Pilot Study of CYP2B6 Genetic Variation to Explore the Contribution of Nitrosamine Activation to Lung Carcinogenesis

Abstract
:1. Introduction
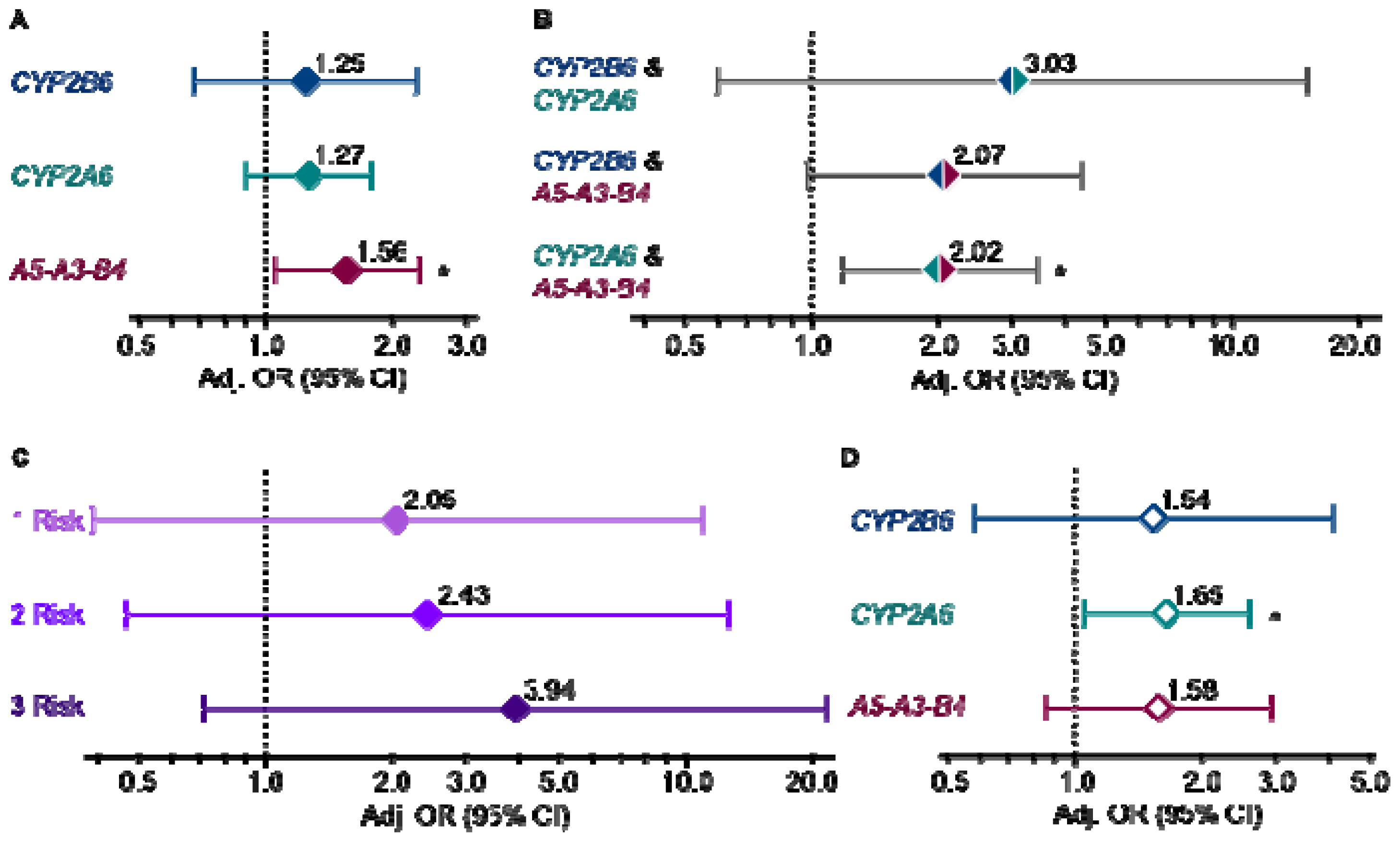
2. Results and Discussion
3. Experimental Section
4. Conclusions
Supplementary Information
ijms-14-08381-s001.pdfAcknowledgments
Conflict of Interest
References
- Wassenaar, C.A.; Dong, Q.; Wei, Q.; Amos, C.I.; Spitz, M.R.; Tyndale, R.F. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J. Natl. Cancer Inst 2011, 103, 1342–1346. [Google Scholar]
- Rossini, A.; de Almeida Simao, T.; Albano, R.M.; Pinto, L.F. CYP2A6 polymorphisms and risk for tobacco-related cancers. Pharmacogenomics 2008, 9, 1737–1752. [Google Scholar]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar]
- Gervot, L.; Rochat, B.; Gautier, J.C.; Bohnenstengel, F.; Kroemer, H.; de Berardinis, V.; Martin, H.; Beaune, P.; de Waziers, I. Human CYP2B6: Expression, inducibility and catalytic activities. Pharmacogenetics 1999, 9, 295–306. [Google Scholar]
- Anttila, S.; Raunio, H.; Hakkola, J. Cytochrome P450-mediated pulmonary metabolism of carcinogens: Regulation and cross-talk in lung carcinogenesis. Am. J. Respir. Cell Mol. Biol 2011, 44, 583–590. [Google Scholar]
- Jalas, J.R.; Hecht, S.S.; Murphy, S.E. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem. Res. Toxicol 2005, 18, 95–110. [Google Scholar]
- Weng, Y.; Fang, C.; Turesky, R.J.; Behr, M.; Kaminsky, L.S.; Ding, X. Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res 2007, 67, 7825–7832. [Google Scholar]
- Dicke, K.E.; Skrlin, S.M.; Murphy, S.E. Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-butanone metabolism by cytochrome P450 2B6. Drug. Metab. Dispos 2005, 33, 1760–1764. [Google Scholar]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol 2005, 18, 61–69. [Google Scholar]
- Lee, A.M.; Jepson, C.; Shields, P.G.; Benowitz, N.; Lerman, C.; Tyndale, R.F. CYP2B6 genotype does not alter nicotine metabolism, plasma levels, or abstinence with nicotine replacement therapy. Cancer Epidemiol. Biomark. Prev 2007, 16, 1312–1314. [Google Scholar]
- Al Koudsi, N.; Tyndale, R.F. Hepatic CYP2B6 is altered by genetic, physiologic, and environmental factors but plays little role in nicotine metabolism. Xenobiotica 2010, 40, 381–392. [Google Scholar]
- Haberl, M.; Anwald, B.; Klein, K.; Weil, R.; Fuss, C.; Gepdiremen, A.; Zanger, U.M.; Meyer, U.A.; Wojnowski, L. Three haplotypes associated with CYP2A6 phenotypes in Caucasians. Pharmacogenet. Genomics 2005, 15, 609–624. [Google Scholar]
- Hoffman, S.M.; Nelson, D.R.; Keeney, D.S. Organization, structure and evolution of the CYP2 gene cluster on human chromosome 19. Pharmacogenetics 2001, 11, 687–698. [Google Scholar]
- Johnstone, E.; Benowitz, N.; Cargill, A.; Jacob, R.; Hinks, L.; Day, I.; Murphy, M.; Walton, R. Determinants of the rate of nicotine metabolism and effects on smoking behavior. Clin. Pharmacol. Ther 2006, 80, 319–330. [Google Scholar]
- Zanger, U.M.; Klein, K.; Saussele, T.; Blievernicht, J.; Hofmann, M.H.; Schwab, M. Polymorphic CYP2B6: Molecular mechanisms and emerging clinical significance. Pharmacogenomics 2007, 8, 743–759. [Google Scholar]
- Lee, A.M.; Jepson, C.; Hoffmann, E.; Epstein, L.; Hawk, L.W.; Lerman, C.; Tyndale, R.F. CYP2B6 genotype alters abstinence rates in a bupropion smoking cessation trial. Biol. Psychiatr 2007, 62, 635–641. [Google Scholar]
- Xu, C.; Ogburn, E.T.; Guo, Y.; Desta, Z. Effects of the CYP2B6*6 allele on catalytic properties and inhibition of CYP2B6 in vitro: Implication for the mechanism of reduced efavirenz metabolism and other CYP2B6 substrates in vivo. Drug Metab. Dispos 2012, 40, 717–725. [Google Scholar]
- Yimer, G.; Amogne, W.; Habtewold, A.; Makonnen, E.; Ueda, N.; Suda, A.; Worku, A.; Haefeli, W.E.; Burhenne, J.; Aderaye, G.; et al. High plasma efavirenz level and CYP2B6*6 are associated with efavirenz-based HAART-induced liver injury in the treatment of naive HIV patients from Ethiopia: A prospective cohort study. Pharmacogenomics J 2011, 12, 499–506. [Google Scholar]
- Rotger, M.; Tegude, H.; Colombo, S.; Cavassini, M.; Furrer, H.; Decosterd, L.; Blievernicht, J.; Saussele, T.; Gunthard, H.F.; Schwab, M.; et al. Predictive value of known and novel alleles of CYP2B6 for efavirenz plasma concentrations in HIV-infected individuals. Clin. Pharmacol. Ther 2007, 81, 557–566. [Google Scholar]
- Desta, Z.; Saussele, T.; Ward, B.; Blievernicht, J.; Li, L.; Klein, K.; Flockhart, D.A.; Zanger, U.M. Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics 2007, 8, 547–558. [Google Scholar]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar]
- Larsen, J.E.; Colosimo, M.L.; Yang, I.A.; Bowman, R.; Zimmerman, P.V.; Fong, K.M. CYP1A1 Ile462Val and MPO G-463A interact to increase risk of adenocarcinoma but not squamous cell carcinoma of the lung. Carcinogenesis 2006, 27, 525–532. [Google Scholar]
- Alexandrie, A.K.; Nyberg, F.; Warholm, M.; Rannug, A. Influence of CYP1A1, GSTM1, GSTT1, and NQO1 genotypes and cumulative smoking dose on lung cancer risk in a Swedish population. Cancer Epidemiol. Biomark. Prev 2004, 13, 908–914. [Google Scholar]
- Ishibe, N.; Wiencke, J.K.; Zuo, Z.F.; McMillan, A.; Spitz, M.; Kelsey, K.T. Susceptibility to lung cancer in light smokers associated with CYP1A1 polymorphisms in Mexican- and African-Americans. Cancer Epidemiol. Biomark. Prev 1997, 6, 1075–1080. [Google Scholar]
- Schabath, M.B.; Spitz, M.R.; Hong, W.K.; Delclos, G.L.; Reynolds, W.F.; Gunn, G.B.; Whitehead, L.W.; Wu, X. A myeloperoxidase polymorphism associated with reduced risk of lung cancer. Lung Cancer 2002, 37, 35–40. [Google Scholar]
- Schwartz, A.G.; Prysak, G.M.; Bock, C.H.; Cote, M.L. The molecular epidemiology of lung cancer. Carcinogenesis 2007, 28, 507–518. [Google Scholar]
- King, B.; Dube, S.; Kaufmann, R.; Shaw, L.; Pechacek, T. Vital signs: Current cigarette smoking among adults aged ≥18 years—United States, 2005–2010. Morb. Mortal. Wkly. Rep 2011, 60, 1207–1212. [Google Scholar]
- Lips, E.H.; Gaborieau, V.; McKay, J.D.; Chabrier, A.; Hung, R.J.; Boffetta, P.; Hashibe, M.; Zaridze, D.; Szeszenia-Dabrowska, N.; Lissowska, J.; et al. Association between a 15q25 gene variant, smoking quantity and tobacco-related cancers among 17,000 individuals. Int. J. Epidemiol 2010, 39, 563–577. [Google Scholar]
- Sellers, E.M.; Ramamoorthy, Y.; Zeman, M.V.; Djordjevic, M.V.; Tyndale, R.F. The effect of methoxsalen on nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) metabolism in vivo. Nicotine Tob. Res 2003, 5, 891–899. [Google Scholar]
- Takeuchi, H.; Saoo, K.; Yokohira, M.; Ikeda, M.; Maeta, H.; Miyazaki, M.; Yamazaki, H.; Kamataki, T.; Imaida, K. Pretreatment with 8-methoxypsoralen, a potent human CYP2A6 inhibitor, strongly inhibits lung tumorigenesis induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in female A/J mice. Cancer Res 2003, 63, 7581–7583. [Google Scholar]
- Bao, Z.; He, X.Y.; Ding, X.; Prabhu, S.; Hong, J.Y. Metabolism of nicotine and cotinine by human cytochrome P450 2A13. Drug Metab. Dispos 2005, 33, 258–261. [Google Scholar]
- Ding, X.; Kaminsky, L.S. Human extrahepatic cytochromes P450: Function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu. Rev. Pharmacol. Toxicol 2003, 43, 149–173. [Google Scholar]
- Cauffiez, C.; Lo-Guidice, J.M.; Quaranta, S.; Allorge, D.; Chevalier, D.; Cenee, S.; Hamdan, R.; Lhermitte, M.; Lafitte, J.J.; Libersa, C.; et al. Genetic polymorphism of the human cytochrome CYP2A13 in a French population: Implication in lung cancer susceptibility. Biochem. Biophys. Res. Commun 2004, 317, 662–669. [Google Scholar]
- Wang, H.; Tan, W.; Hao, B.; Miao, X.; Zhou, G.; He, F.; Lin, D. Substantial reduction in risk of lung adenocarcinoma associated with genetic polymorphism in CYP2A13, the most active cytochrome P450 for the metabolic activation of tobacco-specific carcinogen NNK. Cancer Res 2003, 63, 8057–8061. [Google Scholar]
- Binnington, M.J.; Zhu, A.Z.; Renner, C.C.; Lanier, A.P.; Hatsukami, D.K.; Benowitz, N.L.; Tyndale, R.F. CYP2A6 and CYP2B6 genetic variation and its association with nicotine metabolism in South Western Alaska Native people. Pharmacogenet Genomics 2012, 22, 429–440. [Google Scholar]
- Boles, M.; Rohde, K.; He, H.; Maher, J.E.; Stark, M.J.; Fenaughty, A.; O’Connor, T. Effectiveness of a tobacco quitline in an indigenous population: A comparison between Alaska Native people and other first-time quitline callers who set a quit date. Int. J. Circumpolar Health 2009, 68, 170–181. [Google Scholar]
- Fu, S.S.; Kodl, M.M.; Joseph, A.M.; Hatsukami, D.K.; Johnson, E.O.; Breslau, N.; Wu, B.; Bierut, L. Racial/Ethnic disparities in the use of nicotine replacement therapy and quit ratios in lifetime smokers ages 25 to 44 years. Cancer Epidemiol. Biomark. Prev 2008, 17, 1640–1647. [Google Scholar]
- Murray, R.P.; Connett, J.E.; Buist, A.S.; Gerald, L.B.; Eichenhorn, M.S. Experience of Black participants in the Lung Health Study smoking cessation intervention program. Nicotine Tob. Res 2001, 3, 375–382. [Google Scholar]
- Smith, J.J.; Ferucci, E.D.; Dillard, D.A.; Lanier, A.P. Tobacco use among Alaska Native people in the EARTH study. Nicotine Tob Res 2010, 12, 839–844. [Google Scholar]
- Lanier, A.P.; Kelly, J.J.; Maxwell, J.; McEvoy, T.; Homan, C. Cancer in Alaska Native people, 1969–2003. Alaska Med 2006, 48, 30–59. [Google Scholar]
- Haiman, C.A.; Stram, D.O.; Wilkens, L.R.; Pike, M.C.; Kolonel, L.N.; Henderson, B.E.; Le Marchand, L. Ethnic and racial differences in the smoking-related risk of lung cancer. N. Engl. J. Med 2006, 354, 333–342. [Google Scholar]
- Hofmann, M.H.; Blievernicht, J.K.; Klein, K.; Saussele, T.; Schaeffeler, E.; Schwab, M.; Zanger, U.M. Aberrant splicing caused by single nucleotide polymorphism c.516 G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther 2008, 325, 284–292. [Google Scholar]
- Saccone, N.L.; Culverhouse, R.C.; Schwantes-An, T.H.; Cannon, D.S.; Chen, X.; Cichon, S.; Giegling, I.; Han, S.; Han, Y.; Keskitalo-Vuokko, K.; et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: A meta-analysis and comparison with lung cancer and COPD. PLoS Genet 2010, 6, e1001053. [Google Scholar]

{kind=link}
| Cases, n = 398 | Controls, n = 421 | p value | |
|---|---|---|---|
| Mean Age (sd) | 61.9 (10.7) | 61.4 (7.4) | 0.20 |
| Sex, n (%) | |||
| Male | 236 (59) | 246 (58) | 0.80 |
| Female | 162 (41) | 175 (42) | |
| Smoking Status, n (%) | |||
| Current | 206 (52) | 196 (47) | 0.14 |
| Former | 192 (48) | 225 (53) | |
| FTND nicotine dependence score (sd) a | 4.81 (2.28) | 4.81 (2.56) | 0.96 |
| No. cigarettes per day (sd) | 27.4 (13.2) | 26.8 (14.6) | 0.27 |
| Years smoked (sd) | 36.9 (12.3) | 35.6 (11.8) | 0.22 |
| CYP2B6*6 genotyping, n (%) | |||
| *1/*1 | 221 (56) | 240 (57) | 0.71 |
| *1/*6 | 157 (39) | 156 (37) | |
| *6/*6 | 20 (5) | 25 (6) | |
| CYP2A6 genotype groups b, n (%) | |||
| CYP2A6 NM (normal metabolizer) | 326 (82) | 327 (78) | 0.13 |
| CYP2A6 RM (reduced metabolizer) | 72 (18) | 94 (22) | |
| CHRNA5-A3-B4 genotype groups c, n (%) | |||
| A5-A3-B4 GG or GA | 330 (83) | 372 (88) | 0.03 |
| A5-A3-B4 AA | 68 (17) | 49 (12) | |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wassenaar, C.A.; Dong, Q.; Amos, C.I.; Spitz, M.R.; Tyndale, R.F. Pilot Study of CYP2B6 Genetic Variation to Explore the Contribution of Nitrosamine Activation to Lung Carcinogenesis. Int. J. Mol. Sci. 2013, 14, 8381-8392. https://doi.org/10.3390/ijms14048381
Wassenaar CA, Dong Q, Amos CI, Spitz MR, Tyndale RF. Pilot Study of CYP2B6 Genetic Variation to Explore the Contribution of Nitrosamine Activation to Lung Carcinogenesis. International Journal of Molecular Sciences. 2013; 14(4):8381-8392. https://doi.org/10.3390/ijms14048381
Chicago/Turabian StyleWassenaar, Catherine A., Qiong Dong, Christopher I. Amos, Margaret R. Spitz, and Rachel F. Tyndale. 2013. "Pilot Study of CYP2B6 Genetic Variation to Explore the Contribution of Nitrosamine Activation to Lung Carcinogenesis" International Journal of Molecular Sciences 14, no. 4: 8381-8392. https://doi.org/10.3390/ijms14048381
APA StyleWassenaar, C. A., Dong, Q., Amos, C. I., Spitz, M. R., & Tyndale, R. F. (2013). Pilot Study of CYP2B6 Genetic Variation to Explore the Contribution of Nitrosamine Activation to Lung Carcinogenesis. International Journal of Molecular Sciences, 14(4), 8381-8392. https://doi.org/10.3390/ijms14048381