The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. G-Protein-Coupled Receptor Signaling
1.1. Immune Regulation by Dendritic Cells
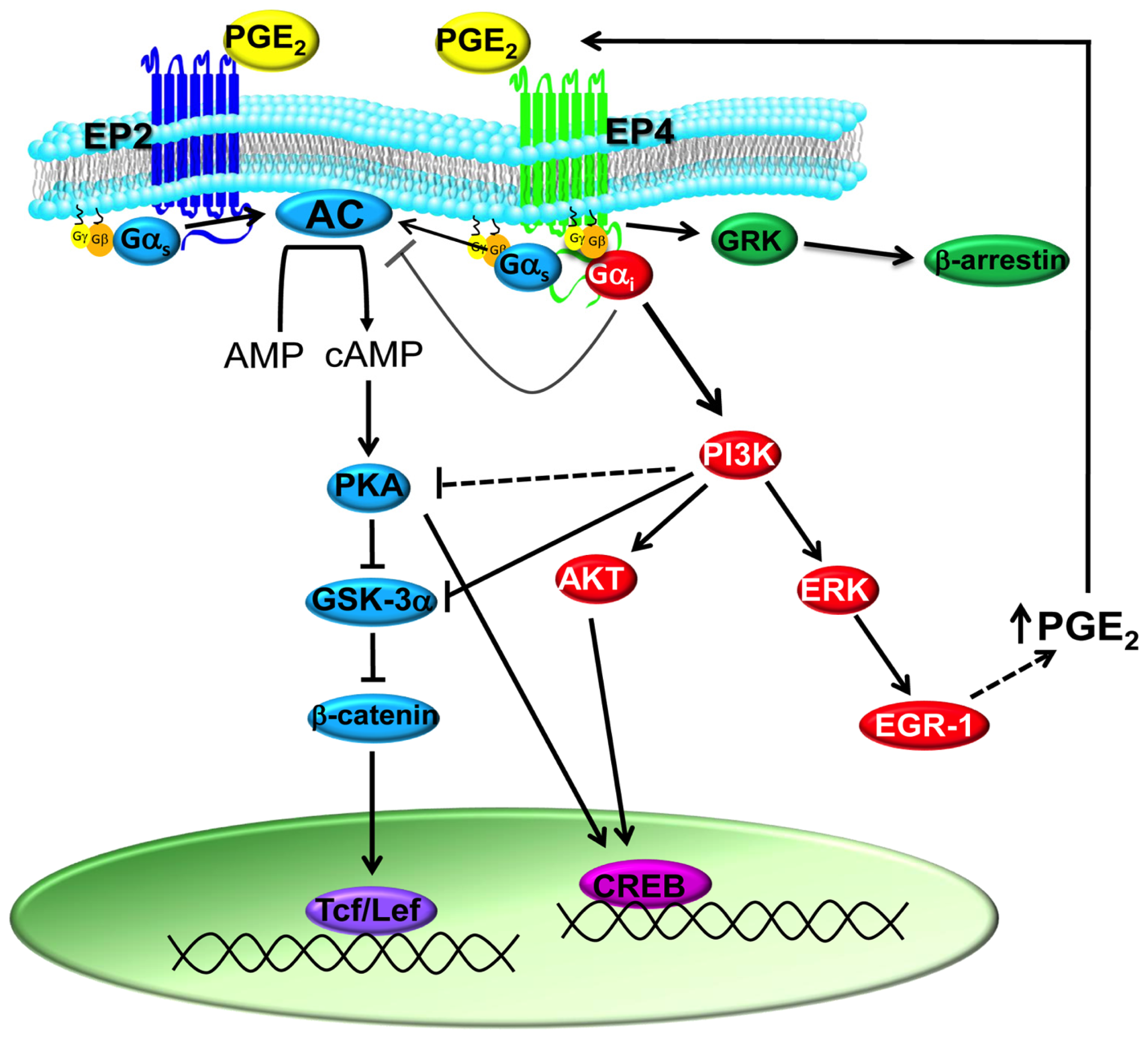
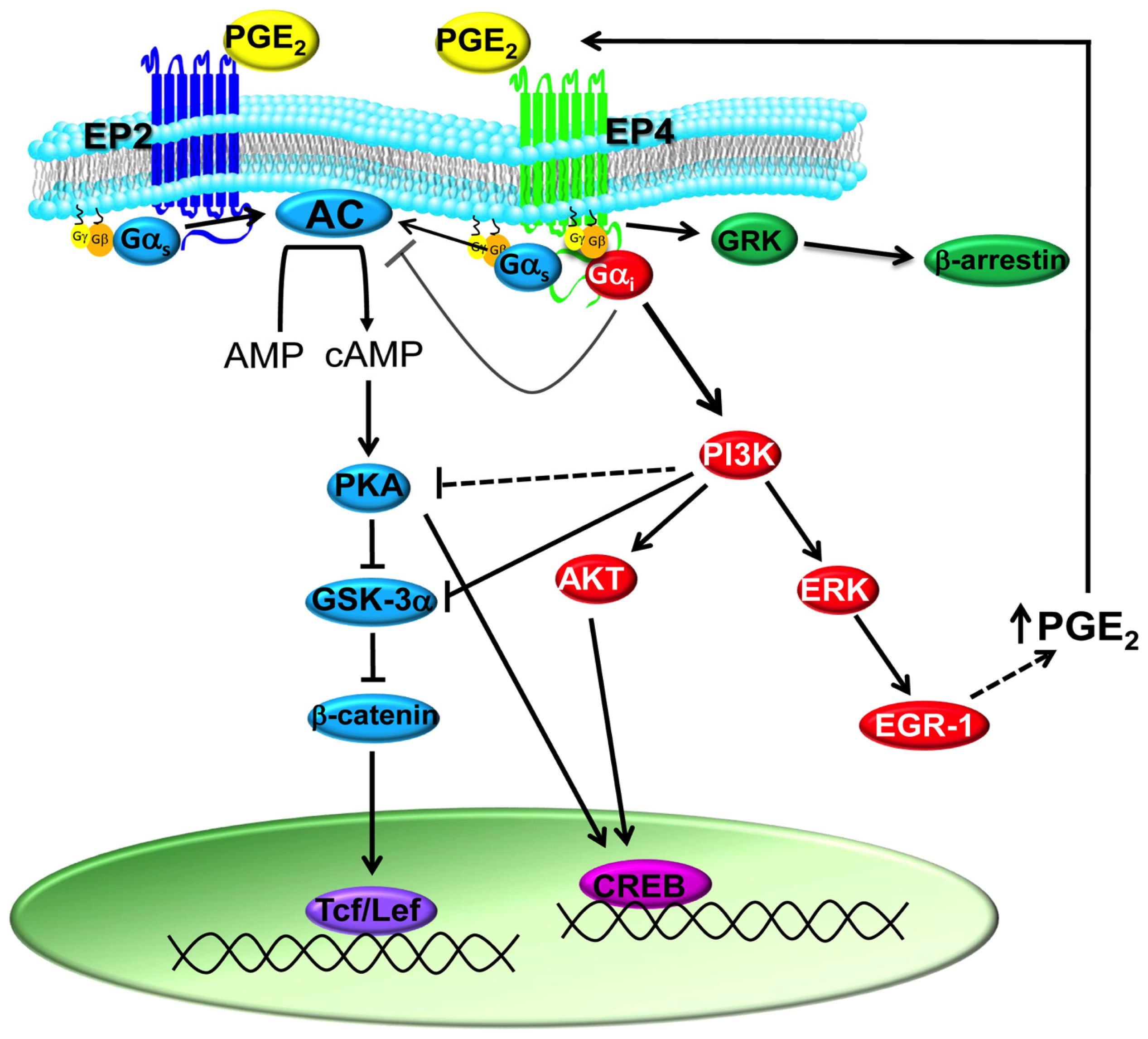
1.2. PGE2 Signaling
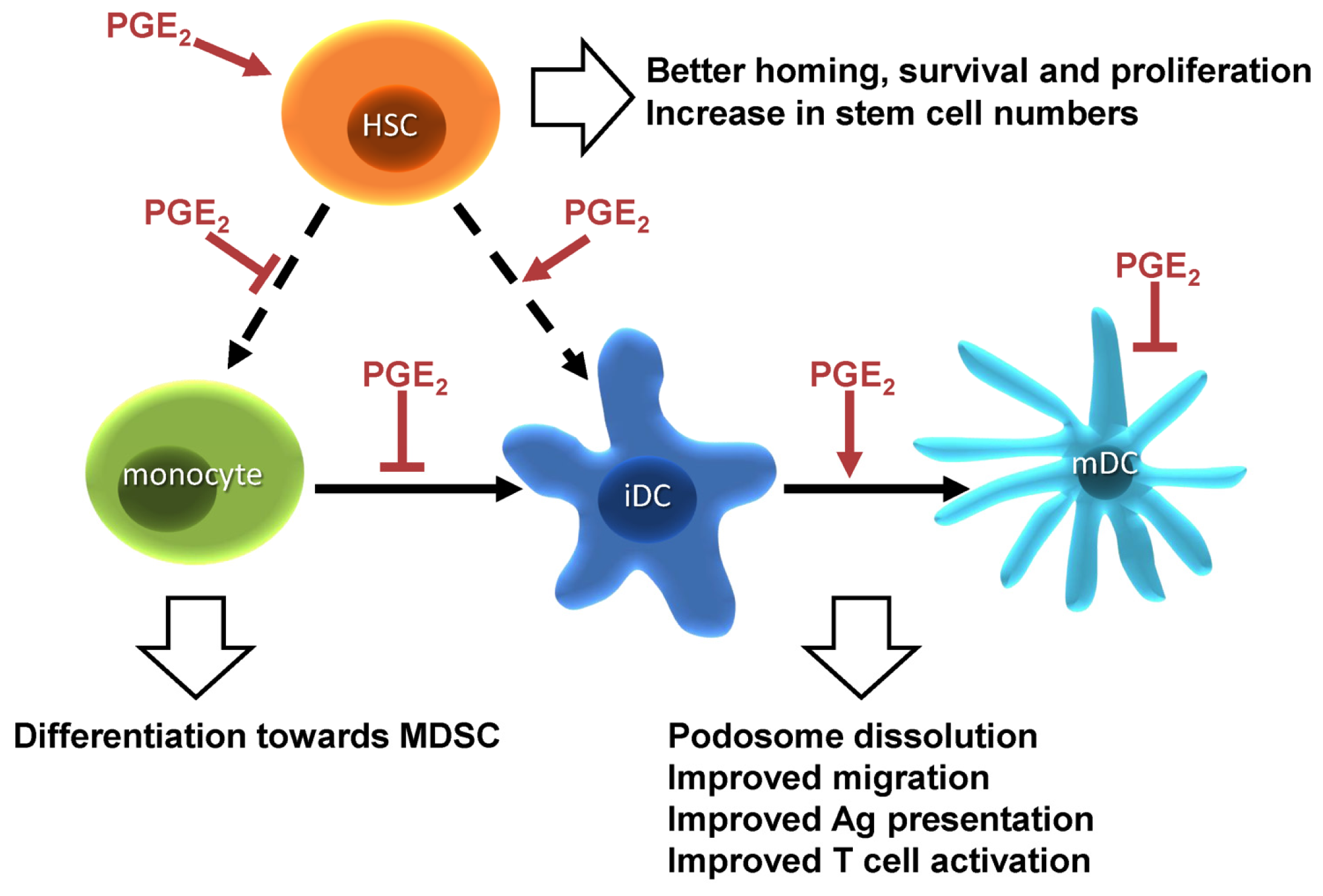
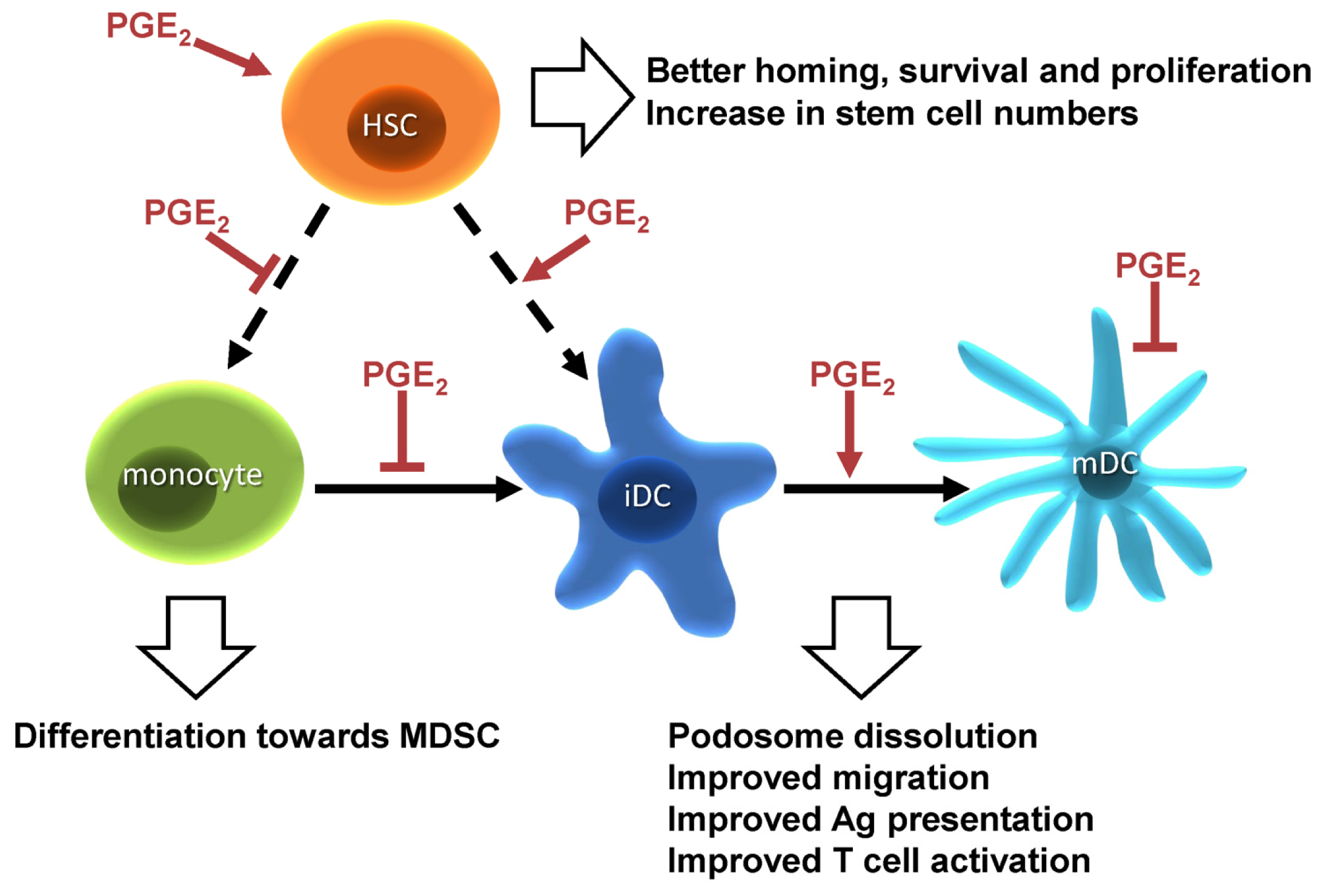
1.3. PGE2 and Hematopoiesis
1.4. PGE2 Responses in Dendritic Cells
2. Outlook
Acknowledgments
Abbreviations
| Gβ | Gγ, heterotrimeric guanine nucleotide-binding proteins |
| Gαs | stimulatory guanine nucleotide binding protein |
| Gαi | inhibitory guanine nucleotide binding protein |
| AC | adenylate cyclase |
| AMP | adenosine monophosphate |
| cAMP | cyclic 3,5-adenosine monophosphate |
| PKA | protein kinase A |
| GSK-3α | glycogen synthase kinase-3 |
| PI3K | phosphatidyl-inositol 3 kinase |
| Akt | also known as Protein Kinase B |
| CREB | cAMP response element–binding protein |
| Tcf | T-cell factor |
| Lef | lymphoid enhance factor |
| ERK | extracellular signal-regulated kinase |
| EGR-1 | early growth response protein 1 |
| GRK | G-protein coupled receptor kinase |
Conflict of Interest
References
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-Transmembrane receptors. Nat. Rev. Mol. Cell Biol 2002, 3, 639–650. [Google Scholar]
- Allen, J.A.; Halverson-Tamboli, R.A.; Rasenick, M.M. Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci 2007, 8, 128–140. [Google Scholar]
- De Keijzer, S.; Serge, A.; van Hemert, F.; Lommerse, P.H.M.; Lamers, G.E.M.; Spaink, H.P.; Schmidt, T.; Snaar-Jagalska, B.E. A spatially restricted increase in receptor mobility is involved in directional sensing during Dictyostelium discoideum chemotaxis. J. Cell Sci 2008, 121, 1750–1757. [Google Scholar]
- De Keijzer, S.; Galloway, J.; Harms, G.S.; Devreotes, P.N.; Iglesias, P.A. Disrupting microtubule network immobilizes amoeboid chemotactic receptor in the plasma membrane. Biochim. Biophys. Acta 2011, 1808, 1701–1708. [Google Scholar]
- Cambi, A.; Joosten, B.; Koopman, M.; de, L.F.; Beeren, I.; Torensma, R.; Fransen, J.A.; Garcia-Parajo, M.; van Leeuwen, F.N.; Figdor, C.G. Organization of the integrin LFA-1 in nanoclusters regulates its activity. Mol. Biol. Cell 2006, 17, 4270–4281. [Google Scholar]
- Ganguly, S.; Pucadyil, T.J.; Chattopadhyay, A. Actin cytoskeleton-dependent dynamics of the human serotonin1A receptor correlates with receptor signaling. Biophys. J 2008, 95, 451–463. [Google Scholar]
- Jaqaman, K.; Kuwata, H.; Touret, N.; Collins, R.; Trimble, W.S.; Danuser, G.; Grinstein, S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell 2011, 146, 593–606. [Google Scholar]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar]
- Steinman, R.M. Some interfaces of dendritic cell biology. APMIS 2003, 111, 675–697. [Google Scholar]
- Figdor, C.G.; de Vries, I.J.; Lesterhuis, W.J.; Melief, C.J. Dendritic cell immunotherapy: Mapping the way. Nat. Med 2004, 10, 475–480. [Google Scholar]
- De Vries, I.J.; Lesterhuis, W.J.; Barentsz, J.O.; Verdijk, P.; van Krieken, J.H.; Boerman, O.C.; Oyen, W.J.; Bonenkamp, J.J.; Boezeman, J.B.; Adema, G.J.; et al. Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat. Biotechnol 2005, 23, 1407–1413. [Google Scholar]
- Thelen, M.; Stein, J.V. How chemokines invite leukocytes to dance. Nat. Immunol 2008, 9, 953–959. [Google Scholar]
- Szatmari, I.; Nagy, L. Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J 2008, 27, 2353–2362. [Google Scholar]
- Thurnher, M. Lipids in dendritic cell biology: Messengers, effectors, and antigens. J. Leukoc. Biol 2007, 81, 154–160. [Google Scholar]
- Martino, A. Sphingosine 1-phosphate as a novel immune regulator of dendritic cells. J. Biosci 2007, 32, 1207–1212. [Google Scholar]
- Szatmari, I.; Rajnavolgyi, E.; Nagy, L. PPARgamma, a lipid-activated transcription factor as a regulator of dendritic cell function. Ann. N. Y. Acad. Sci 2006, 1088, 207–218. [Google Scholar]
- Gualde, N.; Harizi, H. Prostanoids and their receptors that modulate dendritic cell-mediated immunity. Immunol. Cell Biol 2004, 82, 353–360. [Google Scholar]
- Singh, P.; Hoggatt, J.; Hu, P.; Speth, J.M.; Fukuda, S.; Breyer, R.M.; Pelus, L.M. Blockade of prostaglandin E2 signaling through EP1 and EP3 receptors attenuates Flt3L-dependent dendritic cell development from hematopoietic progenitor cells. Blood 2012, 119, 1671–1682. [Google Scholar]
- Legler, D.F.; Krause, P.; Scandella, E.; Singer, E.; Groettrup, M. Prostaglandin E2 is generally required for human dendritic cell migration and exerts its effect via EP2 and EP4 receptors. J. Immunol 2006, 176, 966–973. [Google Scholar]
- Van Helden, S.F.; Krooshoop, D.J.; Broers, K.C.; Raymakers, R.A.; Figdor, C.G.; van Leeuwen, F.N. A critical role for prostaglandin E2 in podosome dissolution and induction of high-speed migration during dendritic cell maturation. J. Immunol 2006, 177, 1567–1574. [Google Scholar]
- Van Helden, S.F.; Oud, M.M.; Joosten, B.; Peterse, N.; Figdor, C.G.; van Leeuwen, F.N. PGE2-mediated podosome loss in dendritic cells is dependent on actomyosin contraction downstream of the RhoA-Rho-kinase axis. J. Cell Sci 2008, 121, 1096–1106. [Google Scholar]
- Khayrullina, T.; Yen, J.H.; Jing, H.; Ganea, D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J. Immunol 2008, 181, 721–735. [Google Scholar]
- Harizi, H.; Juzan, M.; Grosset, C.; Rashedi, M.; Gualde, N. Dendritic cells issued in vitro from bone marrow produce PGE(2) that contributes to the immunomodulation induced by antigen-presenting cells. Cell Immunol 2001, 209, 19–28. [Google Scholar]
- Boullart, A.C.; Aarntzen, E.H.; Verdijk, P.; Jacobs, J.F.; Schuurhuis, D.H.; Benitez-Ribas, D.; Schreibelt, G.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.; et al. Maturation of monocyte-derived dendritic cells with Toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol. Immunother 2008, 57, 1589–1597. [Google Scholar]
- Kalinski, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. Dendritic cells, obtained from peripheral blood precursors in the presence of PGE2, promote Th2 responses. Adv. Exp. Med. Biol 1997, 417, 363–367. [Google Scholar]
- Coleman, R.A.; Smith, W.L.; Narumiya, S. International Union of Pharmacology classification of prostanoid receptors: Properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev 1994, 46, 205–229. [Google Scholar]
- Smith, W.L. Prostanoid biosynthesis and mechanisms of action. Am. J. Physiol 1992, 263, F181–F191. [Google Scholar]
- Samuelsson, B.; Morgenstern, R.; Jakobsson, P.J. Membrane prostaglandin E synthase-1: A novel therapeutic target. Pharmacol. Rev 2007, 59, 207–224. [Google Scholar]
- Tai, H.H.; Ensor, C.M.; Tong, M.; Zhou, H.; Yan, F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002, 68–69, 483–493. [Google Scholar]
- Tai, H.H.; Cho, H.; Tong, M.; Ding, Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: Structure and biological functions. Curr. Pharm. Des 2006, 12, 955–962. [Google Scholar]
- Bhattacharya, M.; Peri, K.; Ribeiro-da-Silva, A.; Almazan, G.; Shichi, H.; Hou, X.; Varma, D.R.; Chemtob, S. Localization of functional prostaglandin E2 receptors EP3 and EP4 in the nuclear envelope. J. Biol. Chem 1999, 274, 15719–15724. [Google Scholar]
- Legler, D.F.; Bruckner, M.; Uetz-von, A.E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol 2009, 42, 198–201. [Google Scholar]
- Fujino, H.; Regan, J.W. EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol. Pharmacol 2006, 69, 5–10. [Google Scholar]
- Matsuoka, Y.; Furuyashiki, T.; Bito, H.; Ushikubi, F.; Tanaka, Y.; Kobayashi, T.; Muro, S.; Satoh, N.; Kayahara, T.; Higashi, M.; et al. Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proc. Natl. Acad. Sci. USA 2003, 100, 4132–4137. [Google Scholar]
- Matsuoka, Y.; Furuyashiki, T.; Yamada, K.; Nagai, T.; Bito, H.; Tanaka, Y.; Kitaoka, S.; Ushikubi, F.; Nabeshima, T.; Narumiya, S. Prostaglandin E receptor EP1 controls impulsive behavior under stress. Proc. Natl. Acad. Sci. USA 2005, 102, 16066–16071. [Google Scholar]
- Hizaki, H.; Segi, E.; Sugimoto, Y.; Hirose, M.; Saji, T.; Ushikubi, F.; Matsuoka, T.; Noda, Y.; Tanaka, T.; Yoshida, N.; et al. Abortive expansion of the cumulus and impaired fertility in mice lacking the prostaglandin E receptor subtype EP2. Proc. Natl. Acad. Sci 1999, 96, 10501–10506. [Google Scholar]
- Takeuchi, K.; Ukawa, H.; Kato, S.; Furukawa, O.; Araki, H.; Sugimoto, Y.; Ichikawa, A.; Ushikubi, F.; Narumiya, S. Impaired duodenal bicarbonate secretion and mucosal integrity in mice lacking prostaglandin E, Äìreceptor subtype EP3. Gastroenterology 1999, 117, 1128–1135. [Google Scholar]
- Yoshida, K.; Oida, H.; Kobayashi, T.; Maruyama, T.; Tanaka, M.; Katayama, T.; Yamaguchi, K.; Segi, E.; Tsuboyama, T.; Matsushita, M.; et al. Stimulation of bone formation and prevention of bone loss by prostaglandin E EP4 receptor activation. Proc. Natl. Acad. Sci. USA 2002, 99, 4580–4585. [Google Scholar]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol 2012, 188, 21–28. [Google Scholar]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol 2012, 90, 579–586. [Google Scholar]
- Yao, C.; Sakata, D.; Esaki, Y.; Li, Y.; Matsuoka, T.; Kuroiwa, K.; Sugimoto, Y.; Narumiya, S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med 2009, 15, 633–640. [Google Scholar]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J. Exp. Med 2009, 206, 535–548. [Google Scholar]
- Kabashima, K.; Sakata, D.; Nagamachi, M.; Miyachi, Y.; Inaba, K.; Narumiya, S. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat. Med 2003, 9, 744–749. [Google Scholar]
- Narumiya, S. Prostanoids in immunity: Roles revealed by mice deficient in their receptors. Life Sci 2003, 74, 391–395. [Google Scholar]
- Honda, A.; Sugimoto, Y.; Namba, T.; Watabe, A.; Irie, A.; Negishi, M.; Narumiya, S.; Ichikawa, A. Cloning and expression of a cDNA for mouse prostaglandin E receptor EP2 subtype. J. Biol. Chem 1993, 268, 7759–7762. [Google Scholar]
- Regan, J.W.; Bailey, T.J.; Pepperl, D.J.; Pierce, K.L.; Bogardus, A.M.; Donello, J.E.; Fairbairn, C.E.; Kedzie, K.M.; Woodward, D.F.; Gil, D.W. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol. Pharmacol 1994, 46, 213–220. [Google Scholar]
- Fujino, H.; Xu, W.; Regan, J.W. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem 2003, 278, 12151–12156. [Google Scholar]
- Fujino, H.; West, K.A.; Regan, J.W. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E-2. J. Biol. Chem 2002, 277, 2614–2619. [Google Scholar]
- Leduc, M.; Breton, B.; Gales, C.; le Gouill, C.; Bouvier, M.; Chemtob, S.; Heveker, N. Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J. Pharmacol. Exp. Ther 2009, 331, 297–307. [Google Scholar]
- Fujino, H.; Salvi, S.; Regan, J.W. Differential regulation of phosphorylation of the cAMP response element-binding protein after activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. Mol. Pharmacol 2005, 68, 251–259. [Google Scholar]
- Naraba, H.; Yokoyama, C.; Tago, N.; Murakami, M.; Kudo, I.; Fueki, M.; Oh-Ishi, S.; Tanabe, T. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J. Biol. Chem 2002, 277, 28601–28608. [Google Scholar]
- Desai, S.; Ashby, B. Agonist-Induced internalization and mitogen-activated protein kinase activation of the human prostaglandin EP4 receptor. FEBS Lett 2001, 501, 156–160. [Google Scholar]
- Neuschafer-Rube, F.; Oppermann, M.; Moller, U.; Boer, U.; Puschel, G.P. Agonist-Induced phosphorylation by G protein-coupled receptor kinases of the EP4 receptor carboxyl-terminal domain in an EP3/EP4 prostaglandin E(2) receptor hybrid. Mol. Pharmacol 1999, 56, 419–428. [Google Scholar]
- Penn, R.B.; Pascual, R.M.; Kim, Y.M.; Mundell, S.J.; Krymskaya, V.P.; Panettieri, R.A., Jr; Benovic, J.L. Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J. Biol. Chem. 2001, 276, 32648–32656. [Google Scholar]
- Desai, S.; April, H.; Nwaneshiudu, C.; Ashby, B. Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: Role of the carboxyl terminus in EP4 receptor sequestration. Mol. Pharmacol 2000, 58, 1279–1286. [Google Scholar]
- North, T.E.; Goessling, W.; Walkley, C.R.; Lengerke, C.; Kopani, K.R.; Lord, A.M.; Weber, G.J.; Bowman, T.V.; Jang, I.-H.; Grosser, T.; et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [Google Scholar]
- Lord, A.M.; North, T.E.; Zon, L.I. Prostaglandin E2: Making more of your marrow. Cell Cycle 2007, 6, 3054–3057. [Google Scholar]
- Goessling, W.; Allen, R.S.; Guan, X.; Jin, P.; Uchida, N.; Dovey, M.; Harris, J.M.; Metzger, M.E.; Bonifacino, A.C.; Stroncek, D.; et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 2011, 8, 445–458. [Google Scholar]
- Hoggatt, J.; Singh, P.; Sampath, J.; Pelus, L.M. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 2009, 113, 5444–5455. [Google Scholar]
- Ikushima, Y.M.; Arai, F.; Hosokawa, K.; Toyama, H.; Takubo, K.; Furuyashiki, T.; Narumiya, S.; Suda, T. Prostaglandin E2 regulates murine hematopoietic stem/progenitor cells directly via EP4 receptor and indirectly through mesenchymal progenitor cells. Blood 2013, 121, 1995–2007. [Google Scholar]
- Porter, R.L.; Georger, M.; Bromberg, O.; McGrath, K.E.; Frisch, B.J.; Becker, M.W.; Calvi, L.M. Prostaglandin E2 increases hematopoietic stem cell survival and accelerates hematopoietic recovery after radiation injury. Stem Cells 2013, 31, 372–383. [Google Scholar]
- Frisch, B.J.; Porter, R.L.; Gigliotti, B.J.; Olm-Shipman, A.J.; Weber, J.M.; O’Keefe, R.J.; Jordan, C.T.; Calvi, L.M. In vivo prostaglandin E2 treatment alters the bone marrow microenvironment and preferentially expands short-term hematopoietic stem cells. Blood 2009, 114, 4054–4063. [Google Scholar]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar]
- Al-Kharusi, M.R.; Smartt, H.J.; Greenhough, A.; Collard, T.J.; Emery, E.D.; Williams, A.C.; Paraskeva, C. LGR5 promotes survival in human colorectal adenoma cells and is upregulated by PGE2: Implications for targeting adenoma stem cells with NSAIDs. Carcinogenesis 2013. [Epub ahead of print].. [Google Scholar]
- Romani, N.; Gruner, S.; Brang, D.; Kampgen, E.; Lenz, A.; Trockenbacher, B.; Konwalinka, G.; Fritsch, P.O.; Steinman, R.M.; Schuler, G. Proliferating dendritic cell progenitors in human blood. J. Exp. Med 1994, 180, 83–93. [Google Scholar]
- Romani, N.; Reider, D.; Heuer, M.; Ebner, S.; Kampgen, E.; Eibl, B.; Niederwieser, D.; Schuler, G. Generation of mature dendritic cells from human blood. An improved method with special regard to clinical applicability. J. Immunol. Methods 1996, 196, 137–151. [Google Scholar]
- Tacken, P.J.; de Vries, I.J.; Torensma, R.; Figdor, C.G. Dendritic-Cell immunotherapy: From ex vivo loading to in vivo targeting. Nat. Rev. Immunol 2007, 7, 790–802. [Google Scholar]
- Cheong, C.; Matos, I.; Choi, J.H.; Dandamudi, D.B.; Shrestha, E.; Longhi, M.P.; Jeffrey, K.L.; Anthony, R.M.; Kluger, C.; Nchinda, G.; et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 2010, 143, 416–429. [Google Scholar]
- Spaggiari, G.M.; Abdelrazik, H.; Becchetti, F.; Moretta, L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: Central role of MSC-derived prostaglandin E2. Blood 2009, 113, 6576–6583. [Google Scholar]
- Obermajer, N.; Kalinski, P. Key role of the positive feedback between PGE(2) and COX2 in the biology of myeloid-derived suppressor cells. Oncoimmunology 2012, 1, 762–764. [Google Scholar]
- Van Helden, S.F.; van den Dries, K.; Oud, M.M.; Raymakers, R.A.; Netea, M.G.; van Leeuwen, F.N.; Figdor, C.G. TLR4-Mediated podosome loss discriminates gram-negative from gram-positive bacteria in their capacity to induce dendritic cell migration and maturation. J. Immunol 2010, 184, 1280–1291. [Google Scholar]
- Scandella, E.; Men, Y.; Gillessen, S.; Forster, R.; Groettrup, M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood 2002, 100, 1354–1361. [Google Scholar]
- Yen, J.H.; Khayrullina, T.; Ganea, D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood 2008, 111, 260–270. [Google Scholar]
- Yen, J.H.; Kocieda, V.P.; Jing, H.; Ganea, D. Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J. Biol. Chem 2011, 286, 38913–38923. [Google Scholar]
- Krause, P.; Bruckner, M.; Uermosi, C.; Singer, E.; Groettrup, M.; Legler, D.F. Prostaglandin E(2) enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4–1BBL on dendritic cells. Blood 2009, 113, 2451–2460. [Google Scholar]
- Doyen, V.; Rubio, M.; Braun, D.; Nakajima, T.; Abe, J.; Saito, H.; Delespesse, G.; Sarfati, M. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J. Exp. Med 2003, 198, 1277–1283. [Google Scholar]
- Krause, P.; Singer, E.; Darley, P.I.; Klebensberger, J.; Groettrup, M.; Legler, D.F. Prostaglandin E2 is a key factor for monocyte-derived dendritic cell maturation: Enhanced T cell stimulatory capacity despite IDO. J. Leukoc. Biol 2007, 82, 1106–1114. [Google Scholar]
- De Smedt, T.; van Mechelen, M.; de Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol 1997, 27, 1229–1235. [Google Scholar]
- Kalinski, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. IL-12-Deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J. Immunol 1997, 159, 28–35. [Google Scholar]
- Kalim, K.W.; Groettrup, M. Prostaglandin E2 inhibits IL-23 and IL-12 production by human monocytes through down-regulation of their common p40 subunit. Mol. Immunol 2013, 53, 274–282. [Google Scholar]
- Poloso, N.J.; Urquhart, P.; Nicolaou, A.; Wang, J.; Woodward, D.F. PGE(2) differentially regulates monocyte-derived dendritic cell cytokine responses depending on receptor usage (EP(2)/EP(4)). Mol. Immunol 2013, 54, 284–295. [Google Scholar]
- Kocieda, V.P.; Adhikary, S.; Emig, F.; Yen, J.H.; Toscano, M.G.; Ganea, D. Prostaglandin E2-induced IL-23p19 subunit is regulated by cAMP-responsive element-binding protein and C/AATT enhancer-binding protein beta in bone marrow-derived dendritic cells. J. Biol. Chem 2012, 287, 36922–36935. [Google Scholar]
- Fabricius, D.; Neubauer, M.; Mandel, B.; Schutz, C.; Viardot, A.; Vollmer, A.; Jahrsdorfer, B.; Debatin, K.M. Prostaglandin E2 inhibits IFN-alpha secretion and Th1 costimulation by human plasmacytoid dendritic cells via E-prostanoid 2 and E-prostanoid 4 receptor engagement. J. Immunol 2010, 184, 677–684. [Google Scholar]
- Fabricius, D.; O’Dorisio, M.S.; Blackwell, S.; Jahrsdorfer, B. Human plasmacytoid dendritic cell function: Inhibition of IFN-alpha secretion and modulation of immune phenotype by vasoactive intestinal peptide. J. Immunol 2006, 177, 5920–5927. [Google Scholar]
- Son, Y.; Ito, T.; Ozaki, Y.; Tanijiri, T.; Yokoi, T.; Nakamura, K.; Takebayashi, M.; Amakawa, R.; Fukuhara, S. Prostaglandin E2 is a negative regulator on human plasmacytoid dendritic cells. Immunology 2006, 119, 36–42. [Google Scholar]
- Strong, P.N.; Goerke, J.; Oberg, S.G.; Kelly, R.B. Beta-Bungarotoxin, a pre-synaptic toxin with enzymatic activity. Proc. Natl. Acad. Sci. USA 1976, 73, 178–182. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Keijzer, S.; Meddens, M.B.M.; Torensma, R.; Cambi, A. The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. Int. J. Mol. Sci. 2013, 14, 6542-6555. https://doi.org/10.3390/ijms14046542
De Keijzer S, Meddens MBM, Torensma R, Cambi A. The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. International Journal of Molecular Sciences. 2013; 14(4):6542-6555. https://doi.org/10.3390/ijms14046542
Chicago/Turabian StyleDe Keijzer, Sandra, Marjolein B. M. Meddens, Ruurd Torensma, and Alessandra Cambi. 2013. "The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle" International Journal of Molecular Sciences 14, no. 4: 6542-6555. https://doi.org/10.3390/ijms14046542
APA StyleDe Keijzer, S., Meddens, M. B. M., Torensma, R., & Cambi, A. (2013). The Multiple Faces of Prostaglandin E2 G-Protein Coupled Receptor Signaling during the Dendritic Cell Life Cycle. International Journal of Molecular Sciences, 14(4), 6542-6555. https://doi.org/10.3390/ijms14046542