Role of cMET in the Development and Progression of Colorectal Cancer

Abstract
:1. Introduction
2. Literature Search Methodology
3. Characteristics of MET and Its Role in CRC
3.1. Molecular Biology of HGF/cMET Axis
3.2. Biological Activity of HGF/cMET Axis
Embryogenesis
Tissue regeneration
Cell proliferation and survival
Cytoskeleton
Scattering and cell motility
3.3. cMET Signaling Pathway and Angiogenesis
3.4. cMET and Other Growth Factor Receptors
3.5. MET Mutation and Deregulation
4. Methods of cMET Assessment
5. MET and the Pathogenesis of Colorectal Cancer
6. MET-Targeting by MicroRNAs
7. cMET as Prognostic Biomarker
8. cMET as Predictive Biomarker
8.1. Anti-HGF Monoclonal Antibodies
8.2. Anti-MET Monoclonal Antibodies
8.3. Tyrosine Kinase Inhibitors
9. Conclusions
Acknowledgements
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Frattini, M.; Balestra, D.; Suardi, S.; Oggionni, M.; Alberici, P.; Radice, P.; Costa, A.; Daidone, M.G.; Leo, E.; Pilotti, S.; et al. Different genetic features associated with colon and rectal carcinogenesis. Clin. Cancer Res 2004, 10, 4015–4021. [Google Scholar]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar]
- Maulik, G.; Shrikhande, A.; Kijima, T.; Ma, P.C.; Morrison, P.T.; Salgia, R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 2002, 13, 41–59. [Google Scholar]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar]
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242. [Google Scholar]
- Gherardi, E.; Stoker, M. Hepatocytes and scatter factor. Nature 1990, 346, 228. [Google Scholar]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-Met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar]
- Park, M.; Dean, M.; Kaul, K.; Braun, M.J.; Gonda, M.A.; Vande Woude, G. Sequence of MET protooncogene Cdna has features characteristic of the tyrosin kinase family growth-factors receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 6379–6383. [Google Scholar]
- Birchmeir, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol 2003, 4, 915–925. [Google Scholar]
- Lorenzato, A.; Olivero, M.; Patanè, S.; Rosso, E.; Oliaro, A.; Comoglio, P.M.; di Renzo, M.F. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res 2002, 62, 7025–7030. [Google Scholar]
- Morishita, R.; Aoki, M.; Hashiya, N.; Yamasaki, K.; Kurinami, H.; Shimizu, S.; Makino, H.; Takesya, Y.; Azuma, J.; Ogihara, T. Therapeutic angiogenesis using hepatocyte growth factor (HGF). Curr. Gene Ther 2004, 4, 199–206. [Google Scholar]
- Mhawech-Fauceglia, P.; Afkhami, M.; Pejovic, T. MET/HGF signaling pathway in ovarian carcinoma: Clinical implications and future direction. Patholog. Res. Int 2012, 2012, e960327. [Google Scholar]
- Fukura, T.; Miki, C.; Inoue, T.; Matsumoto, K.; Suzuki, H. Serum hepatocyte growth factor as an index of disease status of patients with colorectal carcinoma. Br. J. Cancer 1998, 78, 454–459. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; Stefani, A.D.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic mutations of the MET oncogene are selected during metastatic spread in human HNSC carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar]
- Lee, J.H.; Han, S.U.; Cho, H.; Jennings, B.; Gerrard, B.; Dean, M.; Schmidt, L.; Zbar, B.; Vande Woude, G.F. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene 2000, 19, 4947–4953. [Google Scholar]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Res 2006, 66, 283–289. [Google Scholar]
- Matsumoto, K.; Nakamura, T. Hepatocyte growth factor: Renotropic role and potential therapeutics for renal diseases. Kidney Int 2001, 59, 2023–2038. [Google Scholar]
- Duh, F.M.; Scherer, S.W.; Tsui, L.C.; Lerman, M.I.; Zbar, B.; Schmidt, L. Gene structure of the human MET proto-oncogene. Oncogene 1997, 15, 1583–1586. [Google Scholar]
- Liu, Y. The human hepatocyte growth factor receptor gene: Complete structural organization and promoter characterization. Gene 1998, 215, 159–169. [Google Scholar]
- Bardelli, A.; Ponzetto, C.; Comoglio, P.M. Identification of functional domanis in the hepatocyte growth factor and its receptor by molecular engineering. J. Biotechnol 1994, 37, 109–122. [Google Scholar]
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphoring receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar]
- Ding, S.; Merkulova-Rainon, T.; Han, Z.C.; Tobelem, G. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 2003, 101, 4816–4822. [Google Scholar]
- Kajiya, K.; Hirakawa, S.; Ma, B.; Drinnenberg, I.; Detmar, M. Hepatocyte growth factor promotes lymphatic vessel formation and function. Embo. J 2005, 24, 2885–2895. [Google Scholar]
- Jung, W.; Castren, E.; Odenthal, M.; Vande Woude, G.F.; Ishii, T.; Dienes, H.P.; Lindholm, D.; Schirmacher, P. Expression and functional interaction of hepatocyte growth factor-scatter factor and its receptor c-Met in mammalian brain. J. Cell Biol 1994, 126, 485–494. [Google Scholar]
- Okano, J.; Shiota, G.; Kawasaki, H. Expression of hepatocyte growth factor (HGF) and HGF receptor (c-Met) proteins in liver diseases: An immunohistochemical study. Liver 1999, 19, 151–159. [Google Scholar]
- Kmiecik, T.E.; Keller, J.R.; Rosen, E.; Vande Woude, G.F. Hepatocyte Growth Factor is a synergistic factor for the growth of hematopoietic progenitor cells. Blood 1992, 80, 2454–2457. [Google Scholar]
- Liu, Y.; Wilkinson, F.L.; Kirton, J.P.; Jeziorska, M.; Iizasa, H.; Sai, Y.; Nakashima, E.; Heagerty, A.M.; Canfield, A.E.; Alexander, M.Y. Hepatocyte growth factor and c-Met expression in pericytes: Implications for atherosclerotic plaque development. J. Pathol 2007, 212, 12–19. [Google Scholar]
- Seki, T.; Hagiya, M.; Shimonishi, M.; Nakamura, T.; Shimizu, S. Organization of the human hepatocyte growth factor-encoding gene. Gene 1991, 102, 213–219. [Google Scholar]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. C-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 2003, 22, 309–325. [Google Scholar]
- Peruzzi, B.; Bottaro, D.P. Targeting the c-Met Signaling Pathway in Cancer. Clin. Cancer Res 2006, 12, 3657–3660. [Google Scholar]
- Toiyama, Y.; Miki, C.; Inoue, Y.; Okugawa, Y.; Tanaka, K.; Kusunoki, M. Serum hepatocyte growth factor as a prognostic marker for stage II or III colorectal cancer patients. Int. J. Cancer 2009, 125, 1657–1662. [Google Scholar]
- Gu, H.; Neel, B.G. The “Gab” in signal transduction. Trends Cell Biol 2003, 13, 122–130. [Google Scholar]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-Met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar]
- Michalopoulos, G.K.; DeFrances, M.C. Liver regeneration. Science 1997, 276, 60–66. [Google Scholar]
- Borowiak, M.; Garratt, A.N.; Wüstefeld, T.; Strehle, M.; Trautwein, C.; Birchmeier, C. Met provides essential signals for liver regeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 10608–10613. [Google Scholar]
- Jeffers, M.; Koochekpour, S.; Fiscella, M.; Sathyanarayana, B.K.; Vande Woude, G.F. Signaling requirements for oncogenic forms of the Met tyrosine kinase receptor. Oncogene 1998, 17, 2691–2700. [Google Scholar]
- Boccaccio, C.; Andò, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epitelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar]
- Derman, M.P.; Cunha, M.J.; Barros, E.J.; Nigam, S.K.; Cantley, L.G. HGF-mediated chemotaxis and tubulogenesis require activation of the phosphatidylinositol 3-kinase. Am. J. Physiol 1995, 268, F1211–F1217. [Google Scholar]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; vande Woude, G.F. Hepatocyte growth factor/Scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of tumor cell growth, invasion and metastasis by EXEL-2880 (XL 880, GSK 1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 2009, 69, 8009–8016. [Google Scholar]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenografts models. Cancer Sci 2010, 101, 210–215. [Google Scholar]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar]
- Bauer, T.W.; Somcio, R.J.; Fan, F.; Liu, W.; Johnson, M.; Lesslie, D.P.; Evans, D.B.; Gallick, G.E.; Ellis, L.M. Regulatory role of c-Met in insulin like growth factor-I receptor-mediated migration and invasion of human pancreatic carcinoma cells. Mol. Cancer Ther 2006, 5, 1676–1682. [Google Scholar]
- Jo, M.; Stolz, D.B.; Esplen, J.E.; Dorko, K.; Michalopoulos, G.K.; Strom, S.C. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem. 2000, 275, 8806–8811. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar]
- Liska, D.; Chen, C.T.; Bachleitner-Hofmann, T.; Christensen, J.G.; Weiser, M.R. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin. Cancer Res 2011, 17, 472–482. [Google Scholar]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med 2009, 360, 1408–1417. [Google Scholar]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol 2010, 28, 4697–4705. [Google Scholar]
- Bauer, T.W.; Fan, F.; Liu, W.; Johnson, M.; Parikh, N.U.; Parry, G.C.; Callahan, J.; Mazar, A.P.; Gallick, G.E.; Ellis, L.M. Insulin like growth factor-I-mediated migration and invasion of human colon carcinoma cells requires activation of c-Met and urokinase plasminogen activator receptor. Ann. Surg 2005, 241, 748–758. [Google Scholar]
- Varkaris, A.; Gaur, S.; Parikh, N.U.; Song, J.H.; Dayyani, F.; Jin, J.K.; Logothetis, C.J.; Gallick, G.E. Ligand-independent activation of MET through IGF-1/IGF-1R signaling. Int. J. Cancer 2013, 133, 1536–1546. [Google Scholar]
- Chen, Y.Q.; Fisher, J.H.; Wang, M.H. Activation of the RON receptor tyrosine kinase inhibits inducible nitric oxide synthase (iNOS) expression by murine peritoneal exudate macrophages: phosphatidylinositol-3 kinase is required for RON-mediated inhibition of iNOS expression. J. Immunol 1998, 161, 4950–4959. [Google Scholar]
- Wagh, P.K.; Peace, B.E.; Waltz, S.E. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv. Cancer Res 2008, 100, 1–33. [Google Scholar]
- Follenzi, A.; Bakovic, S.; Gual, P.; Stella, M.C.; Longati, P.; Comoglio, P.M. Cross-talk between the proto-oncogenes Met and Ron. Oncogene 2000, 19, 3041–3049. [Google Scholar]
- Schmidt, L.; Duh, F.M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet 1997, 16, 68–73. [Google Scholar]
- Wojta, J.; Nakamura, T.; Fabry, A.; Hufnagl, P.; Beckmann, R.; McGrath, K.; Binder, B.R. Hepatocyte growth factor stimulates expression of plasminogen activator inhibitor type 1 and tissue factor in HepG2 cells. Blood 1994, 84, 151–157. [Google Scholar]
- Fan, S.; Ma, Y.X.; Wang, J.A.; Yuan, R.Q.; Meng, Q.; Cao, Y.; Laterra, J.J.; Goldberg, I.D.; Rosen, E.M. Cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signalling through phosphatidyl inositol 3′ kinase. Oncogene 2000, 19, 2212–2223. [Google Scholar]
- Li, H.W.; Shan, J.X. Effects of hepatocyte growth factor/scatter factor on the invasion of colorectal cancer cells in vitro. World J. Gastroenterol 2005, 11, 3877–3881. [Google Scholar]
- Kermorgant, S.; Aparicio, T.; Dessirier, V.; Lewin, M.J.; Lehy, T. Hepatocyte growth factor induces colonic cancer cell invasiveness via enhanced motility and protease overproduction. Evidence for PI3 kinase and PKC involvement. Carcinogenesis 2001, 22, 1035–1042. [Google Scholar]
- McCarty, K.S., Jr; Szabo, E.; Flowers, J.L.; Cox, E.B.; Leight, G.S.; Miller, L.; Konrath, J.; Soper, J.T.; Budwit, D.A.; Creasman, W.T.; et al. Use of a monoclonal anti-estrogen receptor antibody in the immunohistochemical evaluation of human tumors. Cancer Res 1986, 46, 4244s–4248s. [Google Scholar]
- Cappuzzo, F.; Hirsch, F.R.; Rossi, E.; Bartolini, S.; Ceresoli, G.L.; Bemis, L.; Haney, J.; Witta, S.; Danenberg, K.; Domenichini, I.; et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J. Natl. Cancer Inst 2005, 97, 643–655. [Google Scholar]
- Dziadziuszko, R.; Wynes, M.W.; Singh, S.; Asuncion, B.R.; Ranger-Moore, J.; Konopa, K.; Rzyman, W.; Szostakiewicz, B.; Jassem, J.; Hirsch, F.R. Correlation between MET gene copy number by silver in situ hybridization and protein expression by immunohistochemistry in non-small cell lung cancer. J. Thorac. Oncol 2012, 7, 340–347. [Google Scholar]
- Shousha, S. Oestrogen receptor status of breast carcinoma: Allred/H score conversion table. Histopathology 2008, 53, 346–347. [Google Scholar]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013, 3, 658–673. [Google Scholar]
- Sierra, J.R.; Tsao, M.S. c-Met as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol 2011, 3, S21–S35. [Google Scholar]
- Wolff, A.C.; Hammond, M.E.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol 2007, 25, 118–145. [Google Scholar]
- Lee, H.E.; Kim, M.A.; Lee, H.S.; Jung, E.J.; Yang, H.K.; Lee, B.L.; Bang, Y.J.; Kim, W.H. MET in gastric carcinomas: Comparison between protein expression and gene copy number and impact on clinical outcome. Br. J. Cancer 2012, 107, 325–333. [Google Scholar]
- Takeuchi, H.; Bilchik, A.; Saha, S.; Turner, R.; Wiese, D.; Tanaka, M.; Kuo, C.; Wang, H.J.; Hoon, D.S. c-Met expression level in primary colon cancer: A predictor of tumor invasion and lymph-node metastases. Clin. Cancer Res 2003, 9, 1480–1488. [Google Scholar]
- Kammula, U.S.; Kuntz, E.J.; Francone, T.D.; Zeng, Z.; Shia, J.; Landmann, R.G.; Paty, P.B.; Weiser, M.R. Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett 2007, 248, 219–228. [Google Scholar]
- Reid, J.F.; Sokolova, V.; Zoni, E.; Lampis, A.; Pizzamiglio, S.; Bertan, C.; Zanutto, S.; Perrone, F.; Camerini, T.; Gallino, G.; et al. miRNA profiling in colorectal cancer highlights miR-1 involvement in MET-dependet proliferation. Mol. Cancer Res 2012, 10, 504–515. [Google Scholar]
- Saigusa, S.; Toiyama, Y.; Tanaka, K.; Yokoe, T.; Fujikawa, H.; Matsushita, K.; Okugawa, Y.; Inoue, Y.; Uchida, K.; Mohri, Y.; et al. Inhibition of HGF/c-Met expression prevents distant recurrence of rectal cancer after preoperative chemoradiotherapy. Int. J. Oncol 2012, 40, 583–591. [Google Scholar]
- Zeng, Z.S.; Weiser, M.R.; Kuntz, E.; Chen, C.T.; Khan, S.A.; Forslund, A.; Nash, G.M.; Gimbel, M.; Yamaguchi, Y.; Culliford, A.T., IV; D’Alessio, M.; et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008, 265, 258–269. [Google Scholar]
- Di Renzo, M.F.; Olivero, M.; Giacomini, A.; Porte, H.; Chastre, E.; Mirossay, L.; Nordlinger, B.; Bretti, S.; Bottardi, S.; Giordano, S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin. Cancer Res 1995, 1, 147–154. [Google Scholar]
- Fujita, S.; Sugano, K. Expression of c-Met proto-oncogene in primary colorectal cancer and liver metastases. Jpn. J. Clin. Oncol 1997, 27, 378–383. [Google Scholar]
- Umeki, K.; Shiota, G.; Kawasaki, H. Clinical significance of c-Met oncogene alterations in human colorectal cancer. Oncology 1999, 56, 314–321. [Google Scholar]
- Gao, D.; Vahdat, L.T.; Wong, S.; Chang, J.C.; Mittal, V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012, 72, 4883–4889. [Google Scholar]
- Stein, U.; Walther, W.; Arlt, F.; Schwabe, H.; Smith, J.; Fichtner, I.; Birchmeier, W.; Schlag, P.M. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastases. Nat. Med 2009, 15, 59–67. [Google Scholar]
- Arlt, F.; Stein, U. Colon cancer metastases: MACC1 and Met as metastatic pacemakers. Int. J. Biochem. Cell Biol 2009, 41, 2356–2359. [Google Scholar]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med 2008, 358, 502–511. [Google Scholar]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale; strategies and challenges. Nat. Rev. Drug Discov 2010, 9, 775–789. [Google Scholar]
- Lee, C.T.; Chow, N.H.; Su, P.F.; Lin, S.C.; Lin, P.C.; Lee, J.C. The prognostic significance of RON and MET receptor coexpression in patients with colorectal cancer. Dis. Colon. Rectum 2008, 51, 1268–1274. [Google Scholar]
- Nakayama, Y.; Okazaki, K.; Shibao, K.; Sako, T.; Hirata, K.; Nagata, N.; Kuwano, M.; Itoh, H. Alternative expression of the collagenase and adhesion molecules in the highly metastatic clones of human colonic cancer cell lines. Clin. Exp. Metastasis 1998, 16, 461–469. [Google Scholar]
- De Oliveira, A.T.; Matos, D.; Logullo, A.F.; DA Silva, S.R.; Neto, R.A.; Filho, A.L.; Saad, S.S. MET is highly expressed in advanced stages of colorectal cancer and indicates worse prognosis and mortality. Anticancer Res 2009, 29, 4807–4812. [Google Scholar]
- Osada, S.; Matsui, S.; Komori, S.; Yamada, J.; Sanada, Y.; Ihawa, A.; Tanaka, Y.; Tokuyama, Y.; Okumura, N.; Nonaka, K.; et al. Effect of hepatocyte growth factor on progression of liver metastasis in colorectal cancer. Hepatogastroenterology 2010, 54, 76–80. [Google Scholar]
- Simon, I.; Roepman, P.; Schlicker, A.; Tabernero, J.; Majewski, I.; Aguado, V.M.; Chresta, C.M.; Rosenberg, R.; Nitsche, U.; Macarulla, T.; et al. Association of colorectal cancer intrinsic subtypes with prognosis; chemotherapy response; deficient mismatch repair; and epithelial to mesenchymal transition (EMT). J. Clin. Oncol. 2012, 30. abstr 333. [Google Scholar]
- Raghav, K.; Wang, W.; Overman, M.; Kopetz, S. MET overexpression as a hallmark of the epithelial-mesenchymal transition (EMT) phenotype in colorectal cancer. J. Clin. Oncol. 2012, 30. abstr 334. [Google Scholar]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the treatment of colorectal cancer. N. Engl. J. Med 2007, 357, 2040–2048. [Google Scholar]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.L.; Van Laethem, J.L.; Maurel, J.; Richardson, G.; Wolf, M.; Amado, R.G. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J. Clin. Oncol 2007, 25, 1658–1664. [Google Scholar]
- Amado, R.G.; Wolf, M.; Peeters, M.; van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol 2008, 26, 1626–1634. [Google Scholar]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med 2008, 359, 1757–1765. [Google Scholar]
- De Roock, W.; Claes, B.; Bernasconi, D.; de Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 2010, 11, 753–762. [Google Scholar]
- Oliner, K.S.; Douillard, J.Y.; Siena, S.; Tabernero, J.; Burkes, R.L.; Barugel, M.E.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first-line treatment (tx) for metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2013, 31. abstr 3631. [Google Scholar]
- Suda, K.; Murakami, I.; Katayama, T.; Tomizawa, K.; Osada, H.; Sekido, Y.; Maehara, Y.; Yatabe, Y.; Mitsudomi, T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin. Cancer Res 2010, 16, 5489–5498. [Google Scholar]
- Inno, A.; di Salvatore, M.; Cenci, T.; Martini, M.; Orlandi, A.; Strippoli, A.; Ferrara, A.M.; Bagalà, C.; Cassano, A.; Larocca, L.M.; et al. Is there a role for IGF1R and c-Met pathways in resistance to cetuximab in metastatic colorectal cancer? Clin. Colorectal Cancer 2011, 10, 325–332. [Google Scholar]
- Cappuzzo, F.; Varella-Garcia, M.; Finocchiaro, G.; Skokan, M.; Gajapathy, S.; Carnaghi, C.; Rimassa, L.; Rossi, E.; Ligorio, C.; di Tommaso, L.; et al. Primary resistance to cetuximab therapy in EGFR FISH-positive colorectal cancer patients. Br. J. Cancer 2008, 99, 83–89. [Google Scholar]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; Bencardino, K.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar]
- De Bono, J.; Yap, T. c-Met: An exciting new target for anticancer therapy. Ther. Adv. Med. Oncol 2011, 3, S3–S5. [Google Scholar]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety; pharmacokinetics; and pharmacodynamics of AMG 102; a fully human hepatocyte growth factor-neutralizing monoclonal antibody; in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res 2010, 16, 699–710. [Google Scholar]
- Eng, C.; Tabernero, J.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.P.; Davidenko, I.; Chen, L.; Smethurst, D.; van Cutsem, E. Panitumumab (pmab) plus AMG 102 in patients (pts) with wild-type KRAS metastatic colorectal cancer (mCRC): Updated safety results. J. Clin. Oncol. 2010, 28. abstr 14083. [Google Scholar]
- Eng, C.; van Cutsem, E.; Nowara, E.; Swieboda-Sadlej, A.; Tebbutt, N.C.; Mitchell, E.P.; Davidenko, I.; Oliner, K.; Chen, L.; Huang, J.; et al. A randomized; phase Ib/II trial of rilotumumab (AMG 102; ril) or ganitumumab (AMG 479; gan) with panitumumab (pmab) versus pmab alone in patients (pts) with wild-type (WT) KRAS metastatic colorectal (mCRC): Primary and biomarker analyses. J. Clin. Oncol. 2011, 29. abstr 3500. [Google Scholar]
- Tan, E.; Park, K.; Lim, W.T.; Ahn, M.; Ng, Q.S.; Ahn, J.S.; Tan, D.S.; Sun, J.; Jac, J.; Han, M.; et al. Phase 1b study of ficlatuzumab (AV-299); an anti-hepatocyte growth factor monoclonal antibody; in combination with gefitinib in Asian patients with NSCLC. J. Clin. Oncol. 2011, 29. abstr 7571. [Google Scholar]
- Martens, T.; Schmidt, N.O.; Eckerich, C.; Fillbrandt, R.; Merchant, M.; Schwall, R.; Westphal, M.; Lamszus, K. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin. Cancer Res 2006, 12, 6144–6152. [Google Scholar]
- Bendell, J.C.; Ervin, T.J.; Gallinson, D.; Singh, J.; Wallace, J.A.; Saleh, M.N.; Vallone, M.; Phan, S.C.; Hack, S.P. Treatment rationale and study design for a randomized, double-blind, placebo-controlled phase II study evaluating onartuzumab (MetMAb) in combination with bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal cancer. Clin. Colorectal Cancer 2013, in press. [Google Scholar]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.; Daniel, D.B.; Goldschmidt, J.H.; Blumenschein, G.R.; Krzakowski, M.J.; Robinet, G.; Clement-Duchene, C.; Barlesi, F.; et al. Final efficacy results from OAM4558g, a randomized phase II study evaluating MetMAb or placebo in combination with erlotinib in advanced NSCLC. J. Clin. Oncol. 2011, 29. abstr 7505. [Google Scholar]
- Blumenscheim, G.; Mills, G.; Gonzalez-Angulo, A. Targeting the hepatocyte growth factor—cMET Axis in cancer therapy. J. Clin. Oncol 2012, 30, 3287–3295. [Google Scholar]
- Eder, J.P.; Shapiro, G.I.; Appleman, L.J.; Zhu, A.X.; Miles, D.; Keer, H.; Cancilla, B.; Chu, F.; Hitchcock-Bryan, S.; Sherman, L.; et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin. Cancer Res 2010, 16, 3507–3516. [Google Scholar]
- Hussain, M.; Smith, M.R.; Sweeney, C.; Corn, P.G.; Elfiky, A.; Gordon, M.S.; Haas, N.B.; Harzstark, A.L.; Kurzrock, R.; Lara, P.; et al. Cabozantinib (XL184) in metastatic castration-resistant prostate cancer (mCRPC): Results from a phase II randomized discontinuation trial. J. Clin. Oncol. 2011, 29. abstr 4516. [Google Scholar]
- Bessudo, A.; Bendell, J.C.; Gabrail, N.; Kopp, M.V.; Mueller, L.; Hart, L.L.; Vladimirov, V.I.; Pande, A.U.; Gorbatchevsky, I.; Eng, C. Phase I results of the randomized; placebo controlled; phase I/II study of the novel oral c-Met inhibitor; ARQ 197; irinotecan (cpt-11); and cetuximab (C) in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC) who received front-line systemic therapy. J. Clin. Oncol. 2011, 29. abstr 3582. [Google Scholar]
- Eng, C.; Hart, L.L.; Severtsev, A.; Gladkov, O.; Mueller, L.; Kopp, M.V.; Vladimirov, V.I.; Langdon, R.M.; Kotiv, B.; Barni, S.; et al. A randomized, placebo-controlled, phase I/II study of tivantinib (ARQ 197) in combination with cetuximab and irinotecan in patients (pts) with KRAS wild-type (WT) metastatic colorectal cancer (CRC) who had received previous front-line systemic therapy. J. Clin. Oncol. 2013, 31. abstr 3508. [Google Scholar]
- Raghav, K.P.S.; Eng, C. Role of the MET–HGF axis in colorectal cancer: Precepts and prospects. Colorectal Cancer 2012, 1, 329–341. [Google Scholar]
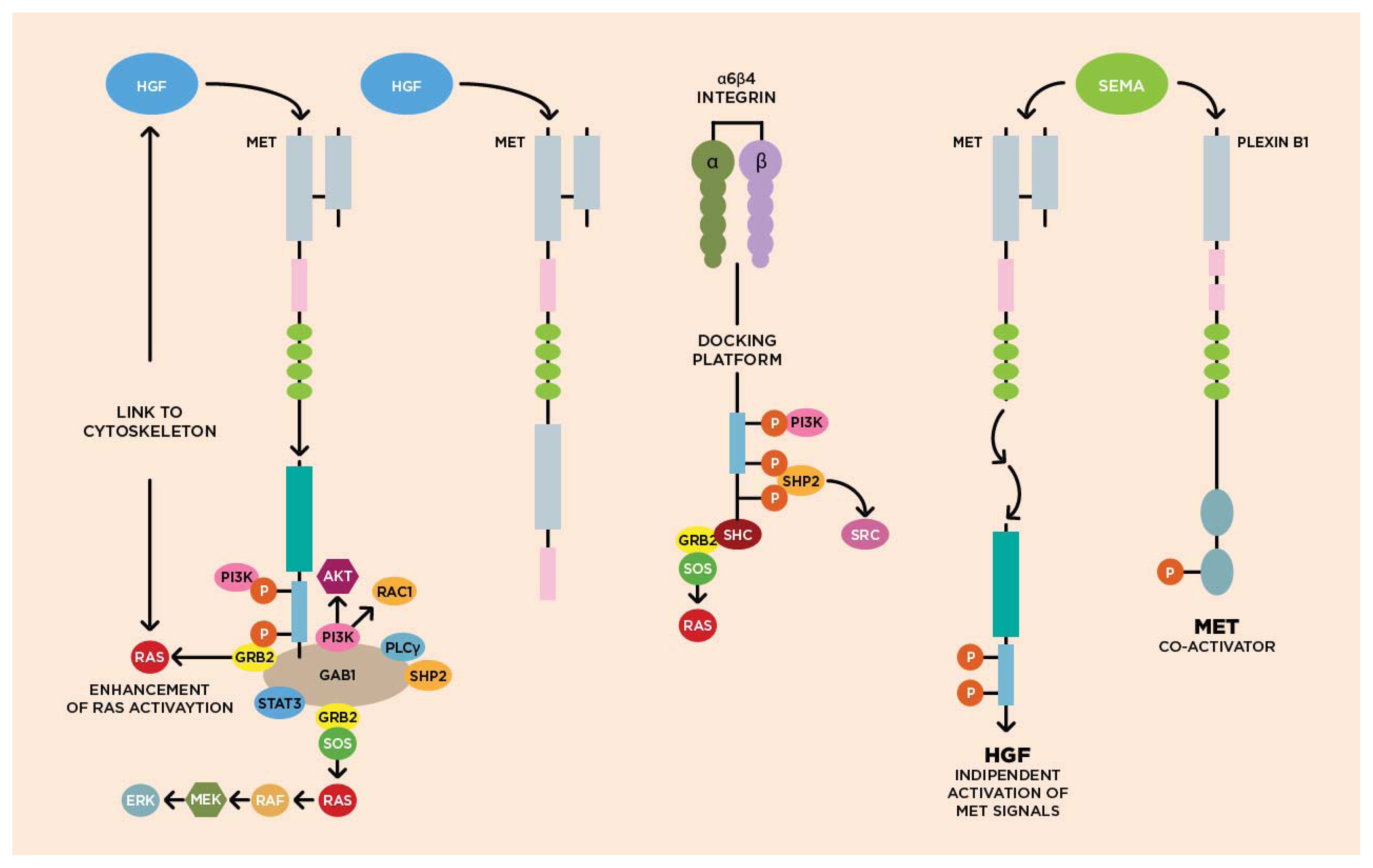

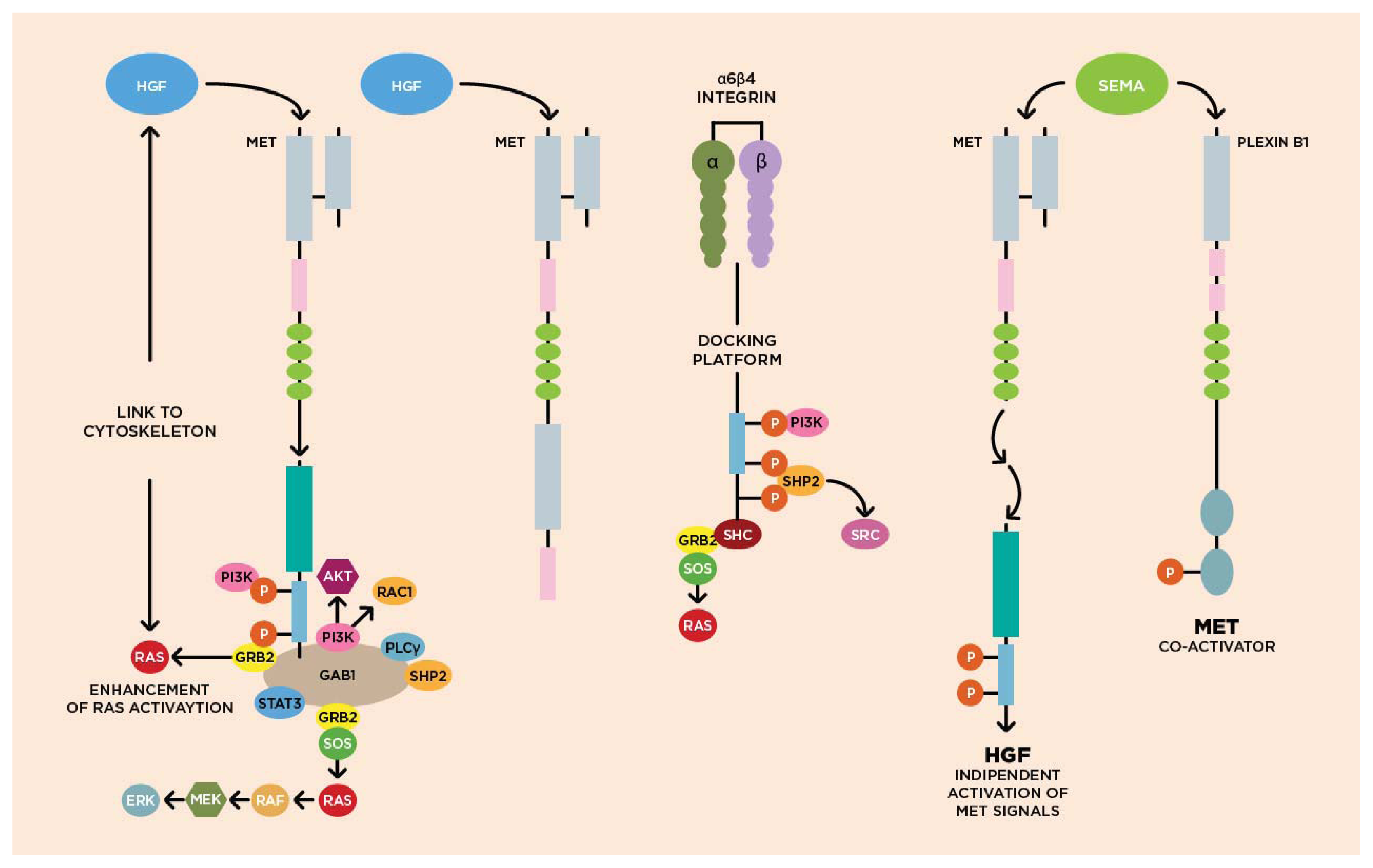{kind=link}
| Cellular/biological process | HGF/cMET pathway involvement |
|---|---|
| Embryogenesis | EMT of myogenic progenitor cells and development of muscular tissue Development of neuronal precursors, liver and placental tissue Regeneration after injury in different epithelial tissues |
| Tissue regeneration | Wound repair of the skin Induction of DNA synthesis and liver regeneration |
| Cell proliferation and survival | Activation of cell proliferation, survival and migration |
| Cytoskeleton | Involvement in cell adhesion, actin reorganization and cell growth |
| Scattering and cell motility | Induction of cell motility, invasion and metastatization |
| Company | Compound | Mechanism of action | Clinical development |
|---|---|---|---|
| Amgen | Rilotumumab | HGF IgG2 Mab | Phase II:CRC |
| Aveo | Ficlatuzumab | HGF IgG1 Mab | Phase II: NSCLC |
| Genetech/Roche | Onartuzumab | MET IgG1 Mab | Phase II: NSCLC Phase II: CRC |
| Pfizer | Crizotinib | MET TKI Other TKI inhibition: ALK, RON, AXL, TIE2 | Phase IV: NSCLC |
| GlaxoSmithKline | Foretinib | MET TKI Other TKI inhibition: VEGFR2, AXL, PDGFR, KIT, FLT3, TIE2 | Phase II: |
| Exelis | Cabozantinib | MET TKI Other TKI inhibition: VEGFR2, RET, KIT, FLT3, TIE2 | Phase II: NSCLC |
| ArQule | Tivantinib | MET TKI | Phase II:CRC |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pérez-Vargas, J.C.S.; Biondani, P.; Maggi, C.; Gariboldi, M.; Gloghini, A.; Inno, A.; Volpi, C.C.; Gualeni, A.V.; Di Bartolomeo, M.; De Braud, F.; et al. Role of cMET in the Development and Progression of Colorectal Cancer. Int. J. Mol. Sci. 2013, 14, 18056-18077. https://doi.org/10.3390/ijms140918056
Pérez-Vargas JCS, Biondani P, Maggi C, Gariboldi M, Gloghini A, Inno A, Volpi CC, Gualeni AV, Di Bartolomeo M, De Braud F, et al. Role of cMET in the Development and Progression of Colorectal Cancer. International Journal of Molecular Sciences. 2013; 14(9):18056-18077. https://doi.org/10.3390/ijms140918056
Chicago/Turabian StylePérez-Vargas, Juan Carlos Samamé, Pamela Biondani, Claudia Maggi, Manuela Gariboldi, Annunziata Gloghini, Alessandro Inno, Chiara Costanza Volpi, Ambra Vittoria Gualeni, Maria Di Bartolomeo, Filippo De Braud, and et al. 2013. "Role of cMET in the Development and Progression of Colorectal Cancer" International Journal of Molecular Sciences 14, no. 9: 18056-18077. https://doi.org/10.3390/ijms140918056
APA StylePérez-Vargas, J. C. S., Biondani, P., Maggi, C., Gariboldi, M., Gloghini, A., Inno, A., Volpi, C. C., Gualeni, A. V., Di Bartolomeo, M., De Braud, F., Castano, A., Bossi, I., & Pietrantonio, F. (2013). Role of cMET in the Development and Progression of Colorectal Cancer. International Journal of Molecular Sciences, 14(9), 18056-18077. https://doi.org/10.3390/ijms140918056