Epidermal Stem Cells and Their Epigenetic Regulation

{kind=link}
Abstract
:1. Introduction of Epidermal Stem Cells
1.1. Identification of Epidermal Stem Cells
1.2. Functional Characteristics of Epidermal Stem Cells
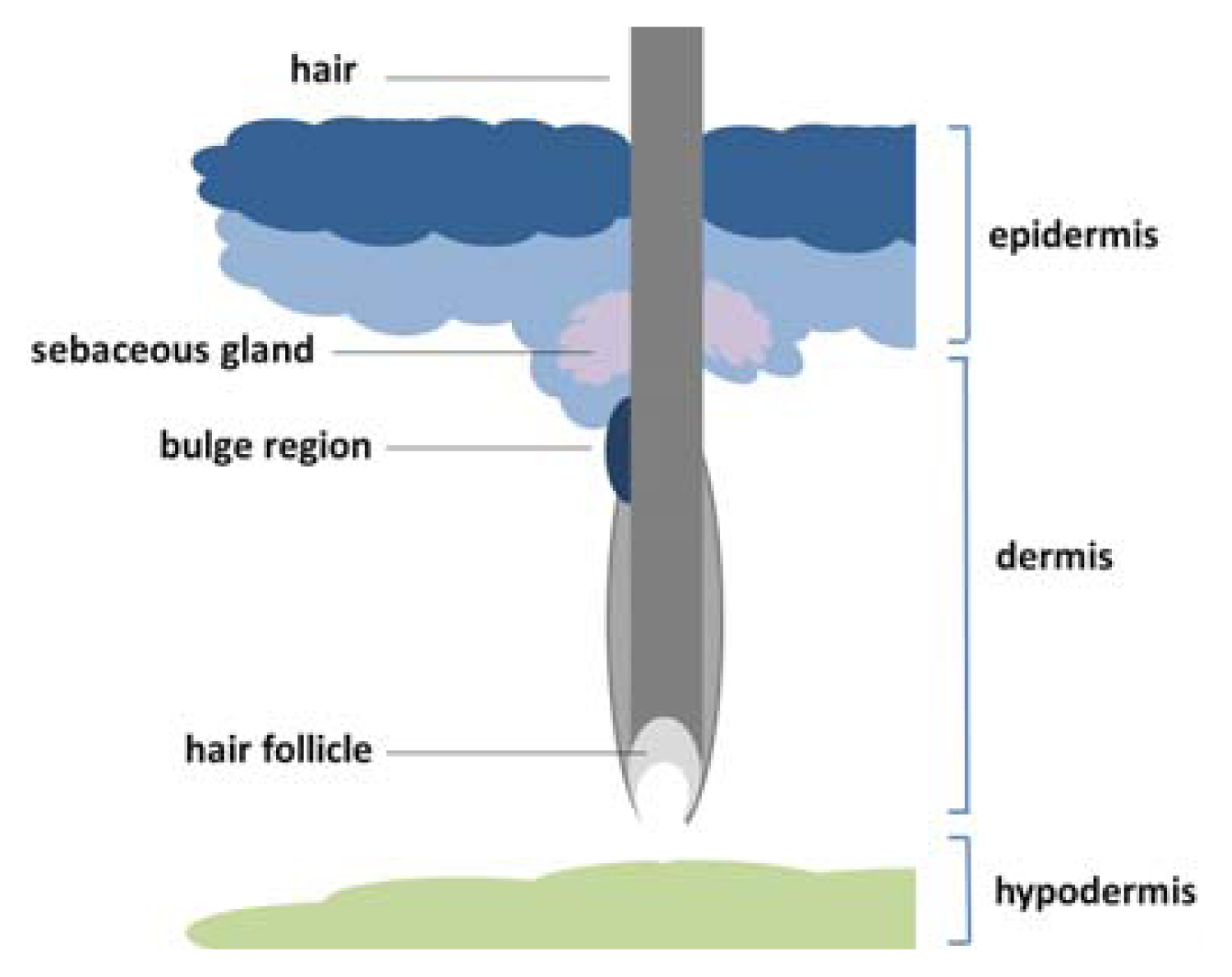
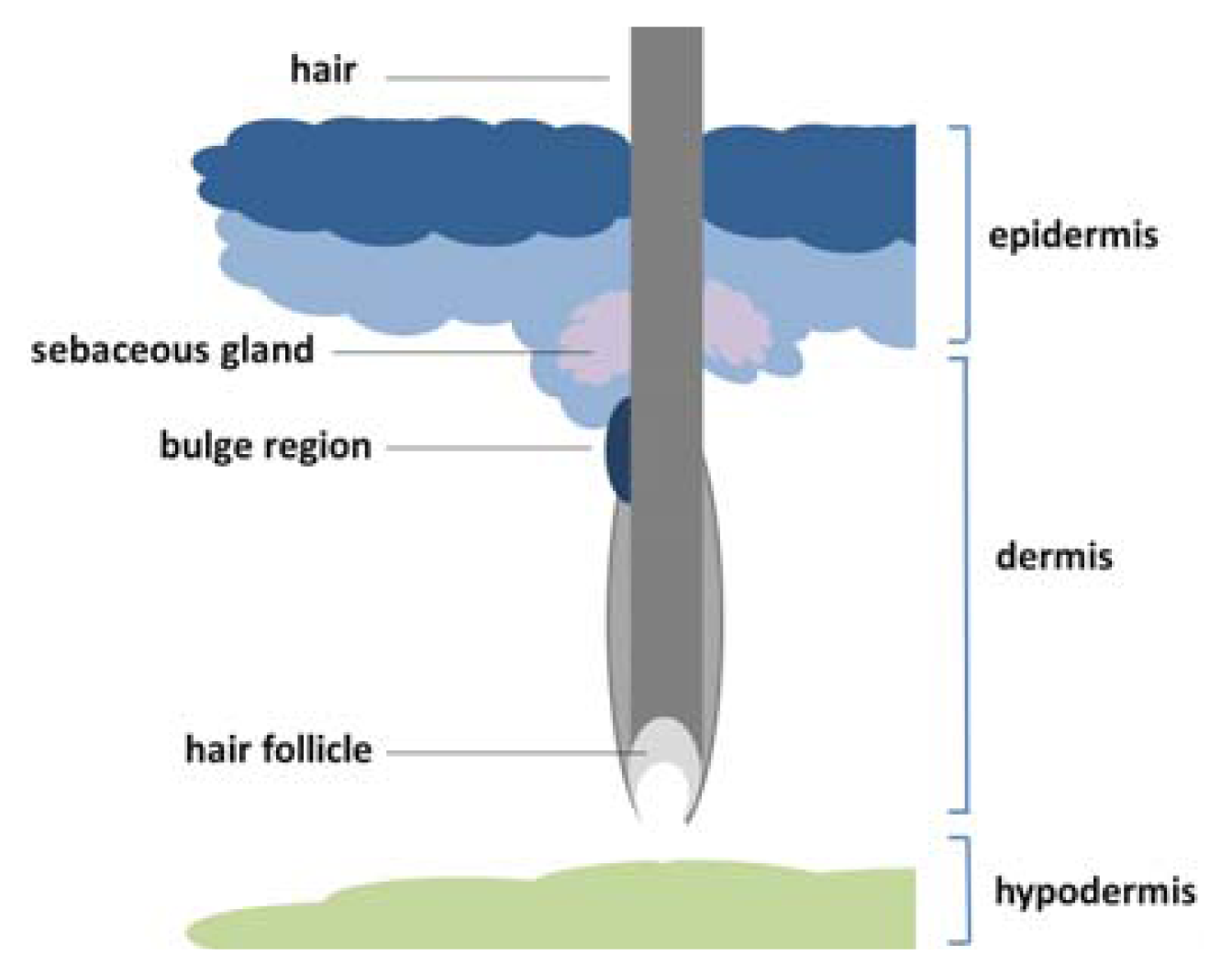
1.3. Location and Classification of Epidermal Stem Cells
1.4. Signaling Pathway Implicated in the Epidermal Stem Cells
2. Epigenetic Regulation of Epidermal Stem Cells
2.1. Histone Modifications and Epidermal Stem Cells
2.2. DNA Modification and Epidermal Stem Cells
2.3. Noncoding RNAs and Epidermal Stem Cells
2.4. Epigenetic Regulations of Epidermal Stem Cells and Skin Disorders
3. Summary
Acknowledgments
Conflicts of Interest
References
- Boehnke, K.; Falkowska-Hansen, B.; Stark, H.J.; Boukamp, P. Stem cells of the human epidermis and their niche: Composition and function in epidermal regeneration and carcinogenesis. Carcinogenesis 2012, 33, 1247–1258. [Google Scholar]
- Wobus, A.M.; Holzhausen, H.; Jakel, P.; Schoneich, J. Characterization of a pluripotent stem cell line derived from a mouse embryo. Exp. Cell. Res. 1984, 152, 212–219. [Google Scholar]
- Aharonowiz, M.; Einstein, O.; Fainstein, N.; Lassmann, H.; Reubinoff, B.; Ben-Hur, T. Neuroprotective effect of transplanted human embryonic stem cell-derived neural precursors in an animal model of multiple sclerosis. PLoS One 2008, 3, e3145. [Google Scholar]
- Sonntag, K.C.; Pruszak, J.; Yoshizaki, T.; van Arensbergen, J.; Sanchez-Pernaute, R.; Isacson, O. Enhanced yield of neuroepithelial precursors and midbrain-like dopaminergic neurons from human embryonic stem cells using the bone morphogenic protein antagonist noggin. Stem Cells 2007, 25, 411–418. [Google Scholar]
- Lumelsky, N.; Blondel, O.; Laeng, P.; Velasco, I.; Ravin, R.; McKay, R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001, 292, 1389–1394. [Google Scholar]
- Kerr, C.L.; Letzen, B.S.; Hill, C.M.; Agrawal, G.; Thakor, N.V.; Sterneckert, J.L.; Gearhart, J.D.; All, A.H. Efficient differentiation of human embryonic stem cells into oligodendrocyte progenitors for application in a rat contusion model of spinal cord injury. Int. J. Neurosci 2010, 120, 305–313. [Google Scholar]
- Hess, P.G. Risk of tumorigenesis in first-in-human trials of embryonic stem cell neural derivatives: Ethics in the face of long-term uncertainty. Account. Res 2009, 16, 175–198. [Google Scholar]
- Kim, E.M.; Stultz, R.; Bonde, S.; Zavazava, N. Embryonic stem cell-derived T cells induce lethal graft-versus-host disease and reject allogenic skin grafts upon thymic selection. Am. J. Transplant 2012, 12, 600–609. [Google Scholar]
- Watt, F.M.; Jensen, K.B. Epidermal stem cell diversity and quiescence. EMBO Mol. Med 2009, 1, 260–267. [Google Scholar]
- Lemaitre, G.; Nissan, X.; Baldeschi, C.; Peschanski, M. Concise review: Epidermal grafting: The case for pluripotent stem cells. Stem Cells 2011, 29, 895–899. [Google Scholar]
- Bickenbach, J.R.; Mackenzie, I.C. Identification and localization of label-retaining cells in hamster epithelia. J. Investig. Dermatol 1984, 82, 618–622. [Google Scholar]
- Bickenbach, J.R. Identification and behavior of label-retaining cells in oral mucosa and skin. J. Dent. Res 1981, 60, 1611–1620. [Google Scholar]
- Packard, D.S., Jr; Skalko, R.G.; Menzies, R.A. Growth retardation and cell death in mouse embryos following exposure to the teratogen bromodeoxyuridine. Exp. Mol. Pathol. 1974, 21, 351–362. [Google Scholar]
- Kanda, T.; Sullivan, K.F.; Wahl, G.M. Histone GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol 1998, 8, 377–385. [Google Scholar]
- Tumbar, T.; Guasch, G.; Greco, V.; Blanpain, C.; Lowry, W.E.; Rendl, M.; Fuchs, E. Defining the epithelial stem cell niche in skin. Science 2004, 303, 359–363. [Google Scholar]
- Bickenbach, J.R.; McCutecheon, J.; Mackenzie, I.C. Rate of loss of tritiated thymidine label in basal cells in mouse epithelial tissues. Cell Tissue Kinet 1986, 19, 325–333. [Google Scholar]
- Claudinot, S.; Nicolas, M.; Oshima, H.; Rochat, A.; Barrandon, Y. Long-term renewal of hair follicles from clonogenic multipotent stem cells. Proc. Natl. Acad. Sci. USA 2005, 102, 14677–14682. [Google Scholar]
- Du, Y.; Li, B.; Wang, E. “Fitting” makes “sensing” simple: Label-free detection strategies based on nucleic Acid aptamers. Acc. Chem. Res 2013, 46, 203–213. [Google Scholar]
- Lavker, R.M.; Sun, T.T. Epidermal stem cells: Properties, markers, and location. Proc. Natl. Acad. Sci. USA 2000, 97, 13473–13475. [Google Scholar]
- Thompson, E.M.; Price, Y.E.; Wright, N.A. Kinetics of enteroendocrine cells with implications for their origin: A study of the cholecystokinin and gastrin subpopulations combining tritiated thymidine labelling with immunocytochemistry in the mouse. Gut 1990, 31, 406–411. [Google Scholar]
- Clayton, E.; Doupe, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar]
- Mascre, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohee, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar]
- Chen, W.; Hara, K.; Tian, Q.; Zhao, K.; Yoshitomi, T. Existence of small slow-cycling Langerhans cells in the limbal basal epithelium that express ABCG2. Exp. Eye Res 2007, 84, 626–634. [Google Scholar]
- Foudi, A.; Hochedlinger, K.; van Buren, D.; Schindler, J.W.; Jaenisch, R.; Carey, V.; Hock, H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol 2009, 27, 84–90. [Google Scholar]
- Rocheteau, P.; Gayraud-Morel, B.; Siegl-Cachedenier, I.; Blasco, M.A.; Tajbakhsh, S. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell 2012, 148, 112–125. [Google Scholar]
- Bonaguidi, M.A.; Wheeler, M.A.; Shapiro, J.S.; Stadel, R.P.; Sun, G.J.; Ming, G.L.; Song, H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 2011, 145, 1142–1155. [Google Scholar]
- Calabro, K.; Curtis, A.; Galarneau, J.R.; Krucker, T.; Bigio, I.J. Gender variations in the optical properties of skin in murine animal models. J. Biomed. Opt. 2011, 16. [Google Scholar] [CrossRef]
- Forni, M.F.; Trombetta-Lima, M.; Sogayar, M.C. Stem cells in embryonic skin development. Biol. Res 2012, 45, 215–222. [Google Scholar]
- Watt, F.M. Epidermal stem cells: Markers, patterning and the control of stem cell fate. Philos. Trans. R. Soc. London Ser. B 1998, 353, 831–837. [Google Scholar]
- Fuchs, E. Finding one’s niche in the skin. Cell Stem Cell 2009, 4, 499–502. [Google Scholar]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol 2009, 10, 207–217. [Google Scholar]
- Fuchs, E. Skin stem cells: Rising to the surface. J. Cell Biol 2008, 180, 273–284. [Google Scholar]
- Horsley, V.; Aliprantis, A.O.; Polak, L.; Glimcher, L.H.; Fuchs, E. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 2008, 132, 299–310. [Google Scholar]
- Jaks, V.; Barker, N.; Kasper, M.; van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgard, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet 2008, 40, 1291–1299. [Google Scholar]
- Horsley, V.; O’Carroll, D.; Tooze, R.; Ohinata, Y.; Saitou, M.; Obukhanych, T.; Nussenzweig, M.; Tarakhovsky, A.; Fuchs, E. Blimp1 defines a progenitor population that governs cellular input to the sebaceous gland. Cell 2006, 126, 597–609. [Google Scholar]
- Watt, F.M.; Estrach, S.; Ambler, C.A. Epidermal Notch signalling: Differentiation, cancer and adhesion. Curr. Opin. Cell Biol 2008, 20, 171–179. [Google Scholar]
- Ishikawa, Y.; Hosogane, M.; Okuyama, R.; Aoyama, S.; Onoyama, I.; Nakayama, K.I.; Nakayama, K. Opposing functions of Fbxw7 in keratinocyte growth, differentiation and skin tumorigenesis mediated through negative regulation of c-Myc and Notch. Oncogene 2012, 32, 1921–1932. [Google Scholar]
- Ambler, C.A.; Watt, F.M. Expression of Notch pathway genes in mammalian epidermis and modulation by beta-catenin. Dev. Dyn 2007, 236, 1595–1601. [Google Scholar]
- Katoh, M. Notch ligand, JAG1, is evolutionarily conserved target of canonical WNT signaling pathway in progenitor cells. Int. J. Mol. Med 2006, 17, 681–685. [Google Scholar]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; di Vignano, A.T.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev 2006, 20, 1028–1042. [Google Scholar]
- Forster, N.; Ellisen, L.W. Notch signaling mediates p63-induced quiescence: A new facet of p63/Notch crosstalk. Cell Cycle 2011, 10, 3632–3633. [Google Scholar]
- Furusawa, C.; Kaneko, K. A dynamical-systems view of stem cell biology. Science 2012, 338, 215–217. [Google Scholar]
- Huang, S.; Eichler, G.; Bar-Yam, Y.; Ingber, D.E. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. Rev. Lett. 2005, 94. [Google Scholar] [CrossRef]
- Shimojo, H.; Ohtsuka, T.; Kageyama, R. Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron 2008, 58, 52–64. [Google Scholar]
- Yu, P.; Xiao, S.; Xin, X.; Song, C.X.; Huang, W.; McDee, D.; Tanaka, T.; Wang, T.; He, C.; Zhong, S. Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Res 2013, 23, 352–364. [Google Scholar]
- Sauvageau, M.; Sauvageau, G. Polycomb group proteins: Multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010, 7, 299–313. [Google Scholar]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar]
- Vandamme, J.; Volkel, P.; Rosnoblet, C.; Le Faou, P.; Angrand, P.O. Interaction proteomics analysis of polycomb proteins defines distinct PRC1 complexes in mammalian cells. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef]
- Hopkin, A.S.; Gordon, W.; Klein, R.H.; Espitia, F.; Daily, K.; Zeller, M.; Baldi, P.; Andersen, B. GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet 2012, 8, e1002829. [Google Scholar]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar]
- Endoh, M.; Endo, T.A.; Endoh, T.; Isono, K.; Sharif, J.; Ohara, O.; Toyoda, T.; Ito, T.; Eskeland, R.; Bickmore, W.A.; et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet 2012, 8, e1002774. [Google Scholar]
- Tavares, L.; Dimitrova, E.; Oxley, D.; Webster, J.; Poot, R.; Demmers, J.; Bezstarosti, K.; Taylor, S.; Ura, H.; Koide, H.; et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell 2012, 148, 664–678. [Google Scholar]
- Luis, N.M.; Morey, L.; Mejetta, S.; Pascual, G.; Janich, P.; Kuebler, B.; Cozutto, L.; Roma, G.; Nascimento, E.; Frye, M.; et al. Regulation of human epidermal stem cell proliferation and senescence requires polycomb- dependent and -independent functions of Cbx4. Cell Stem Cell 2011, 9, 233–246. [Google Scholar]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol 2012, 32, 1433–1441. [Google Scholar]
- Ezhkova, E.; Lien, W.H.; Stokes, N.; Pasolli, H.A.; Silva, J.M.; Fuchs, E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 2011, 25, 485–498. [Google Scholar]
- Herz, H.M.; Mohan, M.; Garrett, A.S.; Miller, C.; Casto, D.; Zhang, Y.; Seidel, C.; Haug, J.S.; Florens, L.; Washburn, M.P.; et al. Polycomb repressive complex 2-dependent and -independent functions of Jarid2 in transcriptional regulation in Drosophila. Mol. Cell. Biol 2012, 32, 1683–1693. [Google Scholar]
- Dietrich, N.; Lerdrup, M.; Landt, E.; Agrawal-Singh, S.; Bak, M.; Tommerup, N.; Rappsilber, J.; Sodersten, E.; Hansen, K. REST-mediated recruitment of polycomb repressor complexes in mammalian cells. PLoS Gent 2012, 8, e1002494. [Google Scholar]
- Benoit, Y.D.; Lepage, M.B.; Khalfaoui, T.; Tremblay, E.; Basora, N.; Carrier, J.C.; Gudas, L.J.; Beaulieu, J.F. Polycomb repressive complex 2 impedes intestinal cell terminal differentiation. J. Cell Sci 2012, 125, 3454–3463. [Google Scholar]
- Shaw, T.; Martin, P. Epigenetic reprogramming during wound healing: Loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep 2009, 10, 881–886. [Google Scholar]
- Herlofsen, S.R.; Bryne, J.C.; Hoiby, T.; Wang, L.; Issner, R.; Zhang, X.; Coyne, M.J.; Boyle, P.; Gu, H.; Meza-Zepeda, L.A.; et al. Genome-wide map of quantified epigenetic changes during in vitro chondrogenic differentiation of primary human mesenchymal stem cells. BMC Genomics 2013, 14, 105. [Google Scholar]
- Cai, L.; Rothbart, S.B.; Lu, R.; Xu, B.; Chen, W.Y.; Tripathy, A.; Rockowitz, S.; Zheng, D.; Patel, D.J.; Allis, C.D.; et al. An H3K36 methylation-engaging tudor motif of polycomb-like proteins mediates PRC2 complex targeting. Mol. Cell 2013, 49, 571–582. [Google Scholar]
- Abed, J.A.; Jones, R.S. H3K36me3 key to polycomb-mediated gene silencing in lineage specification. Nat. Struct. Mol. Biol 2012, 19, 1214–1215. [Google Scholar]
- Li, Z.; Gadue, P.; Chen, K.; Jiao, Y.; Tuteja, G.; Schug, J.; Li, W.; Kaestner, K.H. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 2012, 151, 1608–1616. [Google Scholar]
- Huh, Y.H.; Sherley, J.L. Molecular cloaking of H2A.Z on mortal DNA chromosomes during nonrandom segregation. Stem Cells 2011, 29, 1620–1627. [Google Scholar]
- Illingworth, R.S.; Botting, C.H.; Grimes, G.R.; Bickmore, W.A.; Eskeland, R. PRC1 and PRC2 are not required for targeting of H2A.Z to developmental genes in embryonic stem cells. PLoS One 2012, 7, e34848. [Google Scholar]
- Hu, G.; Cui, K.; Northrup, D.; Liu, C.; Wang, C.; Tang, Q.; Ge, K.; Levens, D.; Crane-Robinson, C.; Zhao, K. H2A.z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell 2013, 12, 180–192. [Google Scholar]
- Ali, M.; Yan, K.; Lalonde, M.E.; Degerny, C.; Rothbart, S.B.; Strahl, B.D.; Cote, J.; Yang, X.J.; Kutateladze, T.G. Tandem PHD fingers of MORF/MOZ acetyltransferases display selectivity for acetylated histone H3 and are required for the association with chromatin. J. Mol. Biol 2012, 424, 328–338. [Google Scholar]
- Aarenstrup, L.; Flindt, E.N.; Otkjaer, K.; Kirkegaard, M.; Andersen, J.S.; Kristiansen, K. HDAC activity is required for p65/RelA-dependent repression of PPARdelta-mediated transactivation in human keratinocytes. J. Investig. Dermatol 2008, 128, 1095–1106. [Google Scholar]
- Robertson, E.D.; Weir, L.; Romanowska, M.; Leigh, I.M.; Panteleyev, A.A. ARNT controls the expression of epidermal differentiation genes through HDAC- and EGFR-dependent pathways. J. Cell Sci 2012, 125, 3320–3332. [Google Scholar]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar]
- Frye, M.; Fisher, A.G.; Watt, F.M. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS One 2007, 2, e763. [Google Scholar]
- Cottle, D.L.; Kretzschmar, K.; Schweiger, P.J.; Quist, S.R.; Gollnick, H.P.; Natsuga, K.; Aoyagi, S.; Watt, F.M. c-MYC-induced sebaceous gland differentiation is controlled by an androgen receptor/p53 axis. Cell Rep 2013, 3, 427–441. [Google Scholar]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar]
- Bestor, T.; Laudano, A.; Mattaliano, R.; Ingram, V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol 1988, 203, 971–983. [Google Scholar]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 2006, 11, 805–814. [Google Scholar]
- Zhang, J.; Gao, Q.; Li, P.; Liu, X.; Jia, Y.; Wu, W.; Li, J.; Dong, S.; Koseki, H.; Wong, J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res 2011, 21, 1723–1739. [Google Scholar]
- Jeltsch, A. On the enzymatic properties of Dnmt1: Specificity, processivity, mechanism of linear diffusion and allosteric regulation of the enzyme. Epigenetics 2006, 1, 63–66. [Google Scholar]
- Xi, S.; Geiman, T.M.; Briones, V.; Tao, Y.G.; Xu, H.; Muegge, K. Lsh participates in DNA methylation and silencing of stem cell genes. Stem Cells 2009, 27, 2691–2702. [Google Scholar]
- Li, J.; Jiang, T.X.; Hughes, M.W.; Wu, P.; Yu, J.; Widelitz, R.B.; Fan, G.; Chuong, C.M. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1. J. Investig. Dermatol 2013, 133, 859. [Google Scholar]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLos One 2010, 5, e8888. [Google Scholar]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Han, J.W.; Kim, S.; Namburi, S.; Hermetz, K.; Kim, J.J.; Rudd, M.K.; et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet 2011, 7, e1002154. [Google Scholar]
- Davis, T.; Vaisvila, R. High sensitivity 5-hydroxymethylcytosine detection in Balb/C brain tissue. J. Vis. Exp. 2011, 48. [Google Scholar] [CrossRef]
- Ruzov, A.; Tsenkina, Y.; Serio, A.; Dudnakova, T.; Fletcher, J.; Bai, Y.; Chebotareva, T.; Pells, S.; Hannoun, Z.; Sullivan, G.; et al. Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res 2011, 21, 1332–1342. [Google Scholar]
- Zhang, F.; Pomerantz, J.H.; Sen, G.; Palermo, A.T.; Blau, H.M. Active tissue-specific DNA demethylation conferred by somatic cell nuclei in stable heterokaryons. Proc. Natl. Acad. Sci. USA 2007, 104, 4395–4400. [Google Scholar]
- Yi, R.; O’Carroll, D.; Pasolli, H.A.; Zhang, Z.; Dietrich, F.S.; Tarakhovsky, A.; Fuchs, E. Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat. Genet 2006, 38, 356–362. [Google Scholar]
- Andl, T.; Murchison, E.P.; Liu, F.; Zhang, Y.; Yunta-Gonzalez, M.; Tobias, J.W.; Andl, C.D.; Seykora, J.T.; Hannon, G.J.; Millar, S.E. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr. Biol 2006, 16, 1041–1049. [Google Scholar]
- Sonkoly, E.; Wei, T.; Pavez Lorie, E.; Suzuki, H.; Kato, M.; Torma, H.; Stahle, M.; Pivarcsi, A. Protein kinase C-dependent upregulation of miR-203 induces the differentiation of human keratinocytes. J. Investig. Dermatol 2010, 130, 124–134. [Google Scholar]
- Nissan, X.; Denis, J.A.; Saidani, M.; Lemaitre, G.; Peschanski, M.; Baldeschi, C. miR-203 modulates epithelial differentiation of human embryonic stem cells towards epidermal stratification. Dev. Biol 2011, 356, 506–515. [Google Scholar]
- Wei, T.; Orfanidis, K.; Xu, N.; Janson, P.; Stahle, M.; Pivarcsi, A.; Sonkoly, E. The expression of microRNA-203 during human skin morphogenesis. Exp. Dermatol 2010, 19, 854–856. [Google Scholar]
- Antonini, D.; Russo, M.T.; de Rosa, L.; Gorrese, M.; Del Vecchio, L.; Missero, C. Transcriptional repression of miR-34 family contributes to p63-mediated cell cycle progression in epidermal cells. J. Investig. Dermatol 2010, 130, 1249–1257. [Google Scholar]
- Wu, N.; Sulpice, E.; Obeid, P.; Benzina, S.; Kermarrec, F.; Combe, S.; Gidrol, X. The miR-17 family links p63 protein to MAPK signaling to promote the onset of human keratinocyte differentiation. PLos One 2012, 7, e45761. [Google Scholar]
- Chikh, A.; Matin, R.N.; Senatore, V.; Hufbauer, M.; Lavery, D.; Raimondi, C.; Ostano, P.; Mello-Grand, M.; Ghimenti, C.; Bahta, A.; et al. iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J 2011, 30, 4261–4273. [Google Scholar]
- Szulwach, K.E.; Li, X.; Smrt, R.D.; Li, Y.; Luo, Y.; Lin, L.; Santistevan, N.J.; Li, W.; Zhao, X.; Jin, P. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol 2010, 189, 127–141. [Google Scholar]
- Wong, C.F.; Tellam, R.L. MicroRNA-26a targets the histone methyltransferase enhancer of zeste homolog 2 during myogenesis. J. Biol. Chem 2008, 283, 9836–9843. [Google Scholar]
- Rybak, A.; Fuchs, H.; Hadian, K.; Smirnova, L.; Wulczyn, E.A.; Michel, G.; Nitsch, R.; Krappmann, D.; Wulczyn, F.G. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat. Cell Biol 2009, 11, 1411–1420. [Google Scholar]
- Hildebrand, J.; Rutze, M.; Walz, N.; Gallinat, S.; Wenck, H.; Deppert, W.; Grundhoff, A.; Knott, A. A comprehensive analysis of microRNA expression during human keratinocyte differentiation in vitro and in vivo. J. Investig. Dermatol 2011, 131, 20–29. [Google Scholar]
- Jurkin, J.; Schichl, Y.M.; Koeffel, R.; Bauer, T.; Richter, S.; Konradi, S.; Gesslbauer, B.; Strobl, H. miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation. J. Immunol 2010, 184, 4955–4965. [Google Scholar]
- Kretz, M.; Webster, D.E.; Flockhart, R.J.; Lee, C.S.; Zehnder, A.; Lopez-Pajares, V.; Qu, K.; Zheng, G.X.; Chow, J.; Kim, G.E.; et al. Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev 2012, 26, 338–343. [Google Scholar]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar]
- Gerdes, M.J.; Yuspa, S.H. The contribution of epidermal stem cells to skin cancer. Stem Cell Rev 2005, 1, 225–231. [Google Scholar]
- Kyrgidis, A.; Tzellos, T.G.; Triaridis, S. Melanoma: Stem cells, sun exposure and hallmarks for carcinogenesis, molecular concepts and future clinical implications. J. Carcinog. 2010, 9. [Google Scholar] [CrossRef]
- Grichnik, J.M. Melanoma, nevogenesis, and stem cell biology. J. Investig. Dermatol 2008, 128, 2365–2380. [Google Scholar]
- Rochette, P.J.; Lacoste, S.; Therrien, J.P.; Bastien, N.; Brash, D.E.; Drouin, R. Influence of cytosine methylation on ultraviolet-induced cyclobutane pyrimidine dimer formation in genomic DNA. Mutat. Res 2009, 665, 7–13. [Google Scholar]
- Gannon, H.S.; Donehower, L.A.; Lyle, S.; Jones, S.N. Mdm2-p53 signaling regulates epidermal stem cell senescence and premature aging phenotypes in mouse skin. Dev. Biol 2011, 353, 1–9. [Google Scholar]
- Dar, A.A.; Majid, S.; Rittsteuer, C.; de Semir, D.; Bezrookove, V.; Tong, S.; Nosrati, M.; Sagebiel, R.; Miller, J.R., III; Kashani-Sabet, M. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J. Natl. Cancer Inst. 2013, 105, 433–442. [Google Scholar]
- Harbst, K.; Staaf, J.; Masback, A.; Olsson, H.; Ingvar, C.; Vallon-Christersson, J.; Ringner, M.; Borg, A.; Jonsson, G. Multiple metastases from cutaneous malignant melanoma patients may display heterogeneous genomic and epigenomic patterns. Melanoma Res 2010, 20, 381–391. [Google Scholar]
- Millington, G.W. Proopiomelanocortin (POMC): The cutaneous roles of its melanocortin products and receptors. Clin. Exp. Dermatol 2006, 31, 407–412. [Google Scholar]
- Xu, N.; Meisgen, F.; Butler, L.M.; Han, G.; Wang, X.J.; Soderberg-Naucler, C.; Stahle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. J. Immunol 2013, 190, 678–688. [Google Scholar]
- Chen, W.; Liu, X.Z.; Oh, J.E.; Shin, K.H.; Kim, R.H.; Jiang, M.; Park, N.H.; Kang, M.K. Grainyhead-like 2 (GRHL2) inhibits keratinocyte differentiation through epigenetic mechanism. Cell Death Dis 2012, 3, e450. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shen, Q.; Jin, H.; Wang, X. Epidermal Stem Cells and Their Epigenetic Regulation. Int. J. Mol. Sci. 2013, 14, 17861-17880. https://doi.org/10.3390/ijms140917861
Shen Q, Jin H, Wang X. Epidermal Stem Cells and Their Epigenetic Regulation. International Journal of Molecular Sciences. 2013; 14(9):17861-17880. https://doi.org/10.3390/ijms140917861
Chicago/Turabian StyleShen, Qi, Hongchuan Jin, and Xian Wang. 2013. "Epidermal Stem Cells and Their Epigenetic Regulation" International Journal of Molecular Sciences 14, no. 9: 17861-17880. https://doi.org/10.3390/ijms140917861
APA StyleShen, Q., Jin, H., & Wang, X. (2013). Epidermal Stem Cells and Their Epigenetic Regulation. International Journal of Molecular Sciences, 14(9), 17861-17880. https://doi.org/10.3390/ijms140917861