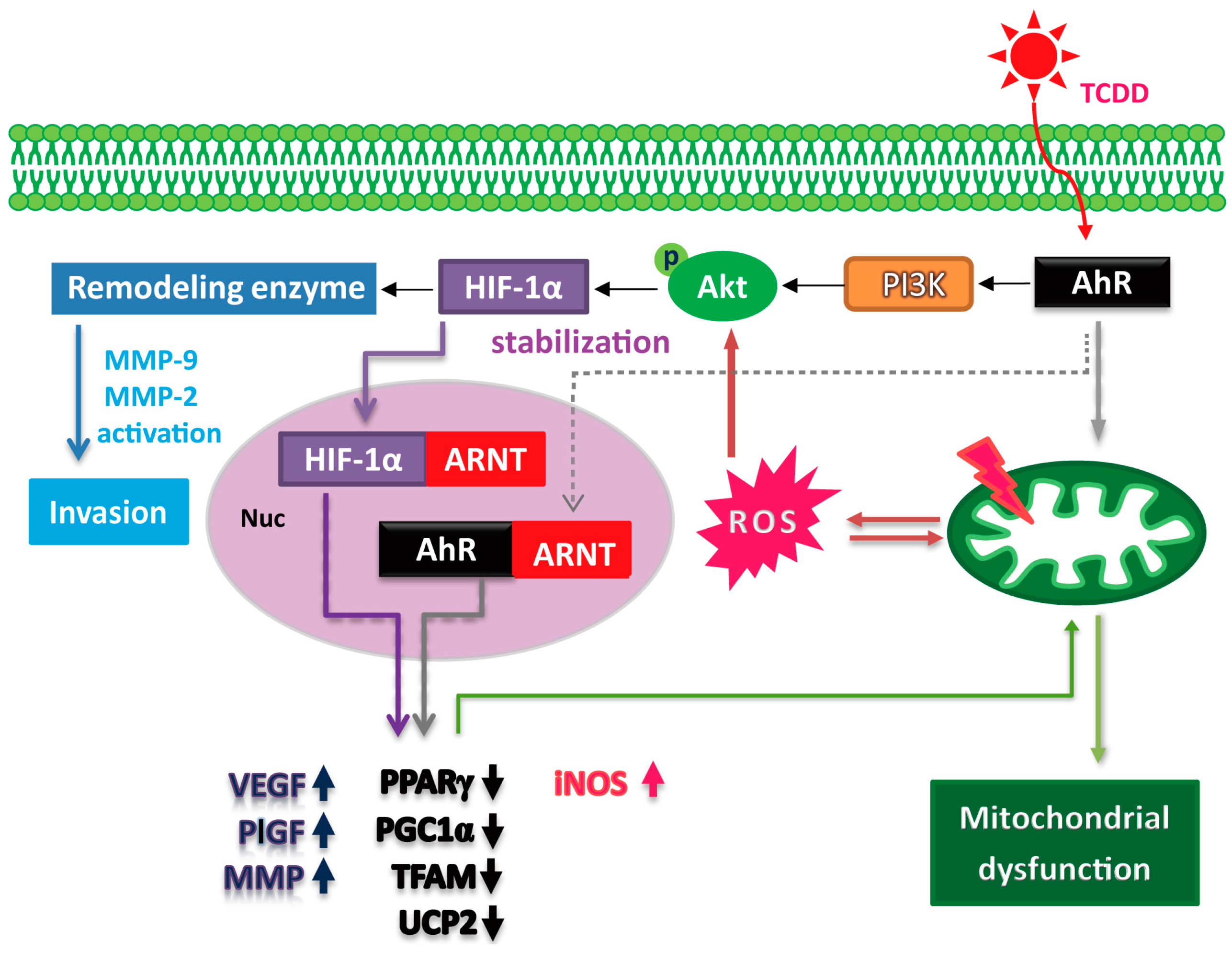
3. Discussion
4. Experimental Section
4.1. Reagents
4.2. Cell Culture
4.3. Western Blots
4.4. Flow Cytometry Analysis for ROS Generation
4.5. Measurement of Nitrite by the Griess Reagent
4.6. RNA Extraction and Real-Time Quantitative PCR
4.7. Cell Invasion Assay
4.8. Gelatin Zymography
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572, 25–30. [Google Scholar] [PubMed]
- Ishimura, R.; Kawakami, T.; Ohsako, S.; Nohara, K.; Tohyama, C. Suppressive effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on vascular remodeling that takes place in the normal labyrinth zone of rat placenta during late gestation. Toxicol. Sci. 2006, 91, 265–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, X.; Zhou, Q.; He, Q.; Kang, J.; Zheng, J.; Wang, K.; Duan, T. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS One 2014, 9, e86549. [Google Scholar]
- Halperin, W.; Vogt, R.; Sweeney, M.H.; Shopp, G.; Fingerhut, M.; Petersen, M. Immunological markers among workers exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Occup. Environ. Med. 1998, 55, 742–749. [Google Scholar] [PubMed]
- Schnorr, T.M.; Lawson, C.C.; Whelan, E.A.; Dankovic, D.A.; Deddens, J.A.; Piacitelli, L.A.; Reefhuis, J.; Sweeney, M.H.; Connally, L.B.; Fingerhut, M.A. Spontaneous abortion, sex ratio, and paternal occupational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Environ. Health Perspect. 2001, 109, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Augustowska, K.; Gregoraszczuk, E.E.; Grochowalski, A.; Milewicz, T.; Mika, M.; Krzysiek, J.; Chrzaszcz, R. Comparison of accumulation and altered steroid secretion by placental tissue treated with TCDD and natural mixture of PCDDs–PCDFs. Reproduction 2003, 126, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Ohsako, S.; Miyabara, Y.; Sakaue, M.; Kawakami, T.; Aoki, Y.; Yonemoto, J.; Tohyama, C. Increased glycogen content and glucose transporter 3 mRNA level in the placenta of Holtzman rats after exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 2002, 178, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Kang, W.Y.; Moon, S.G.; Kim, H.J.; Kim, K.H.; Kim, Y.H.; Hwang, S.H.; Hwang, S.H.; Kim, W. Clinical outcome of veterans with acute coronary syndrome who had been exposed to agent orange. Chonnam. Med. J. 2012, 48, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Eskenazi, B.; Mocarelli, P.; Warner, M.; Chee, W.Y.; Gerthoux, P.M.; Samuels, S.; Needham, L.L.; Patterson, D.G., Jr. Maternal serum dioxin levels and birth outcomes in women of Seveso, Italy. Environ. Health Perspect. 2003, 111, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Kawakami, T.; Ohsako, S.; Tohyama, C. Dioxin-induced toxicity on vascular remodeling of the placenta. Biochem. Pharmacol. 2009, 77, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Ivnitski-Steele, I.; Walker, M.K. Inhibition of neovascularization by environmental agents. Cardiovasc. Toxicol. 2005, 5, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Ohsako, S.; Kawakami, T.; Sakaue, M.; Aoki, Y.; Tohyama, C. Altered protein profile and possible hypoxia in the placenta of 2,3,7,8-tetrachlorodibenzo-p-dioxin-exposed rats. Toxicol. Appl. Pharmacol. 2002, 185, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hagen, T. Oxygen versus reactive oxygen in the regulation of HIF-1α: The balance tips. Biochem. Res. Int. 2012, 2012, 436981. [Google Scholar] [CrossRef]
- Grassman, J.A.; Masten, S.A.; Walker, N.J.; Lucier, G.W. Animal models of human response to dioxins. Environ. Health Perspect. 1998, 106, 761–775. [Google Scholar]
- Hurst, C.H.; DeVito, M.J.; Birnbaum, L.S. Tissue disposition of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in maternal and developing long-evans rats following subchronic exposure. Toxicol. Sci. 2000, 57, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Noguchi, T.; Takekoshi, H.; Suzuki, G.; Nakano, M. Maternal-fetal distribution and transfer of dioxins in pregnant women in Japan, and attempts to reduce maternal transfer with Chlorella (Chlorella pyrenoidosa) supplements. Chemosphere 2005, 61, 1244–1255. [Google Scholar] [CrossRef]
- Nie, M.; Blankenship, A.L.; Giesy, J.P. Interactions between aryl hydrocarbon receptor (AhR) and hypoxia signaling pathways. Environ. Toxicol. Pharmacol. 2001, 10, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Gordon, L.; Wong, N.C.; Moffett, A.; Manuelpillai, U.; Craig, J.M.; Sharkey, A.; Saffery, R. Wide-ranging DNA methylation differences of primary trophoblast cell populations and derived cell lines: Implications and opportunities for understanding trophoblast function. Mol. Hum. Reprod. 2011, 17, 44–353. [Google Scholar] [CrossRef]
- Oreshkova, T.; Dimitrov, R.; Mourdjeva, M. A cross-talk of decidual stromal cells, trophoblast, and immune cells: A prerequisite for the success of pregnancy. Am. J. Reprod. Immunol. 2012, 68, 366–373. [Google Scholar] [CrossRef]
- Fukushima, K.; Tsukimori, K.; Li, D.; Takao, T.; Morokuma, S.; Kato, K.; Seki, H.; Takeda, S.; Matsumura, S.; Wake, N. Effect of transient TCDD exposure on immortalized human trophoblast-derived cell lines. Hum. Exp. Toxicol. 2012, 31, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Takao, T.; Asanoma, K.; Kato, K.; Fukushima, K.; Tsunematsu, R.; Hirakawa, T.; Matsumura, S.; Seki, H.; Takeda, S.; Wake, N. Isolation and characterization of human trophoblast side-population (SP) cells in primary villous cytotrophoblasts and HTR-8/SVneo cell line. PLoS One 2011, 6, e21990. [Google Scholar] [CrossRef]
- Patel, P.H.; Chadalavada, R.S.; Chaganti, R.S.; Motzer, R.J. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin. Cancer. Res. 2006, 12, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Mack, F.; Haase, V.H.; Simon, M.C.; Johnson, R.S. pVHL function is essential for endothelial extracellular matrix deposition. Mol. Cel.l Biol. 2006, 26, 2519–2530. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1α in vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 26765–26771. [Google Scholar] [PubMed]
- Zelzer, E.; Levy, Y.; Kahana, C.; Shilo, B.Z.; Rubinstein, M.; Cohen, B. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO J. 1998, 17, 5085–5094. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Kalra, V.K. Placenta growth factor-induced early growth response 1 (Egr-1) regulates hypoxia-inducible factor-1alpha (HIF-1α) in endothelial cells. J. Biol. Chem. 2010, 285, 20570–20579. [Google Scholar] [CrossRef]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; Ha, J. Reactive oxygen species stabilize hypoxia-inducible factor-1 α protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Stone, P.R.; Chamley, L.W. The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy. Hum. Reprod. Update 2006, 12, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Genbacev, O.; Zhou, Y.; Ludlow, J.W.; Fisher, S.J. Regulation of human placental development by oxygen tension. Science 1997, 277, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Ietta, F.; Wu, Y.; Winter, J.; Xu, J.; Wang, J.; Post, M.; Caniggia, I. Dynamic HIF1A regulation during human placental development. Biol. Reprod. 2006, 75, 112–121. [Google Scholar] [PubMed]
- Villano, C.M.; Murphy, K.A.; Akintobi, A.; White, L.A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol. Appl. Pharmacol. 2006, 210, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, J.; Luo, X.; Wang, X.; Li, M.; Wang, L.; Li, D. Abnormal regulation of chemokine TECK and its receptor CCR9 in the endometriotic milieu is involved in pathogenesis of endometriosis by way of enhancing invasiveness of endometrial stromal cells. Cell Mol. Immunol. 2010, 7, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Mikami, S.; Kikuchi, E.; Kosaka, T.; Miyajima, A.; Nakagawa, K.; Mukai, M.; Okada, Y.; Oya, M. Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by upregulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis 2010, 31, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Ishida, J.; Aoki, I.; Fukamizu, A. Pathophysiology of placentation abnormalities in pregnancy-induced hypertension. Vasc. Health Risk Manag. 2008, 4, 1301–1313. [Google Scholar] [PubMed]
- Geusens, N.; Verlohren, S.; Luyten, C.; Taube, M.; Hering, L.; Vercruysse, L.; Hanssens, M.; Dudenhausen, J.W.; Dechend, R.; Pijnenborg, R. Endovascular trophoblast invasion, spiral artery remodelling and uteroplacental haemodynamics in a transgenic rat model of pre-eclampsia. Placenta 2008, 29, 614–623. [Google Scholar] [CrossRef] [PubMed]
- John, C.S.; Bowen, W.D.; Fisher, S.J.; Lim, B.B.; Geyer, B.C.; Vilner, B.J.; Wahl, R.L. Synthesis, in vitro pharmacologic characterization, and preclinical evaluation of N-[2-(1'-piperidinyl)ethyl]-3-[125I]iodo-4-methoxybenzamide (P[125I]MBA) for imaging breast cancer. Nucl. Med. Biol. 1999, 26, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, K.; Nakayama, M.; Suehara, N.; Wada, Y. Expression of vascular endothelial growth factor, placental growth factor, and their receptors Flt-1 and KDR in human placenta under pathologic conditions. Hum. Pathol. 2002, 33, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Munaut, C.; Lorquet, S.; Pequeux, C.; Blacher, S.; Berndt, S.; Frankenne, F.; Foidart, J.M. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum. Reprod. 2008, 23, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Lash, G.E.; Taylor, C.M.; Trew, A.J.; Cooper, S.; Anthony, F.W.; Wheeler, T.; Baker, P.N. Vascular endothelial growth factor and placental growth factor release in cultured trophoblast cells under different oxygen tensions. Growth Factors 2002, 20, 189–196. [Google Scholar] [CrossRef]
- Waite, L.L.; Person, E.C.; Zhou, Y.; Lim, K.H.; Scanlan, T.S.; Taylor, R.N. Placental peroxisome proliferator-activated receptor-gamma is up-regulated by pregnancy serum. J. Clin. Endocrinol. Metab. 2000, 85, 3808–3814. [Google Scholar] [PubMed]
- Parast, M.M.; Yu, H.; Ciric, A.; Salata, M.W.; Davis, V.; Milstone, D.S. PPARγ regulates trophoblast proliferation and promotes labyrinthine trilineage differentiation. PLoS One 2009, 4, e8055. [Google Scholar] [CrossRef]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef]
- He, X.; Sun, C.; Wang, F.; Shan, A.; Guo, T.; Gu, W.; Cui, B.; Ning, G. Peri-implantation lethality in mice lacking the PGC-1-related coactivator protein. Dev. Dyn. 2012, 241, 975–983. [Google Scholar] [PubMed]
- Ernst, J.; Jann, J.C.; Biemann, R.; Koch, H.M.; Fischer, B. Effects of the environmental contaminants DEHP and TCDD on estradiol synthesis and aryl hydrocarbon receptor and peroxisome proliferator-activated receptor signalling in the human granulosa cell line KGN. Mol. Hum. Reprod. 2014. [Google Scholar] [CrossRef]
- Diani-Moore, S.; Ram, P.; Li, X.; Mondal, P.; Youn, D.Y.; Sauve, A.A.; Rifkind, A.B. Identification of the aryl hydrocarbon receptor target gene TiPARP as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 2010, 285, 38801–38810. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.J.; Hodyl, N.A.; Butler, M.; Clifton, V.L. Localisation and characterisation of uncoupling protein-2 (UCP2) in the human preterm placenta. Placenta 2012, 33, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Liao, T.L.; Wei, Y.H.; Tzeng, C.R.; Kao, S.H. Endocrine disruptor, dioxin (TCDD)-induced mitochondrial dysfunction and apoptosis in human trophoblast-like JAR cells. Mol. Hum. Reprod. 2010, 16, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, T.-L.; Chen, S.-C.; Tzeng, C.-R.; Kao, S.-H. TCDD Induces the Hypoxia-Inducible Factor (HIF)-1α Regulatory Pathway in Human Trophoblastic JAR Cells. Int. J. Mol. Sci. 2014, 15, 17733-17750. https://doi.org/10.3390/ijms151017733
Liao T-L, Chen S-C, Tzeng C-R, Kao S-H. TCDD Induces the Hypoxia-Inducible Factor (HIF)-1α Regulatory Pathway in Human Trophoblastic JAR Cells. International Journal of Molecular Sciences. 2014; 15(10):17733-17750. https://doi.org/10.3390/ijms151017733
Chicago/Turabian StyleLiao, Tien-Ling, Su-Chee Chen, Chii-Reuy Tzeng, and Shu-Huei Kao. 2014. "TCDD Induces the Hypoxia-Inducible Factor (HIF)-1α Regulatory Pathway in Human Trophoblastic JAR Cells" International Journal of Molecular Sciences 15, no. 10: 17733-17750. https://doi.org/10.3390/ijms151017733
APA StyleLiao, T.-L., Chen, S.-C., Tzeng, C.-R., & Kao, S.-H. (2014). TCDD Induces the Hypoxia-Inducible Factor (HIF)-1α Regulatory Pathway in Human Trophoblastic JAR Cells. International Journal of Molecular Sciences, 15(10), 17733-17750. https://doi.org/10.3390/ijms151017733