Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
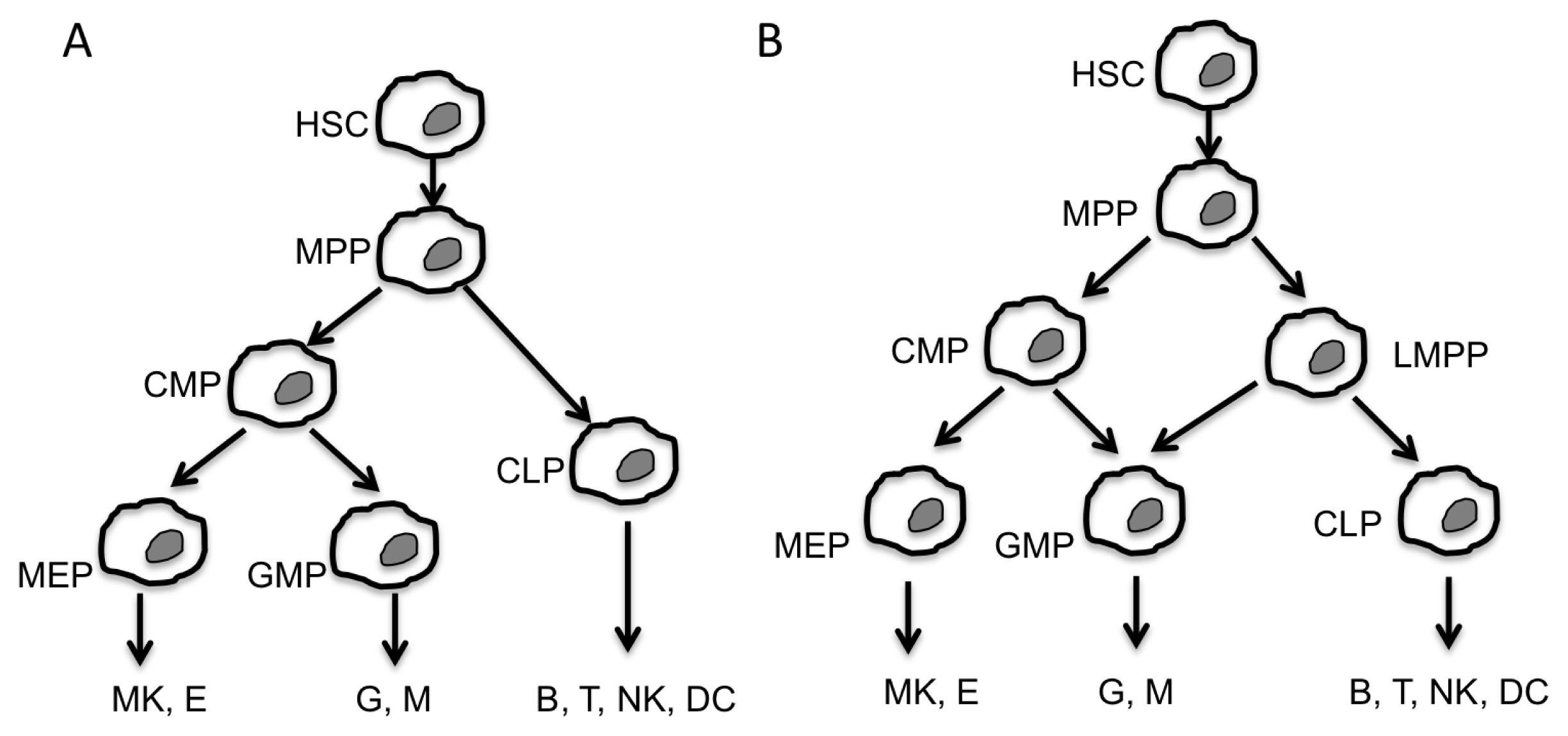
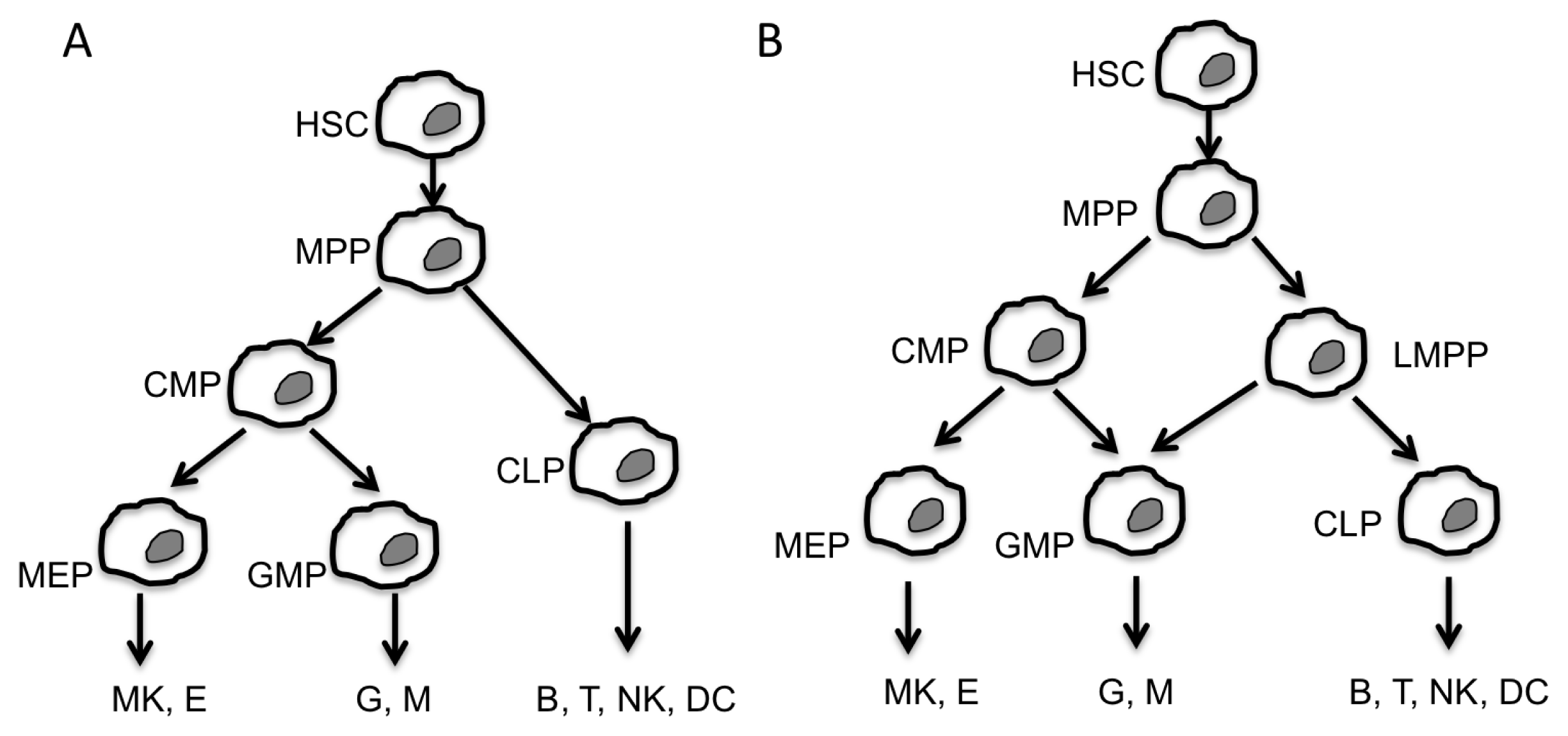
:1. Introduction
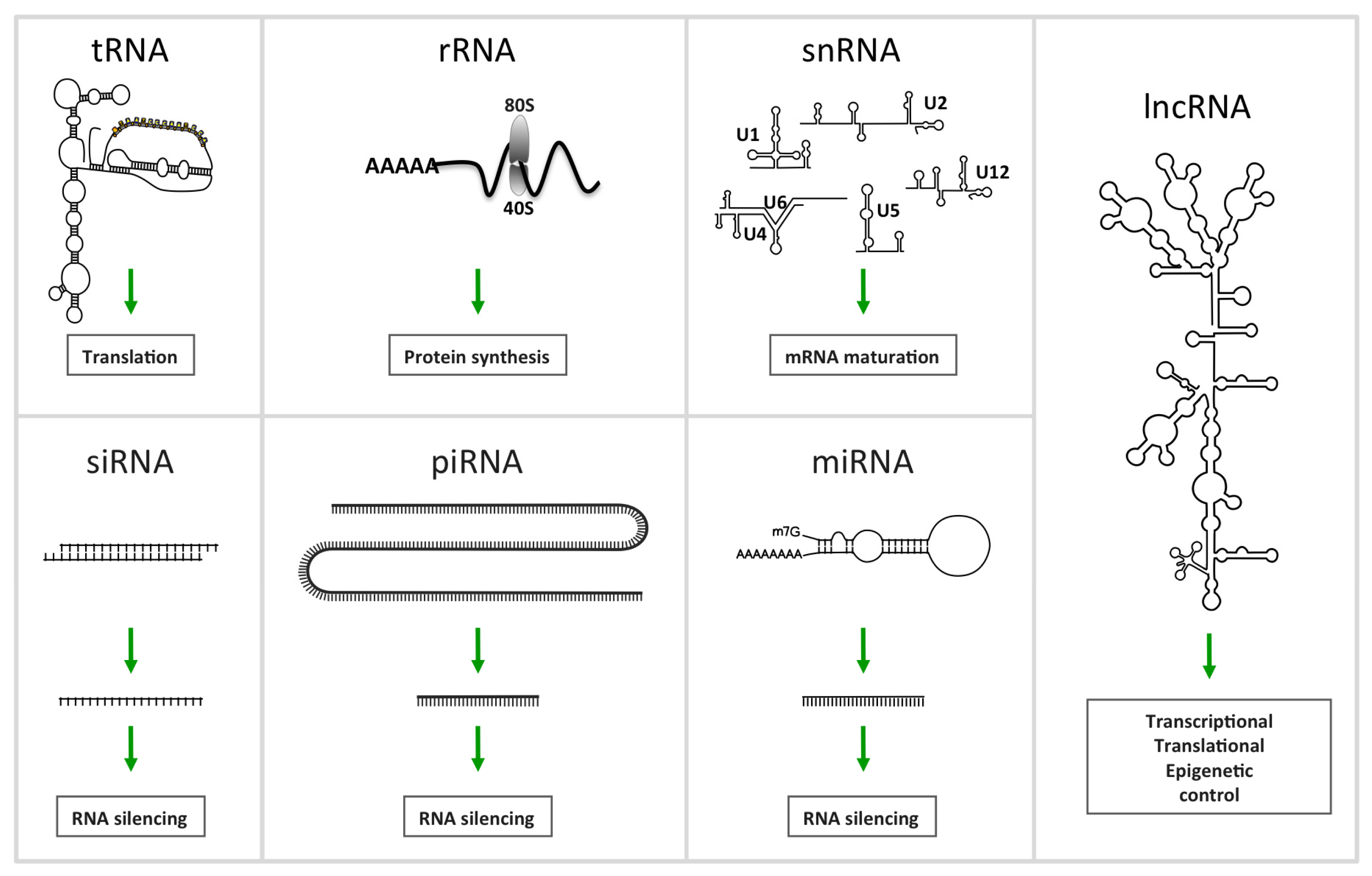
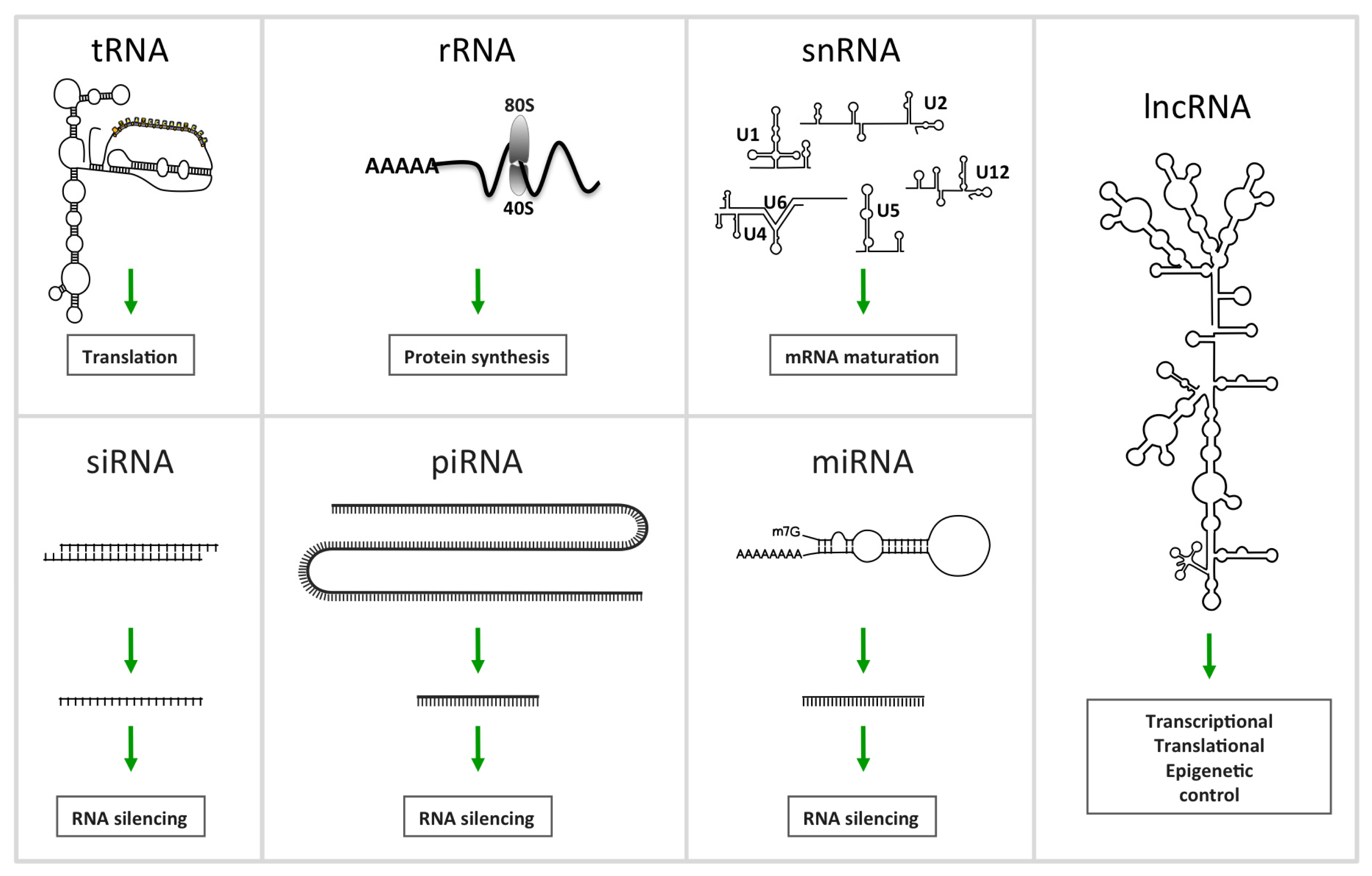
2. Non-Coding RNA
2.1. MiR and Hematopoiesis
2.1.1. MiRNAs, a Family of Regulatory ncRNAs
2.1.2. MiRNAs Regulate the Regulatory Network during Hematopoiesis
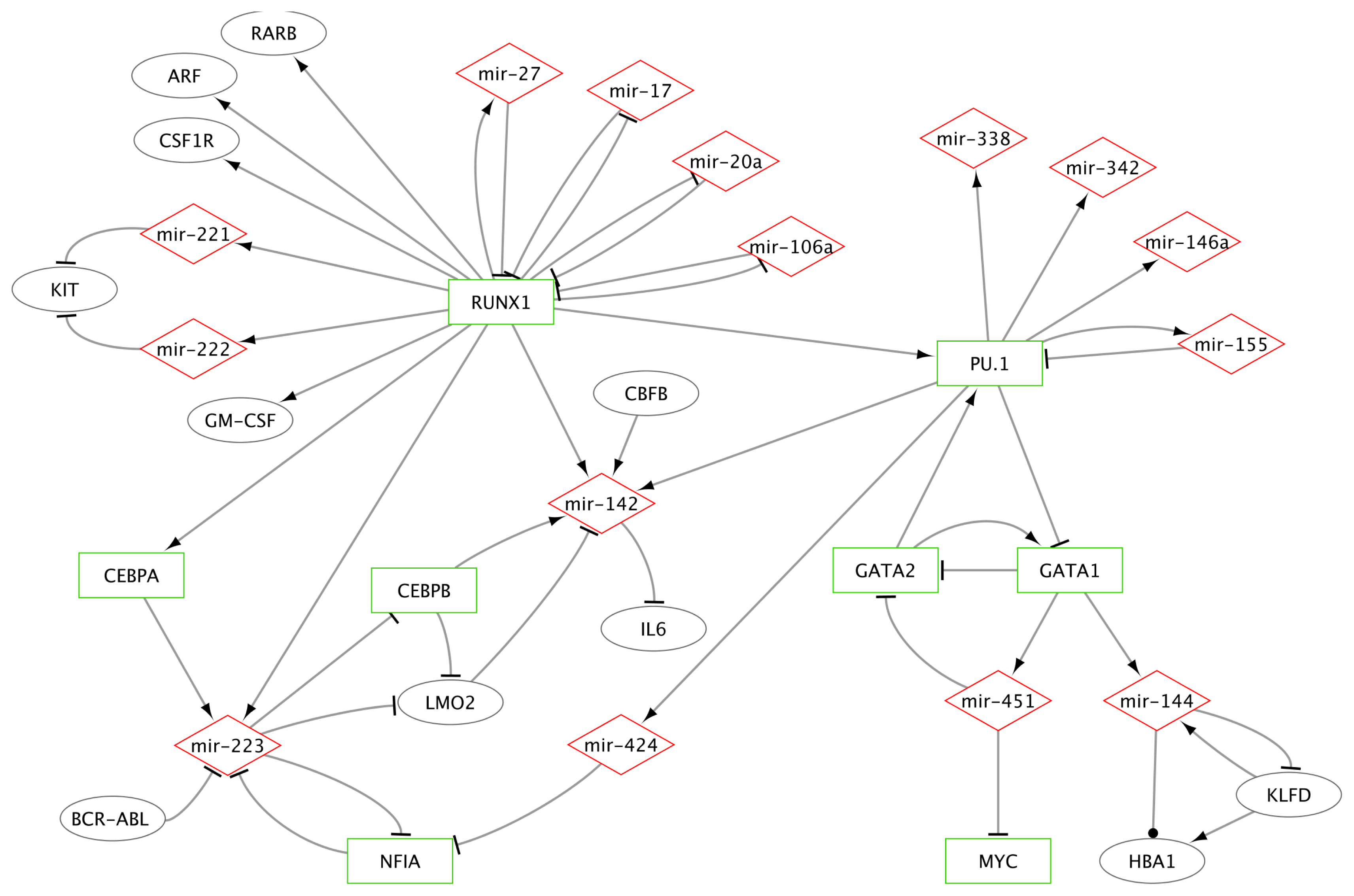
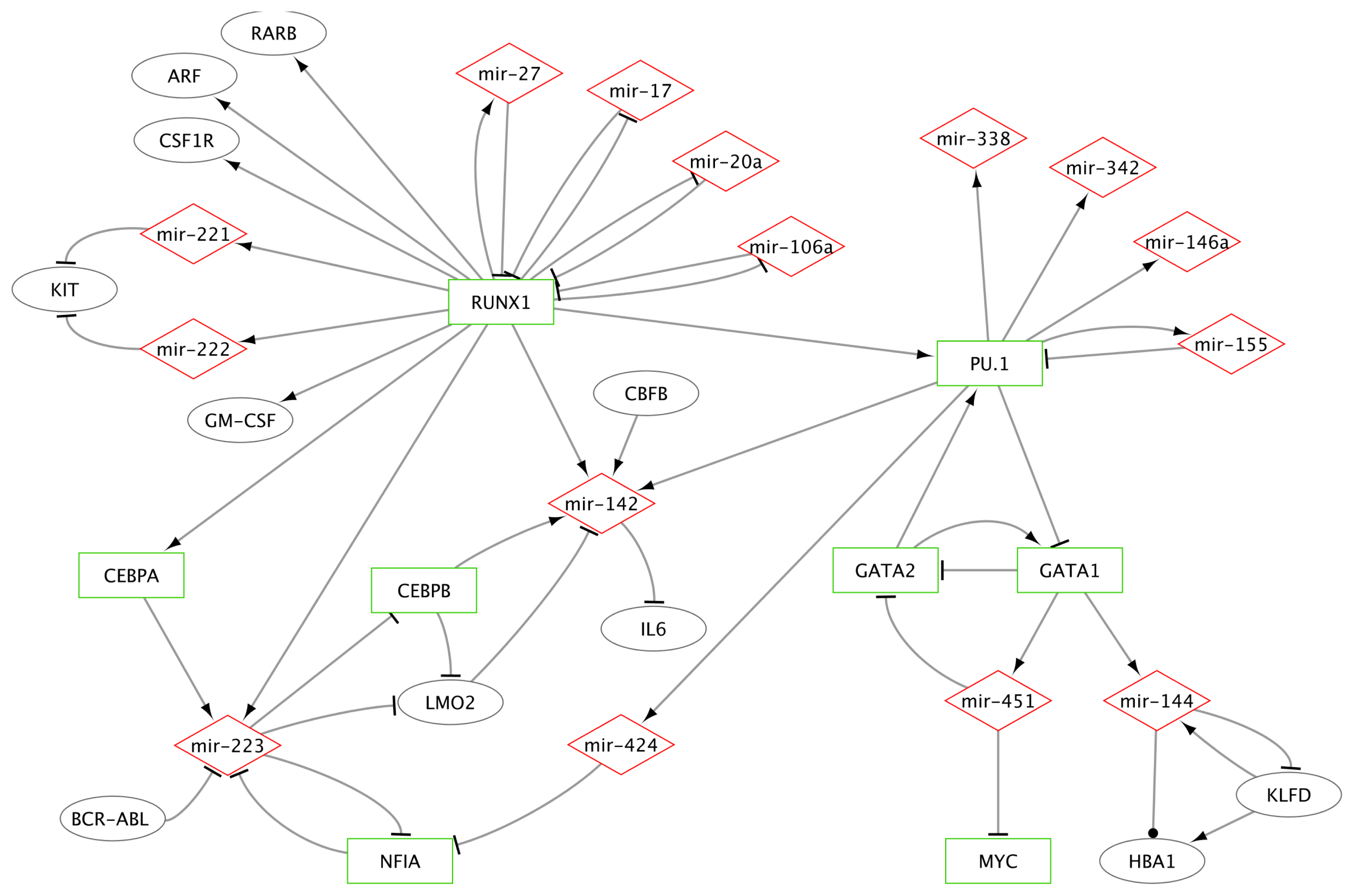
2.1.2.1. MiR-223 Is a Key Hematopoietic miRNA
2.1.2.2. MiRs-223/142/155 and Specific TFs as a Miniature Regulatory Network
2.1.2.3. RUNX1 Connection with miRs-222/221, 17-5p, 20a, 106a and 27a
2.1.2.4. MiR-146a and miR-155 Genes Are Regulated by PU.1
2.1.2.5. Erythroid Specific Expression of miRs-144 and 451
2.2. LncRNA and Hematopoietic Lineage
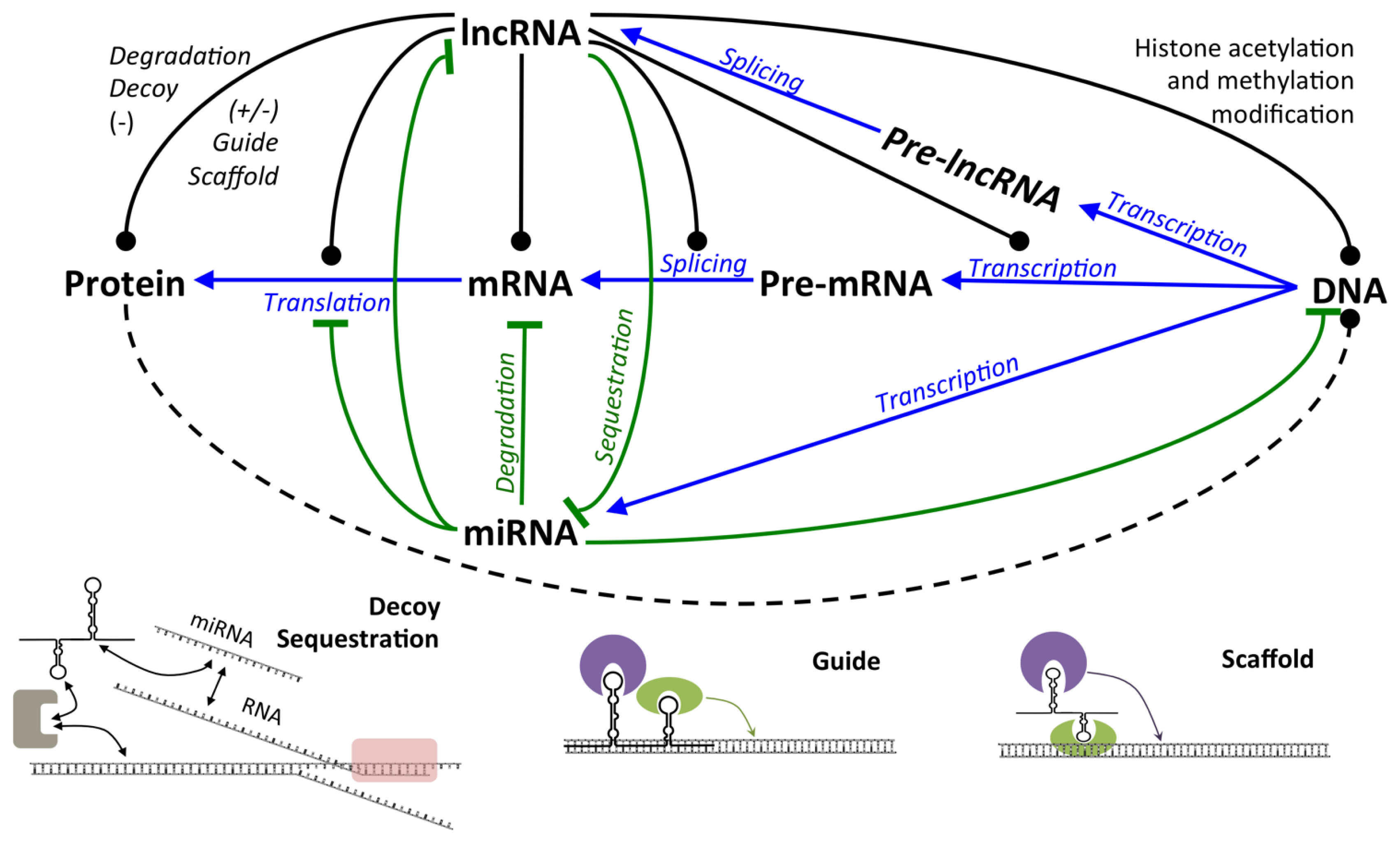
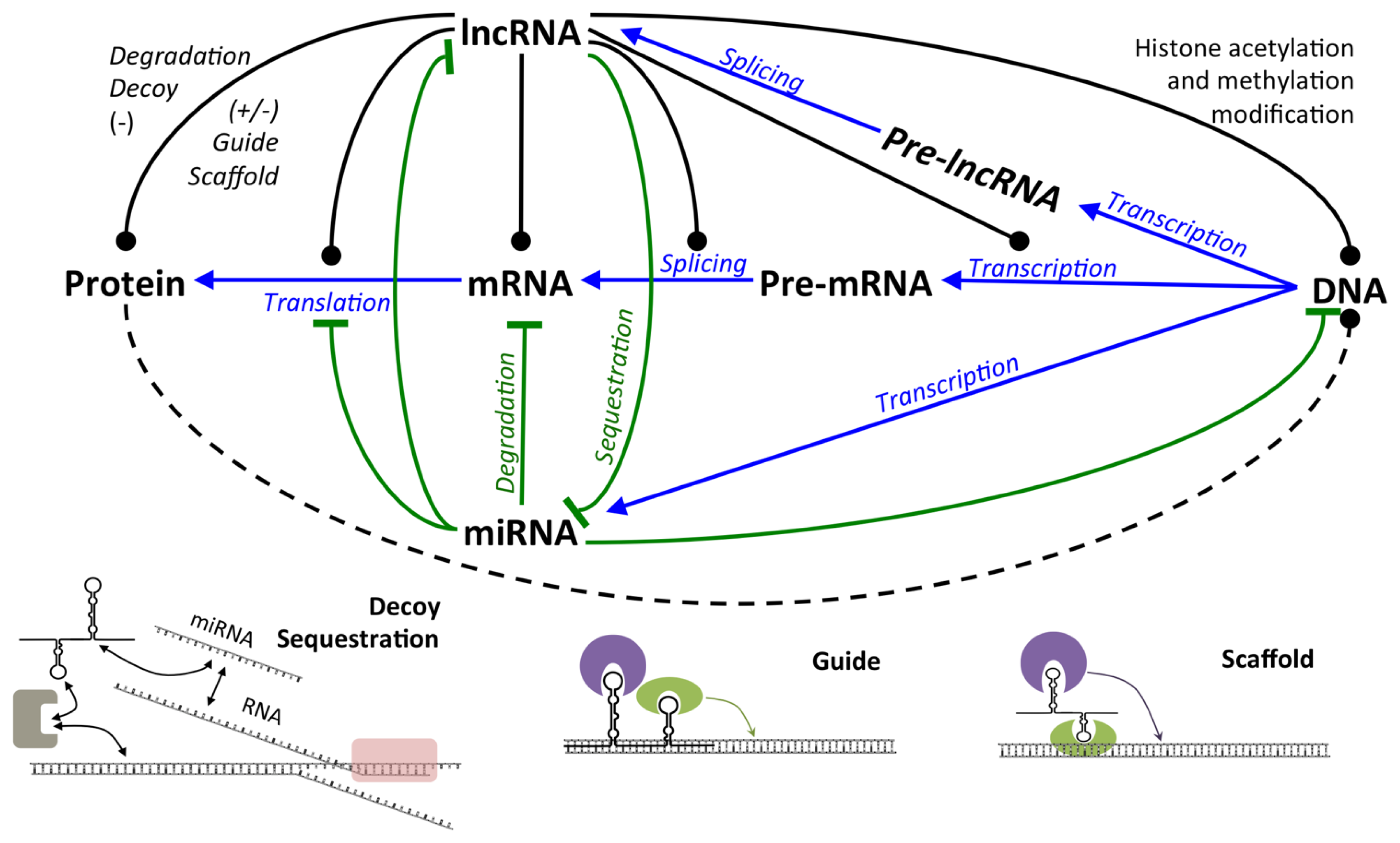
2.2.1. LncRNA
2.2.1.1. Expression and Regulation of lncRNA
2.2.1.2. Mechanisms
2.2.2. LncRNAs in Hematopoiesis
2.2.2.1. LncRNAs in Erythropoiesis
2.2.2.2. LncRNAs in Hematopoietic Differentiation
3. Conclusions
Acknowledgments
Conflict of Interest
References
- Inaba, M.; Yamashita, Y.M. Asymmetric stem cell division: Precizsion for robustness. Cell Stem Cell 2012, 11, 461–469. [Google Scholar]
- Lichtinger, M.; Ingram, R.; Hannah, R.; Muller, D.; Clarke, D.; Assi, S.A.; Lie, A.L.M.; Noailles, L.; Vijayabaskar, M.S.; Wu, M.; et al. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J 2012, 31, 4318–4333. [Google Scholar]
- Papathanasiou, P.; Attema, J.L.; Karsunky, H.; Hosen, N.; Sontani, Y.; Hoyne, G.F.; Tunningley, R.; Smale, S.T.; Weissman, I.L. Self-renewal of the long-term reconstituting subset of hematopoietic stem cells is regulated by Ikaros. Stem Cells 2009, 27, 3082–3092. [Google Scholar]
- Malinge, S.; Thiollier, C.; Chlon, T.M.; Dore, L.C.; Diebold, L.; Bluteau, O.; Mabialah, V.; Vainchenker, W.; Dessen, P.; Winandy, S.; et al. Ikaros inhibits megakaryopoiesis through functional interaction with GATA-1 and NOTCH signaling. Blood 2013, 121, 2440–2451. [Google Scholar]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Reports 2001, 2, 986–991. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Gen 2006, 15, R17–R29. [Google Scholar]
- Pheasant, M.; Mattick, J.S. Raising the estimate of functional human sequences. Genome Res 2007, 17, 1245–1253. [Google Scholar] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 2013, 14, 319. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009, 19, 92–105. [Google Scholar]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar]
- Wang, Z.; Yao, H.; Lin, S.; Zhu, X.; Shen, Z.; Lu, G.; Poon, W.S.; Xie, D.; Lin, M.C.; Kung, H.F. Transcriptional and epigenetic regulation of human microRNAs. Cancer Lett 2013, 331, 1–10. [Google Scholar]
- Kloosterman, W.P.; Plasterk, R.H. The diverse functions of microRNAs in animal development and disease. Dev. Cell 2006, 11, 441–450. [Google Scholar]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol 2006, 13, 1097–1101. [Google Scholar]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 2005, 33, 2697–2706. [Google Scholar]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [Green Version]
- Wu, L.; Fan, J.; Belasco, J.G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar]
- Zardo, G.; Ciolfi, A.; Vian, L.; Starnes, L.M.; Billi, M.; Racanicchi, S.; Maresca, C.; Fazi, F.; Travaglini, L.; Noguera, N.; et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood 2012, 119, 4034–4046. [Google Scholar]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005, 123, 819–831. [Google Scholar]
- Fukao, T.; Fukuda, Y.; Kiga, K.; Sharif, J.; Hino, K.; Enomoto, Y.; Kawamura, A.; Nakamura, K.; Takeuchi, T.; Tanabe, M. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 2007, 129, 617–631. [Google Scholar]
- Masaki, S.; Ohtsuka, R.; Abe, Y.; Muta, K.; Umemura, T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem. Biophys. Res. Commun 2007, 364, 509–514. [Google Scholar]
- Monticelli, S.; Ansel, K.M.; Xiao, C.; Socci, N.D.; Krichevsky, A.M.; Thai, T.H.; Rajewsky, N.; Marks, D.S.; Sander, C.; Rajewsky, K.; et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol 2005, 6, R71. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Choong, M.L.; Yang, H.H.; McNiece, I. MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp. Hematol 2007, 35, 551–564. [Google Scholar]
- Felli, N.; Pedini, F.; Romania, P.; Biffoni, M.; Morsilli, O.; Castelli, G.; Santoro, S.; Chicarella, S.; Sorrentino, A.; Peschle, C.; et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica 2009, 94, 479–486. [Google Scholar]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc 2007, 2, 2366–2382. [Google Scholar]
- Zhou, K.; Yi, S.; Yu, Z.; Li, Z.; Wang, Y.; Zou, D.; Qi, J.; Zhao, Y.; Qiu, L. MicroRNA-223 expression is uniformly down-regulated in B cell lymphoproliferative disorders and is associated with poor survival in patients with chronic lymphocytic leukemia. Leukemia Lymphoma 2012, 53, 1155–1161. [Google Scholar]
- Chen, M.J.; Yokomizo, T.; Zeigler, B.M.; Dzierzak, E.; Speck, N.A. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 2009, 457, 887–891. [Google Scholar]
- Okuda, T.; Takeda, K.; Fujita, Y.; Nishimura, M.; Yagyu, S.; Yoshida, M.; Akira, S.; Downing, J.R.; Abe, T. Biological characteristics of the leukemia-associated transcriptional factor AML1 disclosed by hematopoietic rescue of AML1-deficient embryonic stem cells by using a knock-in strategy. Mol. Cell. Biol 2000, 20, 319–328. [Google Scholar]
- Wang, Q.; Stacy, T.; Binder, M.; Marin-Padilla, M.; Sharpe, A.H.; Speck, N.A. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3444–3449. [Google Scholar]
- Yokomizo, T.; Ogawa, M.; Osato, M.; Kanno, T.; Yoshida, H.; Fujimoto, T.; Fraser, S.; Nishikawa, S.; Okada, H.; Satake, M.; et al. Requirement of Runx1/AML1/PEBP2alphaB for the generation of haematopoietic cells from endothelial cells. Genes Cells 2001, 6, 13–23. [Google Scholar]
- Nishimura, M.; Fukushima-Nakase, Y.; Fujita, Y.; Nakao, M.; Toda, S.; Kitamura, N.; Abe, T.; Okuda, T. VWRPY motif-dependent and -independent roles of AML1/Runx1 transcription factor in murine hematopoietic development. Blood 2004, 103, 562–570. [Google Scholar]
- Ran, D.; Shia, W.J.; Lo, M.C.; Fan, J.B.; Knorr, D.A.; Ferrell, P.I.; Ye, Z.; Yan, M.; Cheng, L.; Kaufman, D.S.; et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood 2013, 121, 2882–2890. [Google Scholar]
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007, 12, 457–466. [Google Scholar]
- Nimer, S.D.; Moore, M.A. Effects of the leukemia-associated AML1-ETO protein on hematopoietic stem and progenitor cells. Oncogene 2004, 23, 4249–4254. [Google Scholar]
- Frank, R.; Zhang, J.; Uchida, H.; Meyers, S.; Hiebert, S.W.; Nimer, S.D. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene 1995, 11, 2667–2674. [Google Scholar]
- Zhang, D.E.; Fujioka, K.; Hetherington, C.J.; Shapiro, L.H.; Chen, H.M.; Look, A.T.; Tenen, D.G. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1). Mol. Cell. Biol 1994, 14, 8085–8095. [Google Scholar]
- Sun, W.; Shen, W.; Yang, S.; Hu, F.; Li, H.; Zhu, T.H. miR-223 and miR-142 attenuate hematopoietic cell proliferation, and miR-223 positively regulates miR-142 through LMO2 isoforms and CEBP-beta. Cell Res 2010, 20, 1158–1169. [Google Scholar]
- Warren, A.J.; Colledge, W.H.; Carlton, M.B.; Evans, M.J.; Smith, A.J.; Rabbitts, T.H. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell 1994, 78, 45–57. [Google Scholar]
- Wadman, I.A.; Osada, H.; Grutz, G.G.; Agulnick, A.D.; Westphal, H.; Forster, A.; Rabbitts, T.H. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J 1997, 16, 3145–3157. [Google Scholar]
- Starnes, L.M.; Sorrentino, A.; Ferracin, M.; Negrini, M.; Pelosi, E.; Nervi, C.; Peschle, C. A transcriptome-wide approach reveals the key contribution of NFI-A in promoting erythroid differentiation of human CD34(+) progenitors and CML cells. Leukemia 2010, 24, 1220–1223. [Google Scholar]
- Starnes, L.M.; Sorrentino, A.; Pelosi, E.; Ballarino, M.; Morsilli, O.; Biffoni, M.; Santoro, S.; Felli, N.; Castelli, G.; de Marchis, M.L.; et al. NFI-A directs the fate of hematopoietic progenitors to the erythroid or granulocytic lineage and controls beta-globin and G-CSF receptor expression. Blood 2009, 114, 1753–1763. [Google Scholar]
- Sun, Y.; Sun, J.; Tomomi, T.; Nieves, E.; Mathewson, N.; Tamaki, H.; Evers, R.; Reddy, P. PU.1-dependent transcriptional regulation of miR-142 contributes to its hematopoietic cell-specific expression and modulation of IL-6. J. Immunol 2013, 190, 4005–4013. [Google Scholar]
- Brioschi, M.; Fischer, J.; Cairoli, R.; Rossetti, S.; Pezzetti, L.; Nichelatti, M.; Turrini, M.; Corlazzoli, F.; Scarpati, B.; Morra, E.; et al. Down-regulation of microRNAs 222/221 in acute myelogenous leukemia with deranged core-binding factor subunits. Neoplasia 2010, 12, 866–876. [Google Scholar]
- Feng, J.; Iwama, A.; Satake, M.; Kohu, K. MicroRNA-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating Runx1. Br. J. Haematol 2009, 145, 412–423. [Google Scholar]
- Ben-Ami, O.; Pencovich, N.; Lotem, J.; Levanon, D.; Groner, Y. A regulatory interplay between miR-27a and Runx1 during megakaryopoiesis. Proc. Natl. Acad. Sci. USA 2009, 106, 238–243. [Google Scholar]
- Rossetti, S.; Sacchi, N. RUNX1: A microRNA hub in normal and malignant hematopoiesis. Int. J. Mol. Sci 2013, 14, 1566–1588. [Google Scholar]
- Wontakal, S.N.; Guo, X.; Will, B.; Shi, M.; Raha, D.; Mahajan, M.C.; Weissman, S.; Snyder, M.; Steidl, U.; Zheng, D.; et al. A large gene network in immature erythroid cells is controlled by the myeloid and B cell transcriptional regulator PU.1. PLoS Genet 2011, 7, e1001392. [Google Scholar]
- Houston, I.B.; Kamath, M.B.; Schweitzer, B.L.; Chlon, T.M.; DeKoter, R.P. Reduction in PU.1 activity results in a block to B-cell development, abnormal myeloid proliferation, and neonatal lethality. Exp. Hematol 2007, 35, 1056–1068. [Google Scholar]
- Anderson, M.K.; Weiss, A.H.; Hernandez-Hoyos, G.; Dionne, C.J.; Rothenberg, E.V. Constitutive expression of PU.1 in fetal hematopoietic progenitors blocks T cell development at the pro-T cell stage. Immunity 2002, 16, 285–296. [Google Scholar]
- Dionne, C.J.; Tse, K.Y.; Weiss, A.H.; Franco, C.B.; Wiest, D.L.; Anderson, M.K.; Rothenberg, E.V. Subversion of T lineage commitment by PU.1 in a clonal cell line system. Dev. Biol 2005, 280, 448–466. [Google Scholar]
- Laiosa, C.V.; Stadtfeld, M.; Xie, H.; de Andres-Aguayo, L.; Graf, T. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBP alpha and PU.1 transcription factors. Immunity 2006, 25, 731–744. [Google Scholar]
- Lefebvre, J.M.; Haks, M.C.; Carleton, M.O.; Rhodes, M.; Sinnathamby, G.; Simon, M.C.; Eisenlohr, L.C.; Garrett-Sinha, L.A.; Wiest, D.L. Enforced expression of Spi-B reverses T lineage commitment and blocks beta-selection. J. Immunol 2005, 174, 6184–6194. [Google Scholar]
- Ghani, S.; Riemke, P.; Schonheit, J.; Lenze, D.; Stumm, J.; Hoogenkamp, M.; Lagendijk, A.; Heinz, S.; Bonifer, C.; Bakkers, J.; et al. Macrophage development from HSCs requires PU.1-coordinated microRNA expression. Blood 2011, 118, 2275–2284. [Google Scholar]
- Rosa, A.; Ballarino, M.; Sorrentino, A.; Sthandier, O.; de Angelis, F.G.; Marchioni, M.; Masella, B.; Guarini, A.; Fatica, A.; Peschle, C.; et al. The interplay between the master transcription factor PU.1 and miR-424 regulates human monocyte/macrophage differentiation. Proc. Natl. Acad. Sci. USA 2007, 104, 19849–19854. [Google Scholar]
- Rhodes, J.; Hagen, A.; Hsu, K.; Deng, M.; Liu, T.X.; Look, A.T.; Kanki, J.P. Interplay of pu.1 and gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Dev. Cell 2005, 8, 97–108. [Google Scholar]
- Zhang, P.; Zhang, X.; Iwama, A.; Yu, C.; Smith, K.A.; Mueller, B.U.; Narravula, S.; Torbett, B.E.; Orkin, S.H.; Tenen, D.G. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000, 96, 2641–2648. [Google Scholar]
- Stachura, D.L.; Chou, S.T.; Weiss, M.J. Early block to erythromegakaryocytic development conferred by loss of transcription factor GATA-1. Blood 2006, 107, 87–97. [Google Scholar]
- Chou, S.T.; Khandros, E.; Bailey, L.C.; Nichols, K.E.; Vakoc, C.R.; Yao, Y.; Huang, Z.; Crispino, J.D.; Hardison, R.C.; Blobel, G.A.; et al. Graded repression of PU.1/Sfpi1 gene transcription by GATA factors regulates hematopoietic cell fate. Blood 2009, 114, 983–994. [Google Scholar]
- Dore, L.C.; Amigo, J.D.; Dos Santos, C.O.; Zhang, Z.; Gai, X.; Tobias, J.W.; Yu, D.; Klein, A.M.; Dorman, C.; Wu, W.; et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3333–3338. [Google Scholar]
- Papapetrou, E.P.; Korkola, J.E.; Sadelain, M. A genetic strategy for single and combinatorial analysis of miRNA function in mammalian hematopoietic stem cells. Stem Cells 2010, 28, 287–296. [Google Scholar]
- Pase, L.; Layton, J.E.; Kloosterman, W.P.; Carradice, D.; Waterhouse, P.M.; Lieschke, G.J. miR-451 regulates zebrafish erythroid maturation in vivo via its target gata2. Blood 2009, 113, 1794–1804. [Google Scholar]
- Minegishi, N.; Suzuki, N.; Yokomizo, T.; Pan, X.; Fujimoto, T.; Takahashi, S.; Hara, T.; Miyajima, A.; Nishikawa, S.; Yamamoto, M. Expression and domain-specific function of GATA-2 during differentiation of the hematopoietic precursor cells in midgestation mouse embryos. Blood 2003, 102, 896–905. [Google Scholar]
- Bruchova, H.; Yoon, D.; Agarwal, A.M.; Mendell, J.; Prchal, J.T. Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp. Hematol 2007, 35, 1657–1667. [Google Scholar]
- Mizuno, Y.; Tokuzawa, Y.; Ninomiya, Y.; Yagi, K.; Yatsuka-Kanesaki, Y.; Suda, T.; Fukuda, T.; Katagiri, T.; Kondoh, Y.; Amemiya, T.; et al. miR-210 promotes osteoblastic differentiation through inhibition of AcvR1b. FEBS Lett 2009, 583, 2263–2268. [Google Scholar]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Carninci, P.; Hayashizaki, Y. Noncoding RNA transcription beyond annotated genes. Curr. Opin. Genetics Dev 2007, 17, 139–144. [Google Scholar]
- Gibb, E.A.; Vucic, E.A.; Enfield, K.S.; Stewart, G.L.; Lonergan, K.M.; Kennett, J.Y.; Becker-Santos, D.D.; MacAulay, C.E.; Lam, S.; Brown, C.J.; et al. Human cancer long non-coding RNA transcriptomes. PLoS One 2011, 6, e25915. [Google Scholar]
- Whitehead, J.; Pandey, G.K.; Kanduri, C. Regulation of the mammalian epigenome by long noncoding RNAs. Biochim. Biophys. Acta 2009, 1790, 936–947. [Google Scholar]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. in press. 2012. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [Green Version]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar]
- Kim, Y.K.; Furic, L.; Parisien, M.; Major, F.; DesGroseillers, L.; Maquat, L.E. Staufen1 regulates diverse classes of mammalian transcripts. EMBO J 2007, 26, 2670–2681. [Google Scholar]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar]
- Kindler, S.; Wang, H.; Richter, D.; Tiedge, H. RNA transport and local control of translation. Annu. Rev. Cell Dev. Biol 2005, 21, 223–245. [Google Scholar]
- Clark, M.B.; Johnston, R.L.; Inostroza-Ponta, M.; Fox, A.H.; Fortini, E.; Moscato, P.; Dinger, M.E.; Mattick, J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res 2012, 22, 885–898. [Google Scholar] [Green Version]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar]
- Wilusz, J.E.; Jnbaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 2012, 26, 2392–2407. [Google Scholar]
- Jalali, S.; Bhartiya, D.; Lalwani, M.K.; Sivasubbu, S.; Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One 2013, 8, e53823. [Google Scholar]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol 2010, 7, 582–585. [Google Scholar]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar]
- Da Sacco, L.; Baldassarre, A.; Masotti, A. Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int. J. Mol. Sci 2012, 13, 97–114. [Google Scholar]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci. Signal. 2010, 3. ra29. [Google Scholar]
- Torarinsson, E.; Sawera, M.; Havgaard, J.H.; Fredholm, M.; Gorodkin, J. Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Res 2006, 16, 885–889. [Google Scholar]
- Espinoza, C.A.; Allen, T.A.; Hieb, A.R.; Kugel, J.F.; Goodrich, J.A. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat. Struct. Mol. Biol 2004, 11, 822–829. [Google Scholar]
- Mariner, P.D.; Walters, R.D.; Espinoza, C.A.; Drullinger, L.F.; Wagner, S.D.; Kugel, J.F.; Goodrich, J.A. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell 2008, 29, 499–509. [Google Scholar]
- Hu, J.; Xu, T.; Zhu, T.; Lou, Q.; Wang, X.; Wu, Y.; Huang, R.; Liu, J.; Liu, H.; Yu, F.; et al. Monoclonal antibodies against accumulation-associated protein affect EPS biosynthesis and enhance bacterial accumulation of Staphylococcus epidermidis. PLoS One 2011, 6, e20918. [Google Scholar]
- Paralkar, V.R.; Weiss, M.J. A new ‘Linc’ between noncoding RNAs and blood development. Genes Dev 2011, 25, 2555–2558. [Google Scholar]
- McGlinn, E.; Yekta, S.; Mansfield, J.H.; Soutschek, J.; Bartel, D.P.; Tabin, C.J. In ovo application of antagomiRs indicates a role for miR-196 in patterning the chick axial skeleton through Hox gene regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 18610–18615. [Google Scholar]
- Crooks, G.M.; Fuller, J.; Petersen, D.; Izadi, P.; Malik, P.; Pattengale, P.K.; Kohn, D.B.; Gasson, J.C. Constitutive HOXA5 expression inhibits erythropoiesis and increases myelopoiesis from human hematopoietic progenitors. Blood 1999, 94, 519–528. [Google Scholar]
- Moore, M.A.; Dorn, D.C.; Schuringa, J.J.; Chung, K.Y.; Morrone, G. Constitutive activation of Flt3 and STAT5A enhances self-renewal and alters differentiation of hematopoietic stem cells. Exp. Hematol 2007, 35, 105–116. [Google Scholar]
- Wang, Q.; Huang, Z.; Xue, H.; Jin, C.; Ju, X.L.; Han, J.D.; Chen, Y.G. MicroRNA miR-24 inhibits erythropoiesis by targeting activin type I receptor ALK4. Blood 2008, 111, 588–595. [Google Scholar]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar]
- Goshen, R.; Rachmilewitz, J.; Schneider, T.; de-Groot, N.; Ariel, I.; Palti, Z.; Hochberg, A.A. The expression of the H-19 and IGF-2 genes during human embryogenesis and placental development. Mol. Reproduct. Dev 1993, 34, 374–379. [Google Scholar]
- Nunez, C.; Bashein, A.M.; Brunet, C.L.; Hoyland, J.A.; Freemont, A.J.; Buckle, A.M.; Murphy, C.; Cross, M.A.; Lucas, G.; Bostock, V.J.; et al. Expression of the imprinted tumour-suppressor gene H19 is tightly regulated during normal haematopoiesis and is reduced in haematopoietic precursors of patients with the myeloproliferative disease polycythaemia vera. J. Pathol 2000, 190, 61–68. [Google Scholar]
- Tessema, M.; Langer, F.; Bock, O.; Seltsam, A.; Metzig, K.; Hasemeier, B.; Kreipe, H.; Lehmann, U. Down-regulation of the IGF-2/H19 locus during normal and malignant hematopoiesis is independent of the imprinting pattern. Int. J. Oncol 2005, 26, 499–507. [Google Scholar]
- Lustig, O.; Ariel, I.; Ilan, J.; Lev-Lehman, E.; De-Groot, N.; Hochberg, A. Expression of the imprinted gene H19 in the human fetus. Mol. Reproduct. Dev 1994, 38, 239–246. [Google Scholar]
- Borensztein, M.; Monnier, P.; Court, F.; Louault, Y.; Ripoche, M.A.; Tiret, L.; Yao, Z.; Tapscott, S.J.; Forne, T.; Montarras, D.; et al. Myod and H19-Igf2 locus interactions are required for diaphragm formation in the mouse. Development 2013, 140, 1231–1239. [Google Scholar]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Upregulated H19 contributes to bladder cancer cell proliferation by regulating ID2 expression. FEBS J 2013, 280, 1709–1716. [Google Scholar]
- Wagner, L.A.; Christensen, C.J.; Dunn, D.M.; Spangrude, G.J.; Georgelas, A.; Kelley, L.; Esplin, M.S.; Weiss, R.B.; Gleich, G.J. EGO, a novel, noncoding RNA gene, regulates eosinophil granule protein transcript expression. Blood 2007, 109, 5191–5198. [Google Scholar]
- Bertani, S.; Sauer, S.; Bolotin, E.; Sauer, F. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol. Cell 2011, 43, 1040–1046. [Google Scholar]
- Jacob, E.; Hod-Dvorai, R.; Ben-Mordechai, O.L.; Boyko, Y.; Avni, O. Dual function of polycomb group proteins in differentiated murine T helper (CD4+) cells. J. Mol. Signal 2011, 6, 5. [Google Scholar]
- Jo, S.; Lee, H.; Kim, S.; Hwang, E.M.; Park, J.Y.; Kang, S.S.; Chung, H. Inhibition of PCGF2 enhances granulocytic differentiation of acute promyelocytic leukemia cell line HL-60 via induction of HOXA7. Biochem. Biophys. Res. Commun 2011, 416, 86–91. [Google Scholar]
- Porcher, C.; Swat, W.; Rockwell, K.; Fujiwara, Y.; Alt, F.W.; Orkin, S.H. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell 1996, 86, 47–57. [Google Scholar]
- Zhang, X.; Lian, Z.; Padden, C.; Gerstein, M.B.; Rozowsky, J.; Snyder, M.; Gingeras, T.R.; Kapranov, P.; Weissman, S.M.; Newburger, P.E. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 2009, 113, 2526–2534. [Google Scholar]
- Zhao, H.; Zhang, X.; Frazao, J.B.; Condino-Neto, A.; Newburger, P.E. HOX antisense lincRNA HOXA-AS2 is an apoptosis repressor in all trans retinoic acid treated NB4 promyelocytic leukemia cells. J. Cell. Biochem. 2013. [Google Scholar] [CrossRef]
- Savarese, F.; Flahndorfer, K.; Jaenisch, R.; Busslinger, M.; Wutz, A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Mol. Cell. Biol 2006, 26, 7167–7177. [Google Scholar]
- Ahn, J.H.; Lee, Y.; Jeon, C.; Lee, S.J.; Lee, B.H.; Choi, K.D.; Bae, Y.S. Identification of the genes differentially expressed in human dendritic cell subsets by cDNA subtraction and microarray analysis. Blood 2002, 100, 1742–1754. [Google Scholar]
- Lee, M.Y.; Kim, W.J.; Kang, Y.J.; Jung, Y.M.; Kang, Y.M.; Suk, K.; Park, J.E.; Choi, E.M.; Choi, B.K.; Kwon, B.S.; et al. Z39Ig is expressed on macrophages and may mediate inflammatory reactions in arthritis and atherosclerosis. J. Leukocyte Biol 2006, 80, 922–928. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morceau, F.; Chateauvieux, S.; Gaigneaux, A.; Dicato, M.; Diederich, M. Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation. Int. J. Mol. Sci. 2013, 14, 14744-14770. https://doi.org/10.3390/ijms140714744
Morceau F, Chateauvieux S, Gaigneaux A, Dicato M, Diederich M. Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation. International Journal of Molecular Sciences. 2013; 14(7):14744-14770. https://doi.org/10.3390/ijms140714744
Chicago/Turabian StyleMorceau, Franck, Sébastien Chateauvieux, Anthoula Gaigneaux, Mario Dicato, and Marc Diederich. 2013. "Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation" International Journal of Molecular Sciences 14, no. 7: 14744-14770. https://doi.org/10.3390/ijms140714744
APA StyleMorceau, F., Chateauvieux, S., Gaigneaux, A., Dicato, M., & Diederich, M. (2013). Long and Short Non-Coding RNAs as Regulators of Hematopoietic Differentiation. International Journal of Molecular Sciences, 14(7), 14744-14770. https://doi.org/10.3390/ijms140714744