Noncoding RNA in Oncogenesis: A New Era of Identifying Key Players

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
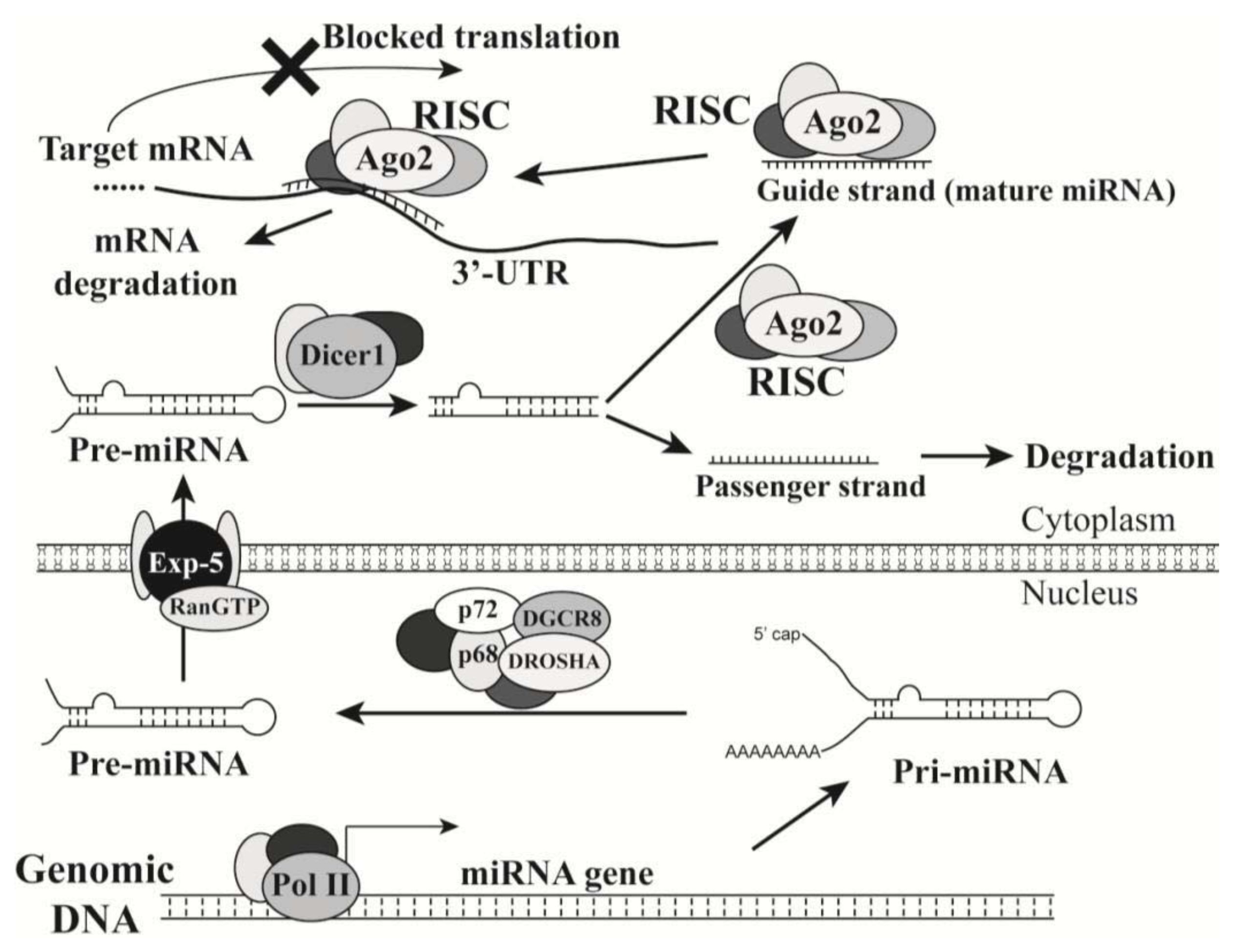
2. MicroRNAs
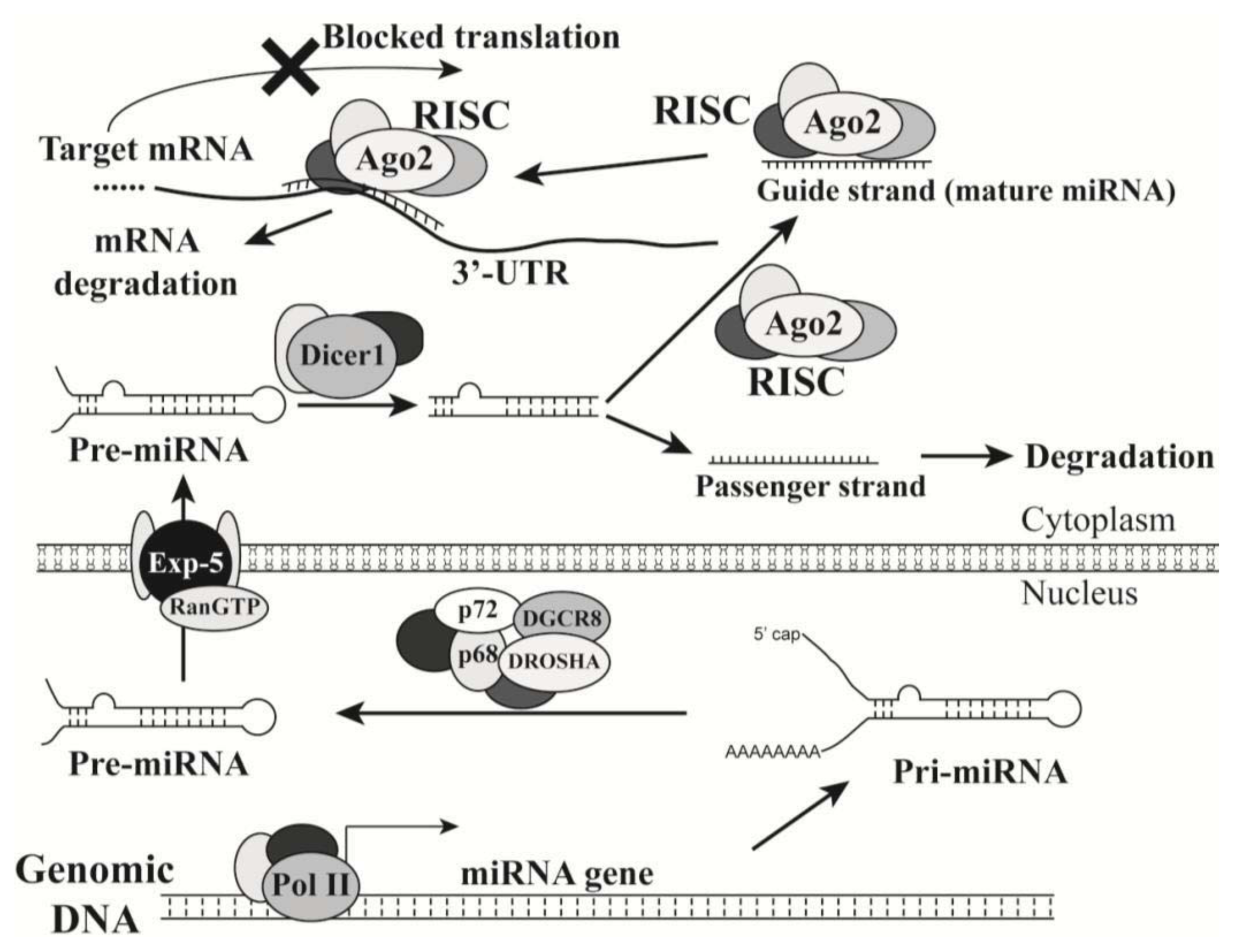
2.1. Introduction of MicroRNAs
2.2. MicroRNAs in Oncogenesis
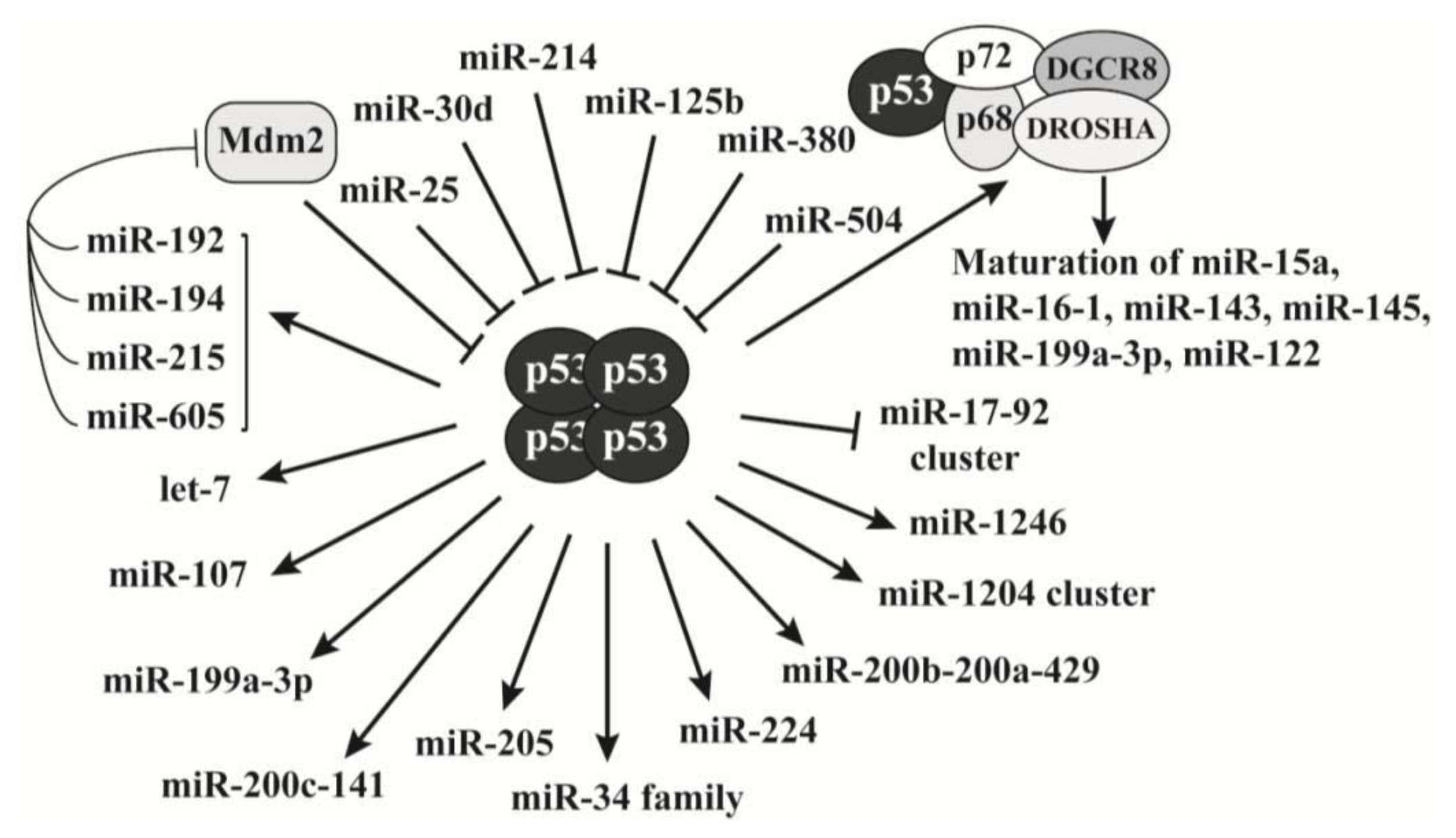
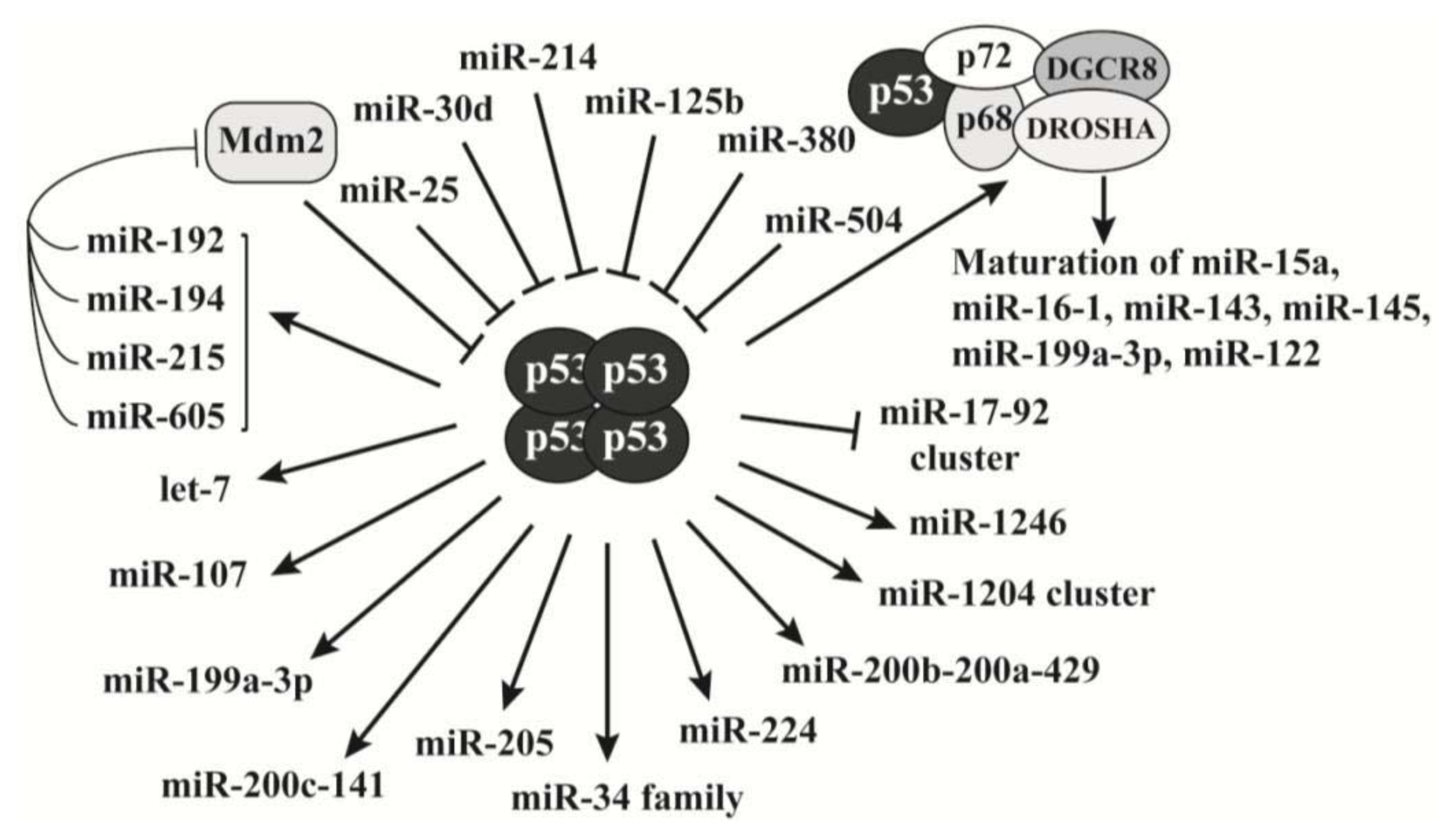
2.3. P53 Is a Key Regulator of MicroRNAs Biogenesis
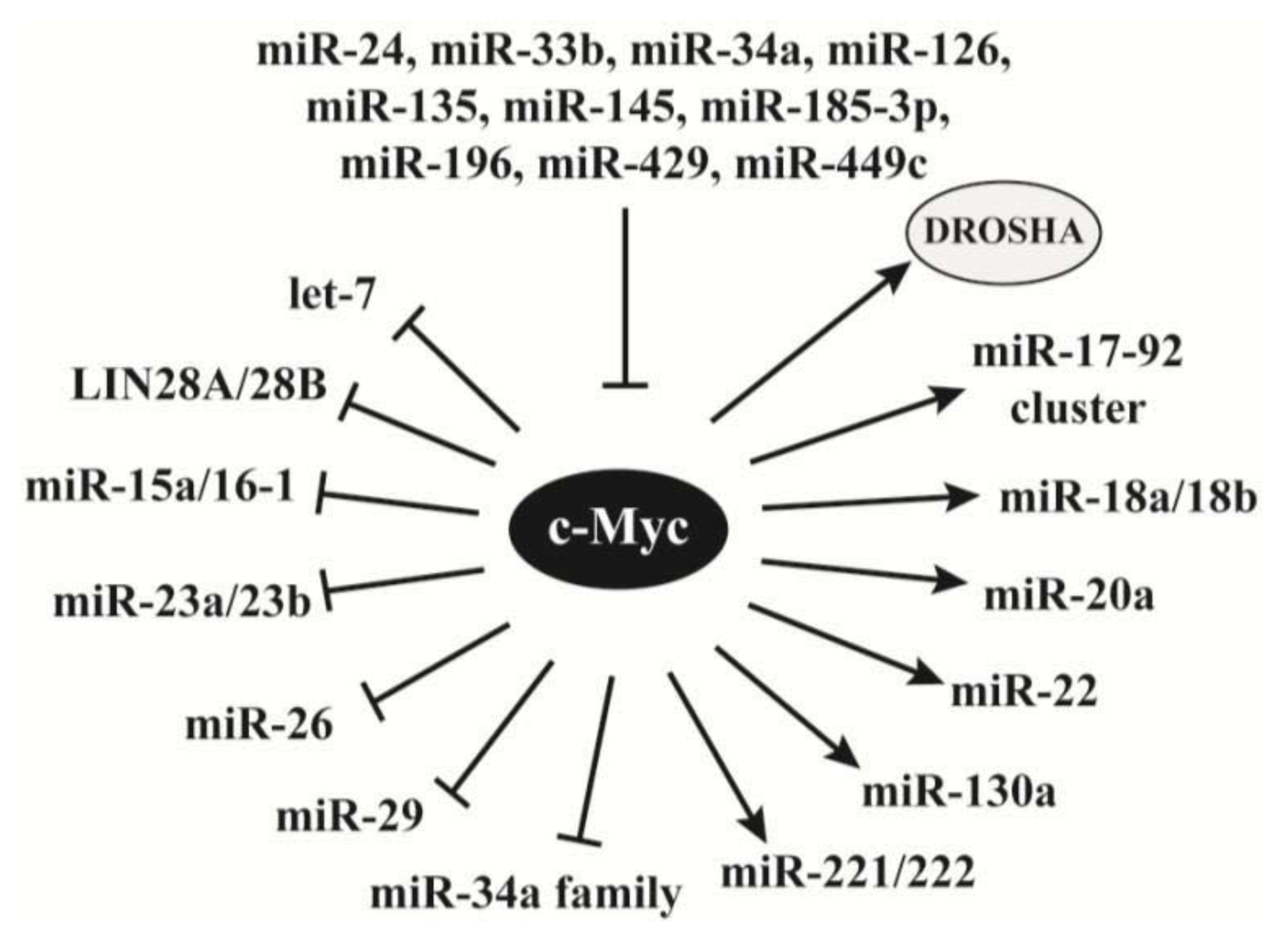
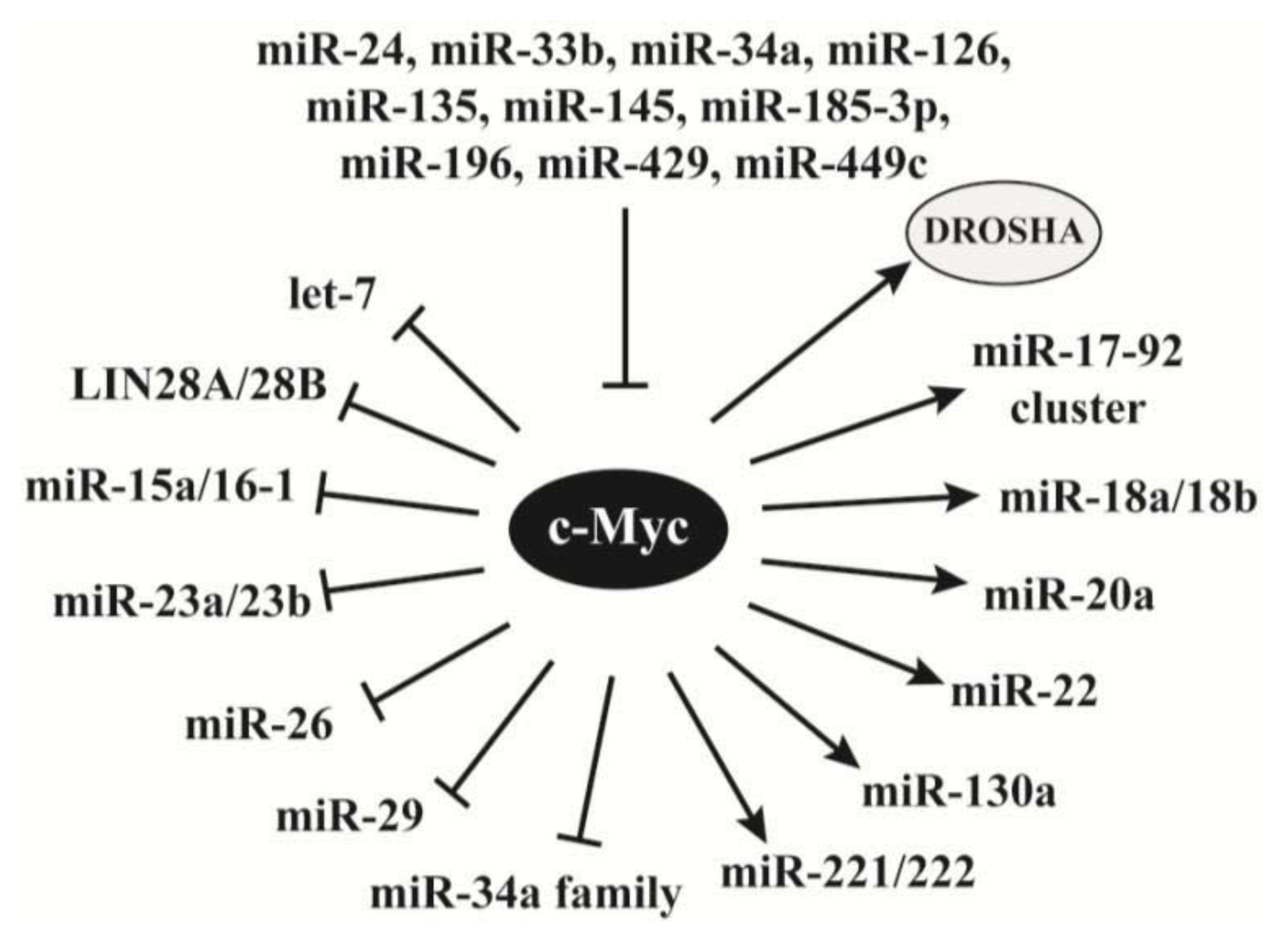
2.4. C-Myc Regulates the Synthesis of miRNAs
3. Long Noncoding RNA
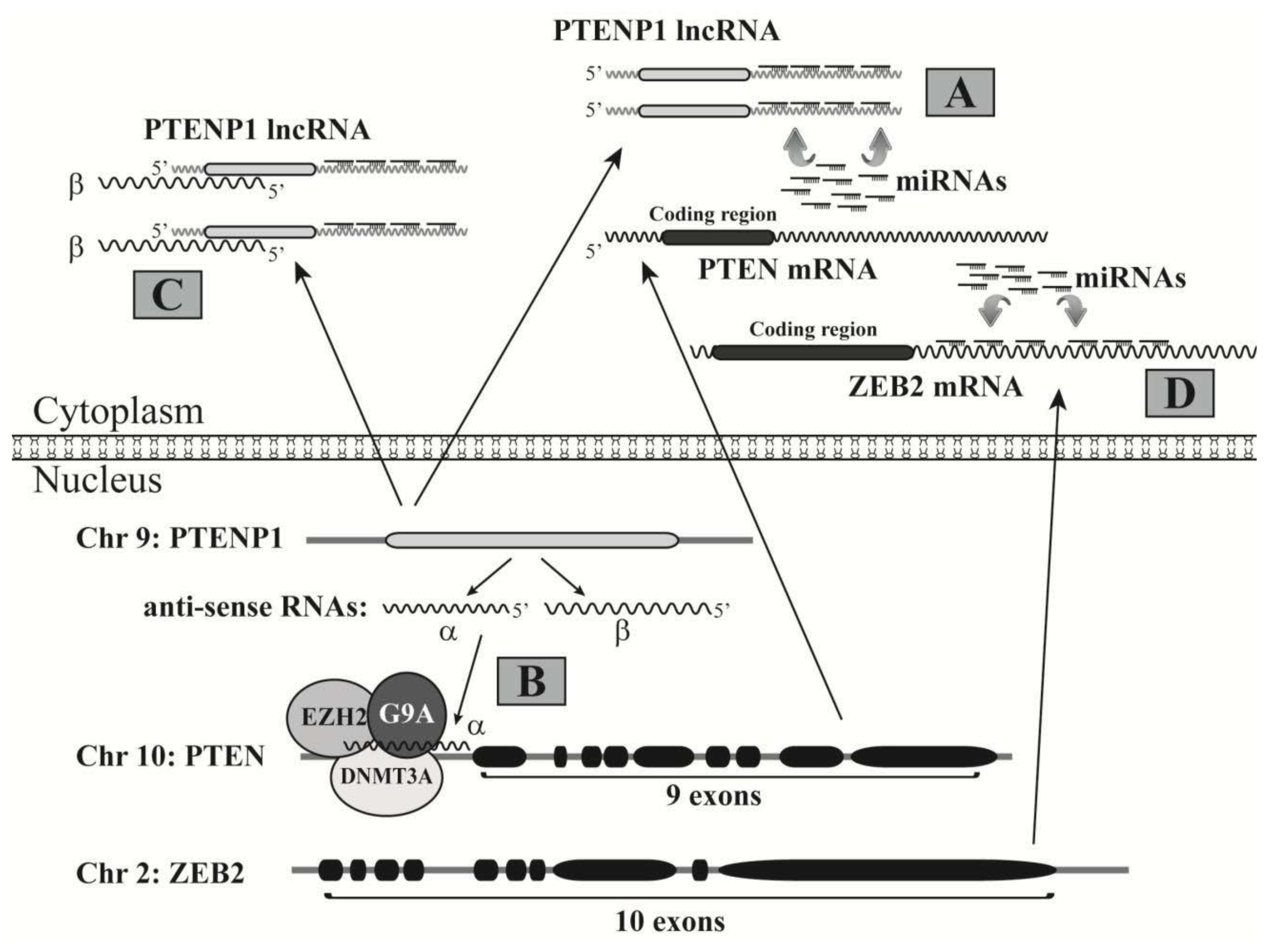
3.1. LncRNAs Regulating Gene Expression
3.2. LncRNAs Acting as miRNA Decoys to Free Target mRNAs
3.3. LncRNAs Regulating mRNA Translation
3.4. LncRNAs Regulating Protein Activities
4. Circular RNAs
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 2005, 308, 1149–1154. [Google Scholar]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev 2009, 23, 1494–1504. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet 2009, 10, 155–159. [Google Scholar]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar]
- Cooper, G.M. Cellular transforming genes. Science 1982, 217, 801–806. [Google Scholar]
- Santos, E.; Tronick, S.R.; Aaronson, S.A.; Pulciani, S.; Barbacid, M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature 1982, 298, 343–347. [Google Scholar]
- Parada, L.F.; Tabin, C.J.; Shih, C.; Weinberg, R.A. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature 1982, 297, 474–478. [Google Scholar]
- Godbout, R.; Dryja, T.P.; Squire, J.; Gallie, B.L.; Phillips, R.A. Somatic inactivation of genes on chromosome 13 is a common event in retinoblastoma. Nature 1983, 304, 451–453. [Google Scholar]
- Murphree, A.L.; Benedict, W.F. Retinoblastoma: Clues to human oncogenesis. Science 1984, 223, 1028–1033. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet 2011, 12, 861–874. [Google Scholar]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol 2005, 6, 376–385. [Google Scholar]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet 2011, 12, 19–31. [Google Scholar]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Guo, L.; Lu, Z. The fate of miRNA* strand through evolutionary analysis: Implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One 2010, 5, e11387. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet 2005, 37, 495–500. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar]
- Chen, X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230. [Google Scholar]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 2006, 20, 515–524. [Google Scholar]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet 2011, 12, 99–110. [Google Scholar]
- Djuranovic, S.; Nahvi, A.; Green, R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 2012, 336, 237–240. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Wang, D.; Qiu, C.; Zhang, H.; Wang, J.; Cui, Q.; Yin, Y. Human microRNA oncogenes and tumor suppressors show significantly different biological patterns: From functions to targets. PLoS One 2010, 5, e13067. [Google Scholar]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar]
- Diederichs, S.; Haber, D.A. Sequence variations of microRNAs in human cancer: Alterations in predicted secondary structure do not affect processing. Cancer Res 2006, 66, 6097–6104. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene 2006, 25, 2537–2545. [Google Scholar]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar]
- Rosenfeld, N.; Aharonov, R.; Meiri, E.; Rosenwald, S.; Spector, Y.; Zepeniuk, M.; Benjamin, H.; Shabes, N.; Tabak, S.; Levy, A.; et al. MicroRNAs accurately identify cancer tissue origin. Nat. Biotechnol 2008, 26, 462–469. [Google Scholar]
- Xi, Y.; Nakajima, G.; Gavin, E.; Morris, C.G.; Kudo, K.; Hayashi, K.; Ju, J. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA 2007, 13, 1668–1674. [Google Scholar]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar]
- Chan, M.; Liaw, C.S.; Ji, S.M.; Tan, H.H.; Wong, C.Y.; Thike, A.A.; Tan, P.H.; Ho, G.H.; Lee, A.S. Identification of circulating microRNA signatures for breast cancer detection. Clin. Cancer Res 2013, 19, 4477–4487. [Google Scholar]
- Chen, J.; Yao, D.; Li, Y.; Chen, H.; He, C.; Ding, N.; Lu, Y.; Ou, T.; Zhao, S.; Li, L.; et al. Serum microRNA expression levels can predict lymph node metastasis in patients with early-stage cervical squamous cell carcinoma. Int. J. Mol. Med 2013, 32, 557–567. [Google Scholar]
- Xiao, Y.F.; Yong, X.; Fan, Y.H.; Lu, M.H.; Yang, S.M.; Hu, C.J. microRNA detection in feces, sputum, pleural effusion and urine: Novel tools for cancer screening (Review). Oncol. Rep 2013, 30, 535–544. [Google Scholar]
- Xiang, J.; Wu, J. Feud or Friend? The role of the miR-17-92 cluster in tumorigenesis. Curr. Genomics 2010, 11, 129–135. [Google Scholar]
- Ota, A.; Tagawa, H.; Karnan, S.; Tsuzuki, S.; Karpas, A.; Kira, S.; Yoshida, Y.; Seto, M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 2004, 64, 3087–3095. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar]
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar]
- Engelmann, D.; Putzer, B.M. The dark side of E2F1: In transit beyond apoptosis. Cancer Res 2012, 72, 571–575. [Google Scholar]
- Biswas, A.K.; Johnson, D.G. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res 2012, 72, 13–17. [Google Scholar]
- Hossain, A.; Kuo, M.T.; Saunders, G.F. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol. Cell. Biol 2006, 26, 8191–8201. [Google Scholar]
- Louie, M.C.; Zou, J.X.; Rabinovich, A.; Chen, H.W. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol. Cell. Biol 2004, 24, 5157–5171. [Google Scholar]
- Qin, L.; Liao, L.; Redmond, A.; Young, L.; Yuan, Y.; Chen, H.; O’Malley, B.W.; Xu, J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol. Cell. Biol 2008, 28, 5937–5950. [Google Scholar]
- Yu, Z.; Wang, C.; Wang, M.; Li, Z.; Casimiro, M.C.; Liu, M.; Wu, K.; Whittle, J.; Ju, X.; Hyslop, T.; et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J. Cell Biol 2008, 182, 509–517. [Google Scholar]
- Yu, Z.; Willmarth, N.E.; Zhou, J.; Katiyar, S.; Wang, M.; Liu, Y.; McCue, P.A.; Quong, A.A.; Lisanti, M.P.; Pestell, R.G. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 8231–8236. [Google Scholar]
- Li, H.; Yang, B.B. Stress response of glioblastoma cells mediated by miR-17-5p targeting PTEN and the passenger strand miR-17-3p targeting MDM2. Oncotarget 2012, 3, 1653–1668. [Google Scholar]
- Shan, S.W.; Fang, L.; Shatseva, T.; Rutnam, Z.J.; Yang, X.; Du, W.; Lu, W.Y.; Xuan, J.W.; Deng, Z.; Yang, B.B. Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. J. Cell Sci 2013, 126, 1517–1530. [Google Scholar]
- Cao, P.; Deng, Z.; Wan, M.; Huang, W.; Cramer, S.D.; Xu, J.; Lei, M.; Sui, G. MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1alpha/HIF-1beta. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.J.; Ruan, H.J.; He, X.J.; Ma, Y.Y.; Jiang, X.T.; Xia, Y.J.; Ye, Z.Y.; Tao, H.Q. MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 2010, 46, 2295–2303. [Google Scholar]
- Hu, Z.; Lin, Y.; Chen, H.; Mao, Y.; Wu, J.; Zhu, Y.; Xu, X.; Li, S.; Zheng, X.; Xie, L. MicroRNA-101 suppresses motility of bladder cancer cells by targeting c-Met. Biochem. Biophys. Res. Commun 2013, 435, 82–87. [Google Scholar]
- Wang, R.; Wang, H.B.; Hao, C.J.; Cui, Y.; Han, X.C.; Hu, Y.; Li, F.F.; Xia, H.F.; Ma, X. MiR-101 is involved in human breast carcinogenesis by targeting Stathmin1. PLoS One 2012, 7, e46173. [Google Scholar]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar]
- Sellers, W.R.; Loda, M. The EZH2 polycomb transcriptional repressor—A marker or mover of metastatic prostate cancer? Cancer Cell 2002, 2, 349–350. [Google Scholar]
- Friedman, J.M.; Liang, G.; Liu, C.C.; Wolff, E.M.; Tsai, Y.C.; Ye, W.; Zhou, X.; Jones, P.A. The putative tumor suppressor microRNA-101 modulates the cancer epigenome by repressing the polycomb group protein EZH2. Cancer Res 2009, 69, 2623–2629. [Google Scholar]
- Banerjee, R.; Mani, R.S.; Russo, N.; Scanlon, C.S.; Tsodikov, A.; Jing, X.; Cao, Q.; Palanisamy, N.; Metwally, T.; Inglehart, R.C.; et al. The tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2 overexpression in invasive squamous cell carcinoma. Oncogene 2011, 30, 4339–4349. [Google Scholar]
- Kottakis, F.; Polytarchou, C.; Foltopoulou, P.; Sanidas, I.; Kampranis, S.C.; Tsichlis, P.N. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol. Cell 2011, 43, 285–298. [Google Scholar]
- Qazi, A.M.; Gruzdyn, O.; Semaan, A.; Seward, S.; Chamala, S.; Dhulipala, V.; Sethi, S.; Ali-Fehmi, R.; Philip, P.A.; Bouwman, D.L.; et al. Restoration of E-cadherin expression in pancreatic ductal adenocarcinoma treated with microRNA-101. Surgery 2012, 152, 704–711, , discussion 711–713.. [Google Scholar]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Zhang, J.; Chen, X.; eVere White, R.W.; Kung, H.J.; Evans, C.P.; Gao, A.C. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J. Biol. Chem 2012, 287, 1527–1537. [Google Scholar]
- Boyerinas, B.; Park, S.M.; Hau, A.; Murmann, A.E.; Peter, M.E. The role of let-7 in cell differentiation and cancer. Endocr. Relat. Cancer 2010, 17, F19–F36. [Google Scholar]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar]
- Hunten, S.; Siemens, H.; Kaller, M.; Hermeking, H. The p53/microRNA network in cancer: Experimental and bioinformatics approaches. Adv. Exp. Med. Biol 2013, 774, 77–101. [Google Scholar]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Chang, I.; Tanaka, Y.; Gupta, A.; Dahiya, R. MicroRNA-34a suppresses malignant transformation by targeting c-Myc transcriptional complexes in human renal cell carcinoma. Carcinogenesis 2012, 33, 294–300. [Google Scholar]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS One 2012, 7, e29722. [Google Scholar]
- Cole, K.A.; Attiyeh, E.F.; Mosse, Y.P.; Laquaglia, M.J.; Diskin, S.J.; Brodeur, G.M.; Maris, J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res 2008, 6, 735–742. [Google Scholar]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010, 17, 193–199. [Google Scholar]
- Wong, C.S.; Sceneay, J.; House, C.M.; Halse, H.M.; Liu, M.C.; George, J.; Hunnam, T.C.; Parker, B.S.; Haviv, I.; Ronai, Z.; et al. Vascular normalization by loss of Siah2 results in increased chemotherapeutic efficacy. Cancer Res 2012, 72, 1694–1704. [Google Scholar]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Yamakuchi, M.; Lotterman, C.D.; Bao, C.; Hruban, R.H.; Karim, B.; Mendell, J.T.; Huso, D.; Lowenstein, C.J. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6334–6339. [Google Scholar]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol 2011, 13, 317–323. [Google Scholar]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.S.; et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J. Exp. Med 2011, 208, 875–883. [Google Scholar]
- Yan, H.L.; Xue, G.; Mei, Q.; Wang, Y.Z.; Ding, F.X.; Liu, M.F.; Lu, M.H.; Tang, Y.; Yu, H.Y.; Sun, S.H. Repression of the miR-17–92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J 2009, 28, 2719–2732. [Google Scholar]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar]
- Kohlstedt, K.; Trouvain, C.; Boettger, T.; Shi, L.; Fisslthaler, B.; Fleming, I. AMP-activated protein kinase regulates endothelial cell angiotensin-converting enzyme expression via p53 and the post-transcriptional regulation of microRNA-143/145. Circ. Res 2013, 112, 1150–1158. [Google Scholar]
- Wang, J.; He, Q.; Han, C.; Gu, H.; Jin, L.; Li, Q.; Mei, Y.; Wu, M. p53-facilitated miR-199a-3p regulates somatic cell reprogramming. Stem Cells 2012, 30, 1405–1413. [Google Scholar]
- Manfe, V.; Biskup, E.; Rosbjerg, A.; Kamstrup, M.; Skov, A.G.; Lerche, C.M.; Lauenborg, B.T.; Odum, N.; Gniadecki, R. miR-122 regulates p53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS One 2012, 7, e29541. [Google Scholar]
- Michael, D.; Oren, M. The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol 2003, 13, 49–58. [Google Scholar]
- Pichiorri, F.; Suh, S.S.; Rocci, A.; De Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M., Jr; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010, 18, 367–381. [Google Scholar]
- Khella, H.W.; Bakhet, M.; Allo, G.; Jewett, M.A.; Girgis, A.H.; Latif, A.; Girgis, H.; von Both, I.; Bjarnason, G.A.; Yousef, G.M. miR-192, miR-194 and miR-215: A convergent microRNA network suppressing tumor progression in renal cell carcinoma. Carcinogenesis 2013. [Google Scholar] [CrossRef]
- Senanayake, U.; Das, S.; Vesely, P.; Alzoughbi, W.; Frohlich, L.F.; Chowdhury, P.; Leuschner, I.; Hoefler, G.; Guertl, B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis 2012, 33, 1014–1021. [Google Scholar]
- Xiao, J.; Lin, H.; Luo, X.; Wang, Z. miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J 2011, 30, 524–532. [Google Scholar]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar]
- Craig, V.J.; Cogliatti, S.B.; Imig, J.; Renner, C.; Neuenschwander, S.; Rehrauer, H.; Schlapbach, R.; Dirnhofer, S.; Tzankov, A.; Muller, A. Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011, 117, 6227–6236. [Google Scholar] [Green Version]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16–1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med 2008, 14, 1271–1277. [Google Scholar]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar]
- Schmitt, M.J.; Margue, C.; Behrmann, I.; Kreis, S. MiRNA-29: A microRNA family with tumor-suppressing and immune-modulating properties. Curr. Mol. Med 2013, 13, 572–585. [Google Scholar]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.U.; Straka, E.; Su, L.; Burki, E.A.; et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res 2008, 68, 8535–8540. [Google Scholar]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res 2009, 15, 6479–6483. [Google Scholar]
- Jung, Y.J.; Kim, J.W.; Park, S.J.; Min, B.Y.; Jang, E.S.; Kim, N.Y.; Jeong, S.H.; Shin, C.M.; Lee, S.H.; Park, Y.S.; et al. c-Myc-mediated overexpression of miR-17-92 suppresses replication of hepatitis B virus in human hepatoma cells. J. Med. Virol 2013, 85, 969–978. [Google Scholar]
- Kim, J.W.; Mori, S.; Nevins, J.R. Myc-induced microRNAs integrate Myc-mediated cell proliferation and cell fate. Cancer Res 2010, 70, 4820–4828. [Google Scholar]
- Medina, R.; Zaidi, S.K.; Liu, C.G.; Stein, J.L.; van Wijnen, A.J.; Croce, C.M.; Stein, G.S. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res 2008, 68, 2773–2780. [Google Scholar]
- Visone, R.; Russo, L.; Pallante, P.; de Martino, I.; Ferraro, A.; Leone, V.; Borbone, E.; Petrocca, F.; Alder, H.; Croce, C.M.; et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr. Relat. Cancer 2007, 14, 791–798. [Google Scholar]
- Fornari, F.; Gramantieri, L.; Ferracin, M.; Veronese, A.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Giovannini, C.; Croce, C.M.; Bolondi, L.; et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene 2008, 27, 5651–5661. [Google Scholar]
- Garofalo, M.; di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar]
- Wang, X.; Zhao, X.; Gao, P.; Wu, M. c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Goodrich, J.A.; Kugel, J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol 2006, 7, 612–616. [Google Scholar]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res 2012, 22, 1760–1774. [Google Scholar]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012, 22, 1775–1789. [Google Scholar]
- Banfai, B.; Jia, H.; Khatun, J.; Wood, E.; Risk, B.; Gundling, W.E., Jr; Kundaje, A.; Gunawardena, H.P.; Yu, Y.; Xie, L.; et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res 2012, 22, 1646–1657. [Google Scholar]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafreniere, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar]
- Clemson, C.M.; McNeil, J.A.; Willard, H.F.; Lawrence, J.B. XIST RNA paints the inactive X chromosome at interphase: Evidence for a novel RNA involved in nuclear/chromosome structure. J. Cell Biol 1996, 132, 259–275. [Google Scholar]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar]
- Tian, D.; Sun, S.; Lee, J.T. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 2010, 143, 390–403. [Google Scholar]
- Chureau, C.; Chantalat, S.; Romito, A.; Galvani, A.; Duret, L.; Avner, P.; Rougeulle, C. Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum. Mol. Genet 2011, 20, 705–718. [Google Scholar]
- Lee, J.T.; Davidow, L.S.; Warshawsky, D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet 1999, 21, 400–404. [Google Scholar]
- Lee, J.T. Disruption of imprinted X inactivation by parent-of-origin effects at Tsix. Cell 2000, 103, 17–27. [Google Scholar]
- Ogawa, Y.; Lee, J.T. Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol. Cell 2003, 11, 731–743. [Google Scholar]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar]
- Yang, Z.; Zhou, L.; Wu, L.M.; Lai, M.C.; Xie, H.Y.; Zhang, F.; Zheng, S.S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol 2011, 18, 1243–1250. [Google Scholar]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res 2011, 39, 2119–2128. [Google Scholar]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res 2011, 71, 6320–6326. [Google Scholar]
- Kim, K.; Jutooru, I.; Chadalapaka, G.; Johnson, G.; Frank, J.; Burghardt, R.; Kim, S.; Safe, S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2013, 32, 1616–1625. [Google Scholar]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar]
- Zhang, X.; Lian, Z.; Padden, C.; Gerstein, M.B.; Rozowsky, J.; Snyder, M.; Gingeras, T.R.; Kapranov, P.; Weissman, S.M.; Newburger, P.E. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 2009, 113, 2526–2534. [Google Scholar]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, 39. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar]
- Miyagawa, R.; Tano, K.; Mizuno, R.; Nakamura, Y.; Ijiri, K.; Rakwal, R.; Shibato, J.; Masuo, Y.; Mayeda, A.; Hirose, T.; et al. Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles. RNA 2012, 18, 738–751. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 2013, 73, 1180–1189. [Google Scholar]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011, 147, 773–788. [Google Scholar]
- Guo, F.; Li, Y.; Liu, Y.; Wang, J.; Li, G. Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 224–229. [Google Scholar]
- Yamada, K.; Kano, J.; Tsunoda, H.; Yoshikawa, H.; Okubo, C.; Ishiyama, T.; Noguchi, M. Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci 2006, 97, 106–112. [Google Scholar]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar]
- Xu, C.; Yang, M.; Tian, J.; Wang, X.; Li, Z. MALAT-1: A long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int. J. Oncol 2011, 39, 169–175. [Google Scholar]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 2012, 26, 2392–2407. [Google Scholar]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar]
- Pasmant, E.; Laurendeau, I.; Heron, D.; Vidaud, M.; Vidaud, D.; Bieche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 2007, 67, 3963–3969. [Google Scholar]
- Yap, K.L.; Li, S.; Munoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar]
- Mourtada-Maarabouni, M.; Hedge, V.L.; Kirkham, L.; Farzaneh, F.; Williams, G.T. Growth arrest in human T-cells is controlled by the non-coding RNA growth-arrest-specific transcript 5 (GAS5). J. Cell Sci 2008, 121, 939–946. [Google Scholar]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal 2010, 3. [Google Scholar] [CrossRef]
- Conti, E.; Izaurralde, E. Nonsense-mediated mRNA decay: Molecular insights and mechanistic variations across species. Curr. Opin. Cell Biol 2005, 17, 316–325. [Google Scholar]
- Zhang, Z.; Zhu, Z.; Watabe, K.; Zhang, X.; Bai, C.; Xu, M.; Wu, F.; Mo, Y.Y. Negative regulation of lncRNA GAS5 by miR-21. Cell Death Differ. 2013. [Google Scholar] [CrossRef]
- Pan, X.; Wang, Z.X.; Wang, R. MicroRNA-21: A novel therapeutic target in human cancer. Cancer Biol. Ther 2010, 10, 1224–1232. [Google Scholar]
- Smedley, D.; Sidhar, S.; Birdsall, S.; Bennett, D.; Herlyn, M.; Cooper, C.; Shipley, J. Characterization of chromosome 1 abnormalities in malignant melanomas. Genes Chromosomes Cancer 2000, 28, 121–125. [Google Scholar]
- Nupponen, N.N.; Carpten, J.D. Prostate cancer susceptibility genes: Many studies, many results, no answers. Cancer Metastasis Rev 2001, 20, 155–164. [Google Scholar]
- Stange, D.E.; Radlwimmer, B.; Schubert, F.; Traub, F.; Pich, A.; Toedt, G.; Mendrzyk, F.; Lehmann, U.; Eils, R.; Kreipe, H.; et al. High-resolution genomic profiling reveals association of chromosomal aberrations on 1q and 16p with histologic and genetic subgroups of invasive breast cancer. Clin. Cancer Res 2006, 12, 345–352. [Google Scholar]
- Morrison, L.E.; Jewell, S.S.; Usha, L.; Blondin, B.A.; Rao, R.D.; Tabesh, B.; Kemper, M.; Batus, M.; Coon, J.S. Effects of ERBB2 amplicon size and genomic alterations of chromosomes 1, 3, and 10 on patient response to trastuzumab in metastatic breast cancer. Genes Chromosomes Cancer 2007, 46, 397–405. [Google Scholar]
- Peters, U.; Jiao, S.; Schumacher, F.R.; Hutter, C.M.; Aragaki, A.K.; Baron, J.A.; Berndt, S.I.; Bezieau, S.; Brenner, H.; Butterbach, K.; et al. Identification of genetic susceptibility loci for colorectal tumors in a genome-wide meta-analysis. Gastroenterology 2013, 144, 799–807. [Google Scholar]
- Nakamura, Y.; Takahashi, N.; Kakegawa, E.; Yoshida, K.; Ito, Y.; Kayano, H.; Niitsu, N.; Jinnai, I.; Bessho, M. The GAS5 (growth arrest-specific transcript 5) gene fuses to BCL6 as a result of t(1;3)(q25;q27) in a patient with B-cell lymphoma. Cancer Genet Cytogenet 2008, 182, 144–149. [Google Scholar]
- Tani, H.; Torimura, M.; Akimitsu, N. The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS One 2013, 8, e55684. [Google Scholar]
- Smith, C.M.; Steitz, J.A. Classification of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5′-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol. Cell. Biol 1998, 18, 6897–6909. [Google Scholar]
- Tanaka, R.; Satoh, H.; Moriyama, M.; Satoh, K.; Morishita, Y.; Yoshida, S.; Watanabe, T.; Nakamura, Y.; Mori, S. Intronic U50 small-nucleolar-RNA (snoRNA) host gene of no protein-coding potential is mapped at the chromosome breakpoint t(3;6)(q27;q15) of human B-cell lymphoma. Genes Cells 2000, 5, 277–287. [Google Scholar]
- Raho, G.; Barone, V.; Rossi, D.; Philipson, L.; Sorrentino, V. The gas 5 gene shows four alternative splicing patterns without coding for a protein. Gene 2000, 256, 13–17. [Google Scholar]
- Hirose, T.; Steitz, J.A. Position within the host intron is critical for efficient processing of box C/D snoRNAs in mammalian cells. Proc. Natl. Acad. Sci. USA 2001, 98, 12914–12919. [Google Scholar]
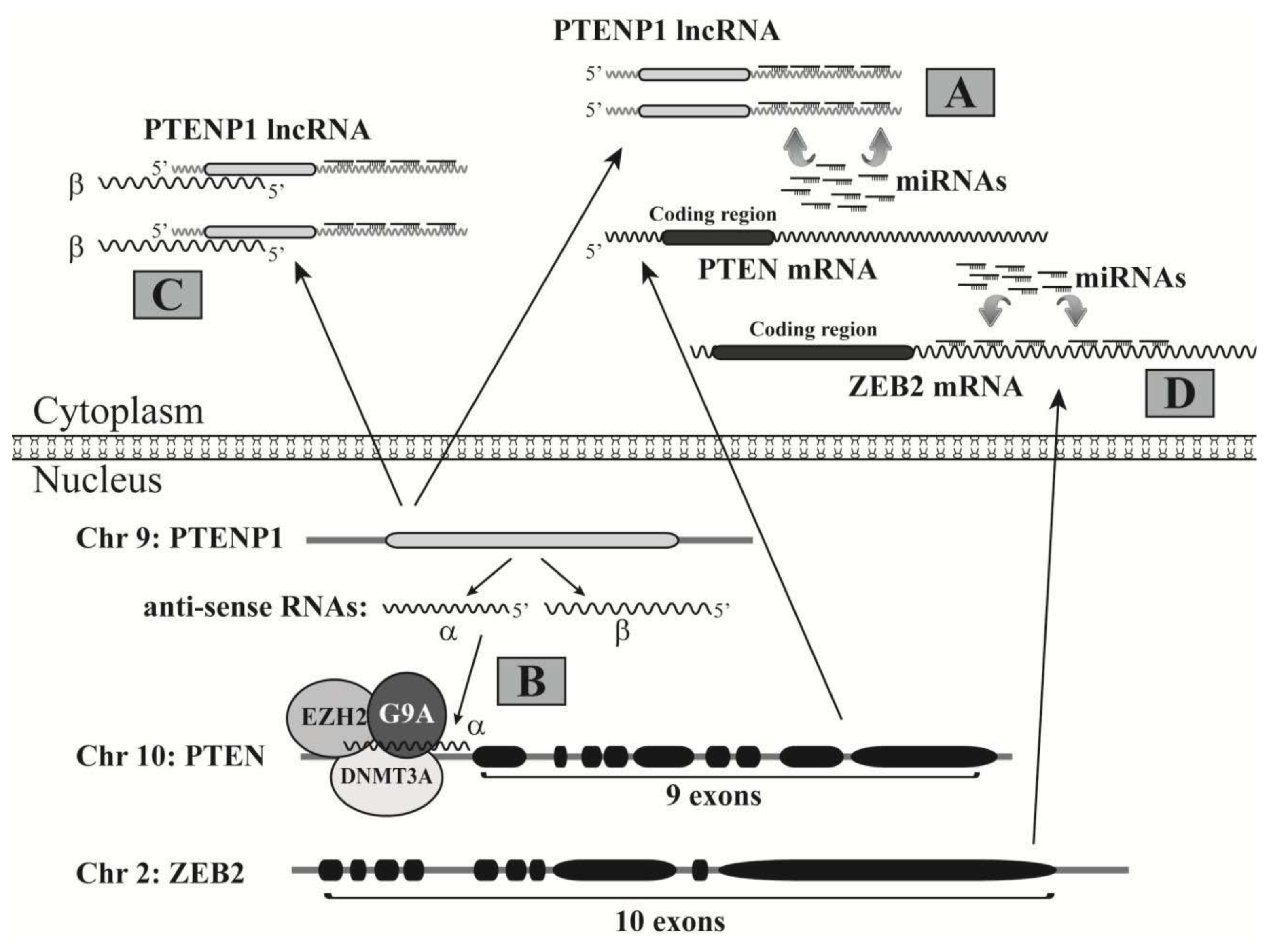
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.O.; Corcoran, M.; Grander, D.; Morris, K.V. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol 2013, 20, 440–446. [Google Scholar]
- Bond, A.M.; Vangompel, M.J.; Sametsky, E.A.; Clark, M.F.; Savage, J.C.; Disterhoft, J.F.; Kohtz, J.D. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci 2009, 12, 1020–1027. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar]
- Sheik Mohamed, J.; Gaughwin, P.M.; Lim, B.; Robson, P.; Lipovich, L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA 2010, 16, 324–337. [Google Scholar]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar]
- Pesole, G.; Liuni, S.; Grillo, G.; Saccone, C. Structural and compositional features of untranslated regions of eukaryotic mRNAs. Gene 1997, 205, 95–102. [Google Scholar]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev 2009, 23, 1327–1337. [Google Scholar]
- Liu, Z.L.; Wang, H.; Liu, J.; Wang, Z.X. MicroRNA-21 (miR-21) expression promotes growth, metastasis, and chemo- or radioresistance in non-small cell lung cancer cells by targeting PTEN. Mol. Cell. Biochem 2013, 372, 35–45. [Google Scholar]
- Vanin, E.F. Processed pseudogenes: Characteristics and evolution. Annu. Rev. Genet 1985, 19, 253–272. [Google Scholar]
- Fujii, G.H.; Morimoto, A.M.; Berson, A.E.; Bolen, J.B. Transcriptional analysis of the PTEN/MMAC1 pseudogene, psiPTEN. Oncogene 1999, 18, 1765–1769. [Google Scholar]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar]
- Lund, A.H.; van Lohuizen, M. Epigenetics and cancer. Genes Dev 2004, 18, 2315–2335. [Google Scholar]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 2011, 147, 382–395. [Google Scholar]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar]
- Nakagawa, S.; Ip, J.Y.; Shioi, G.; Tripathi, V.; Zong, X.; Hirose, T.; Prasanth, K.V. Malat1 is not an essential component of nuclear speckles in mice. RNA 2012, 18, 1487–1499. [Google Scholar]
- Zhang, A.; Zhou, N.; Huang, J.; Liu, Q.; Fukuda, K.; Ma, D.; Lu, Z.; Bai, C.; Watabe, K.; Mo, Y.Y. The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell Res 2013, 23, 340–350. [Google Scholar]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 2007, 318, 798–801. [Google Scholar]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 2010, 38, 5797–5806. [Google Scholar]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar]
- Jauliac, S.; Lopez-Rodriguez, C.; Shaw, L.M.; Brown, L.F.; Rao, A.; Toker, A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol 2002, 4, 540–544. [Google Scholar]
- Yoeli-Lerner, M.; Yiu, G.K.; Rabinovitz, I.; Erhardt, P.; Jauliac, S.; Toker, A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol. Cell 2005, 20, 539–550. [Google Scholar]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar]
- Sharma, S.; Findlay, G.M.; Bandukwala, H.S.; Oberdoerffer, S.; Baust, B.; Li, Z.; Schmidt, V.; Hogan, P.G.; Sacks, D.B.; Rao, A. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc. Natl. Acad. Sci. USA 2011, 108, 11381–11386. [Google Scholar]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012, 7, e30733. [Google Scholar]
- Capel, B.; Swain, A.; Nicolis, S.; Hacker, A.; Walter, M.; Koopman, P.; Goodfellow, P.; Lovell-Badge, R. Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell 1993, 73, 1019–1030. [Google Scholar]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar]
- Hansen, T.B.; Wiklund, E.D.; Bramsen, J.B.; Villadsen, S.B.; Statham, A.L.; Clark, S.J.; Kjems, J. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 2011, 30, 4414–4422. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deng, G.; Sui, G. Noncoding RNA in Oncogenesis: A New Era of Identifying Key Players. Int. J. Mol. Sci. 2013, 14, 18319-18349. https://doi.org/10.3390/ijms140918319
Deng G, Sui G. Noncoding RNA in Oncogenesis: A New Era of Identifying Key Players. International Journal of Molecular Sciences. 2013; 14(9):18319-18349. https://doi.org/10.3390/ijms140918319
Chicago/Turabian StyleDeng, Guorui, and Guangchao Sui. 2013. "Noncoding RNA in Oncogenesis: A New Era of Identifying Key Players" International Journal of Molecular Sciences 14, no. 9: 18319-18349. https://doi.org/10.3390/ijms140918319
APA StyleDeng, G., & Sui, G. (2013). Noncoding RNA in Oncogenesis: A New Era of Identifying Key Players. International Journal of Molecular Sciences, 14(9), 18319-18349. https://doi.org/10.3390/ijms140918319