Phospholipases of Mineralization Competent Cells and Matrix Vesicles: Roles in Physiological and Pathological Mineralizations





Abstract
:
Contents
- 1. Introduction 5040
- 1.1. Bone Biology and Physiological Mineralization 5040
- 1.2. Ectopic Calcifications and Defective Mineralizations 5042
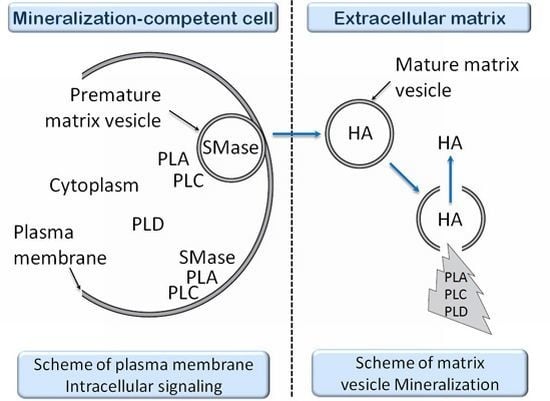
- 1.3. Matrix Vesicles and Early Stages of Mineralization 5043
- 1.4. Dietary Lipids and Bone Health 5044
- 1.5. Groups of Phospholipases and Possible Roles during Mineralization 5044
- 2. Phospholipases A1 5047
- 2.1. Groups, Subgroups and Specificity 5047
- 3. Phospholipases A2 5048
- 3.1. Groups, Subgroups and Specificity 5048
- 3.2. Presence of PLA2s in Chondrocytes and Possible Roles 5049
- 3.3 Presence of PLA2s in Osteoblasts and Possible Roles 5050
- 3.4. Presence of PLA2s in osteoclasts and Possible Roles 5051
- 3.5. Presence of PLA2s in Smooth Muscle Cells and Possible Roles 5052
- 3.6. The Expressions of PLA2s under Pathological Conditions 5052
- 3.7. Transgenic Knockout Animal for PLA2 Enzymes as Models for Bone Formation and Mineralization Diseases 5053
- 3.8. Inhibitors of PLA2 as Drug Therapy 5053
- 3.9. Effects Mediated by Arachidonic Acid and Its Pathways at Cellular Level 5054
- 3.9.1. Effects Mediated by PGE2 5055
- 3.9.2. Effects Mediated by PGF2α and PGD2 5056
- 3.10. Effects Mediated by Lysophospholipids and Their Pathways at Cellular Level 5057
- 3.11. The Effects of PLA Metabolites at Matrix Vesicle Level 5060
- 4. Non-Specific Phospholipase C 5060
- 4.1. Groups, Subgroups and Specificity 5060
- 4.2. Presence of PC-PLC in Chondrocytes and in Osteoblasts and Its Possible Role 5061
- 4.3. Presence of PC-PLC in Osteoclasts and Possible Roles 5061
- 4.4. Presence of PC-PLC in Smooth Muscle Cells and Possible Roles 5061
- 4.5. The Effect of PLC Metabolites in Matrix Vesicles 5061
- 5. PI-Specific Phospholipase C 5062
- 5.1. Groups, Subgroups and Specificity 5062
- 5.2. PI-PLC in Tissues 5063
- 5.3. Presence of PI-PLC in Chondrocytes and Possible Roles. 5064
- 5.4. Presence of PI-PLC in Osteoblasts 5065
- 5.4.1. Endothelin-1 Induced Signaling Pathway 5065
- 5.4.2. Basic FGF Induced Signaling Pathway 5066
- 5.4.3. Platelet-Derived Growth Factor Induced Signaling Pathway 5066
- 5.4.4. Parathyroid Hormone Induced Signaling Pathway 5066
- 5.4.5. PGD2 Induced-Signaling Pathway 5066
- 5.4.6. PGE2 Induced-Signaling Pathway 5067
- 5.4.7. PGF2 Induced-Signaling Pathway 5067
- 5.4.8. Vitamin D-Induced Signaling Pathway 5067
- 5.4.9. Interleukin-1-Induced Signaling Pathway 5067
- 5.4.10. Miscelanous Ligand Binding Stimulated PI-PLC in Osteoblasts 5068
- 5.4.11. Purinergic and Serotonin-2 B Receptors 5068
- 5.5. Presence of PI-PLC in Osteoclasts 5068
- 5.5.1. Calcitonin Induced Signaling Pathway 5069
- 5.5.2. Intracellular Ca2+ Induced Signaling Pathway 5069
- 5.5.3. Osteoprotegrin Induced Signaling Pathway 5070
- 5.5.4. RANK Induced Signaling Pathways 5071
- 5.5.5. Parathyroid Hormone Induced Signaling Pathway 5071
- 5.6. Presence of PI-PLC in Smooth Muscle Cells and Possible Roles 5072
- 5.7. Presence of PI-PLC in Odontoblasts and Possible Roles 5072
- 5.8. Genetic Models 5073
- 6. PLC-Related but Catalytically Inactive Protein 5073
- 7. Sphingomyelinase 5074
- 7.1. Groups, Subgroups and Specificity 5074
- 7.2. Presence of Sphingomyelinase in Chondrocytes and Possible Roles 5074
- 7.3. Presence of Sphingomyelinase in Osteoblasts and Possible Roles 5074
- 7.4. Presence of Sphyngomyelinase in Osteoclasts and Possible Roles 5075
- 7.5. Genetic Models 5075
- 7.6. Effects of Sphyngomyelinase Metabolites at Matrix Vesicle Level 5075
- 8. Phospholipase D 5076
- 8.1. Groups, Subgroups and Specificity 5076
- 8.2. Presence of PLD in Chondrocytes and Possible Roles 5077
- 8.3. Presence of PLD in Osteoblasts and Possible Roles 5079
- 8.4. Presence of PLD in Osteoclasts and Possible Roles 5080
- 8.5. Genetic Models 5080
- 8.6. Effects of PLD Metabolite at Matrix Vesicle Level 5080
- 9. Non-HKD Enzymes—GPI-PLD 5081
- 9.1. Groups, Subgroups and Specificity 5081
- 9.2. Presence of GPI-PLD in Chondrocytes and Possible Roles 5081
- 9.3. Presence of GPI-PLD in Osteoblasts and Possible Roles 5082
- 10. Non-HKD Enzymes—Autotaxin 5082
- 10.1. Groups, Subgroups and Specificity 5082
- 10.2. Presence of ATX in Chondrocytes and Possible Roles. 5083
- 10.3. Presence of ATX in Osteoblasts and Possible Roles 5083
- 10.4. Presence of ATX in Osteoclasts and Possible Roles. 5084
- 10.5. Presence of ATX in Smooth Muscle Cells and Possible Roles 5084
- 11. Concluding Remarks 5084
- Acknowledgments 5084
- References 5084
Abbreviations
| 1α,25-(OH)2D3 | 1α,25-dihydroxyvitamin D3 |
| 24R,25(OH)2D3 | 24R,25-dihydroxyvitamin D3 |
| AA | arachidonic acid |
| ATX | autotaxin |
| BM | bone marrow |
| Ca2+e | extracellular Ca2+ |
| Ca2+i | intracellular Ca2+ |
| CaR | calcium-sensing receptor |
| CIA | collagen-induced arthritis |
| cPLA2 | cytosolic Ca2+-dependent PLA2 |
| COX | cyclooxygenase |
| DAG | diacylglycerol |
| DHT | 5α-dihydrotestosterone |
| ECM | extracellular matrix |
| ERK | extracellular signal-regulated kinase |
| ET | endothelin |
| FGF | fibroblast growth factor |
| GPCR | G-protein-coupled receptor |
| GPI-PLD | glycosyl-PI specific PLD |
| HA | hydroxyapatite |
| IL | interleukin |
| IP3 | inositol 1,4,5-trisphosphate |
| iPLA2 | Ca2+-independent PLA2 |
| LOX | lipooxygenase |
| LPA | lysophosphatidic acid |
| LPC | lysophosphatidylcholine |
| LPE | lysophosphatidylethanolamine |
| LPG | lysophosphatidylglycerol |
| LPI | lysophosphatidylinositol |
| LPL | lysophospholipid |
| LPS | lysophosphatidylserine |
| LRRc17 | leucine-rich repeat-containing 17 |
| MAP | mitogen activated protein |
| MPP | metalloproteinase |
| MV | matrix vesicle |
| NF-κB | nuclear factor κB |
| NFAT | nuclear factor of activated T cell |
| NPP | ectonucleotide pyrophosphatase phosphodiesterase |
| NSAID | non-steroidal anti-inflammatory drug |
| OA | osteoarthritis |
| OPG | osteoprotegerin |
| PA | phosphatidic acid |
| PAF | platelet-activating factor |
| PAF-AH | PAF-acetylhydrolase |
| PBMC | peripheral blood mononuclear cell |
| PC | phosphatidylcholine |
| PChol | phosphocholine |
| PE | phosphatidylethanolamine |
| PEA | phosphoethanolamine |
| PGD2 | prostaglandin D2 |
| PGE1 | prostaglandin E1 |
| PGE2 | prostaglandin E2 |
| PGF2 | prostaglandin F2 |
| PH | pleckstrin homology |
| PHOSPHO1 | phosphatase orphan 1 |
| PI | phosphatidylinositol |
| PI-PLC | PI-specific |
| PIP2 | PI 4,5-bisphosphate |
| PIP3 | PI 3,4,5-trisphosphate |
| PKC | protein kinase C |
| PLA1 | phospholipase A1 |
| PLA2 | phospholipase A2 |
| PLC | phospholipase C |
| PLD | phospholipase D |
| Pi | inorganic phosphate |
| PPi | inorganic pyrophosphate |
| PRIP | PLC-related but catalytically inactive protein |
| PS | phosphatidylserine |
| PS-PLA1 | PS-specific PLA1 |
| PTH | parathyroid hormone |
| PTX | pertussis toxin |
| PUFA | polyunsaturated fatty acid |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor κB ligand |
| Runx2 | runt-related transcription factor 2 |
| SH | src homology |
| SM | sphingomyelin |
| SMase | sphingomyelinase |
| SMPD3 | sphingomyeline phosphodiesterase-3 |
| sPLA2 | secreted PLA2 |
| S1P | sphingosine-1-phosphate |
| STAT | signal transducer and activator of transcription |
| TNAP | tissue-non specific alkaline phosphatase |
| TNF | tumor necrosis factor |
| VSMC | vascular smooth muscle cell |
1. Introduction
1.1. Bone Biology and Physiological Mineralization
1.2. Ectopic Calcifications and Defective Mineralizations
1.3. Matrix Vesicles and Early Stages of Mineralization
1.4. Dietary Lipids and Bone Health
1.5. Groups of Phospholipases and Possible Roles during Mineralization
2. Phospholipases A1
2.1. Groups, Subgroups and Specificity
3. Phospholipases A2
3.1. Groups, Subgroups and Specificity
3.2. Presence of PLA2s in Chondrocytes and Possible Roles
3.3 Presence of PLA2s in Osteoblasts and Possible Roles
3.4. Presence of PLA2s in osteoclasts and Possible Roles
3.5. Presence of PLA2s in Smooth Muscle Cells and Possible Roles
3.6. The Expressions of PLA2s under Pathological Conditions
3.7. Transgenic Knockout Animal for PLA2 Enzymes as Models for Bone Formation and Mineralization Diseases
3.8. Inhibitors of PLA2 as Drug Therapy
3.9. Effects Mediated by Arachidonic Acid and Its Pathways at Cellular Level
3.9.1. Effects Mediated by PGE2
3.9.2. Effects Mediated by PGF2α and PGD2
3.10. Effects Mediated by Lysophospholipids and Their Pathways at Cellular Level
3.11. The Effects of PLA Metabolites at Matrix Vesicle Level
4. Non-Specific Phospholipase C
4.1. Groups, Subgroups and Specificity
4.2. Presence of PC-PLC in Chondrocytes and in Osteoblasts and Its Possible Role
4.3. Presence of PC-PLC in Osteoclasts and Possible Roles
4.4. Presence of PC-PLC in Smooth Muscle Cells and Possible Roles
4.5. The Effect of PLC Metabolites in Matrix Vesicles
5. PI-Specific Phospholipase C
5.1. Groups, Subgroups and Specificity
5.2. PI-PLC in Tissues
5.3. Presence of PI-PLC in Chondrocytes and Possible Roles
5.4. Presence of PI-PLC in Osteoblasts
5.4.1. Endothelin-1 Induced Signaling Pathway
5.4.2. Basic FGF Induced Signaling Pathway
5.4.3. Platelet-Derived Growth Factor Induced Signaling Pathway
5.4.4. Parathyroid Hormone Induced Signaling Pathway
5.4.5. PGD2 Induced-Signaling Pathway
5.4.6. PGE2 Induced-Signaling Pathway
5.4.7. PGF2 Induced-Signaling Pathway
5.4.8. Vitamin D-Induced Signaling Pathway
5.4.9. Interleukin-1-Induced Signaling Pathway
5.4.10. Miscelanous Ligand Binding Stimulated PI-PLC in Osteoblasts
5.4.11. Purinergic and Serotonin-2 B Receptors
5.5. Presence of PI-PLC in Osteoclasts
5.5.1. Calcitonin Induced Signaling Pathway
5.5.2. Intracellular Ca2+ Induced Signaling Pathway
5.5.3. Osteoprotegrin Induced Signaling Pathway
5.5.4. RANK Induced Signaling Pathways
5.5.5. Parathyroid Hormone Induced Signaling Pathway
5.6. Presence of PI-PLC in Smooth Muscle Cells and Possible Roles
5.7. Presence of PI-PLC in Odontoblasts and Possible Roles
5.8. Genetic Models
6. PLC-Related but Catalytically Inactive Protein
7. Sphingomyelinase
7.1. Groups, Subgroups and Specificity
7.2. Presence of Sphingomyelinase in Chondrocytes and Possible Roles
7.3. Presence of Sphingomyelinase in Osteoblasts and Possible Roles
7.4. Presence of Sphyngomyelinase in Osteoclasts and Possible Roles
7.5. Genetic Models
7.6. Effects of Sphyngomyelinase Metabolites at Matrix Vesicle Level
8. Phospholipase D
8.1. Groups, Subgroups and Specificity
8.2. Presence of PLD in Chondrocytes and Possible Roles
8.3. Presence of PLD in Osteoblasts and Possible Roles
8.4. Presence of PLD in Osteoclasts and Possible Roles
8.5. Genetic Models
8.6. Effects of PLD Metabolite at Matrix Vesicle Level
9. Non-HKD Enzymes—GPI-PLD
9.1. Groups, Subgroups and Specificity
9.2. Presence of GPI-PLD in Chondrocytes and Possible Roles
9.3. Presence of GPI-PLD in Osteoblasts and Possible Roles
10. Non-HKD Enzymes—Autotaxin
10.1. Groups, Subgroups and Specificity
10.2. Presence of ATX in Chondrocytes and Possible Roles
10.3. Presence of ATX in Osteoblasts and Possible Roles
10.4. Presence of ATX in Osteoclasts and Possible Roles
10.5. Presence of ATX in Smooth Muscle Cells and Possible Roles
11. Concluding Remarks
Acknowledgments
References
- Del Fattore, A.; Teti, A.; Rucci, N. Bone cells and the mechanisms of bone remodelling. Front. Biosci 2012, 4, 2302–2321. [Google Scholar]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn 2006, 235, 176–190. [Google Scholar]
- Parfitt, A.M. Bone-forming cells in clinical conditions. In Bone; Hall, B.K., Ed.; Telford Press: Calwell, NJ, USA, 1990; Volume 1, pp. 351–429. [Google Scholar]
- Banks, W.J. The ossification process of the developing antler in the white-tailed deer (Odocoileus virginianus). Calcif. Tissue Res 1974, 14, 257–274. [Google Scholar]
- Ekanayake, S.; Hall, B.K. The development of acellularity of the vertebral bone of the Japanese medaka, Oryzias latipes (Teleostei; Cyprinidontidae). J. Morphol 1987, 193, 253–261. [Google Scholar]
- Witten, P.E.; Huysseune, A.; Franz-Odendaal, T.A.; Fedak, T.; Vickaryous, M.; Cole, A.; Hall, B.K. Acellular teleost bone: Primitive or derived, dead or alive? Palaeontol. Newslett 2004, 55, 37–41. [Google Scholar]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar]
- Noonan, K.J.; Hunziker, E.B.; Nessler, J.; Buckwalter, J.A. Changes in cell, matrix compartment, and fibrillar collagen volumes between growth-plate zones. J. Orthop. Res 1998, 16, 500–508. [Google Scholar]
- Pawelek, J.M.; Chakraborty, A.K. The cancer cell—Leukocyte fusion theory of metastasis. Adv. Cancer Res 2008, 101, 397–444. [Google Scholar]
- Orimo, H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J. Nihon Med. Sch 2010, 77, 4–12. [Google Scholar]
- Anderson, H.C. The role of matrix vesicles in physiological and pathological calcification. Curr. Opin. Orthop 2007, 18, 428–433. [Google Scholar]
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci 2010, 1192, 190–200. [Google Scholar]
- Fleish, H.; Neuman, W. Mechanisms of calcification: Role of collagen, polyphosphates, and phosphatase. Am. J. Physiol 1961, 200, 1296–1300. [Google Scholar]
- Hessle, L.; Johnson, K.A.; Anderson, H.C.; Narisawa, S.; Sali, A.; Goding, J.W.; Terkeltaub, R.; Millan, J.L. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. USA 2002, 99, 9445–9449. [Google Scholar]
- Murshed, M.; Harmey, D.; Millán, J.L.; McKee, M.D.; Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev 2005, 19, 1093–1104. [Google Scholar]
- Price, P.A.; Toroian, D.; Lim, J.E. Mineralization by inhibitor exclusion: The calcification of collagen with fetuin. J. Biol. Chem 2009, 284, 17092–17101. [Google Scholar]
- Tanaka, H.; Fukagawa, M. Hormonal regulation of phosphate balance. Clin. Calcium 2012, 22, 1477–1485. [Google Scholar]
- Komaba, H.; Fukagawa, M. The role of FGF23 in CKD—With or without Klotho. Nat. Rev. Nephrol 2012, 8, 484–490. [Google Scholar]
- Fukumoto, S. The role of bone in phosphate metabolism. Mol. Cell. Endocrinol 2009, 310, 63–70. [Google Scholar]
- Liu, S.; Quarles, L.D. How fibroblast growth factor 23 works. J. Am. Soc. Nephrol 2007, 18, 1637–1647. [Google Scholar]
- Anderson, H.C.; Mulhall, D.; Garimella, R. Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab. Invest 2010, 90, 1549–1557. [Google Scholar]
- Doherty, T.M.; Fitzpatrick, L.A.; Inoue, D.; Qiao, J.H.; Fishbein, M.C.; Detrano, R.C.; Shah, P.K.; Rajavashisth, T.B. Molecular, endocrine, and genetic mechanisms of arterial calcification. Endocr. Rev 2004, 25, 629–672. [Google Scholar]
- Magne, D.; Julien, M.; Vinatier, C.; Merhi-Soussi, F.; Weiss, P.; Guicheux, J. Cartilage formation in growth plate and arteries: From physiology to pathology. Bioessays 2005, 27, 708–716. [Google Scholar]
- Shao, J.S.; Cai, J.; Towler, D.A. Molecular mechanisms of vascular calcification: Lessons learned from the aorta. Arterioscler. Thromb. Vasc. Biol 2006, 26, 1423–1430. [Google Scholar]
- Rutsch, F.; Böyer, P.; Nitschke, Y.; Ruf, N.; Lorenz-Depierieux, B.; Wittkampf, T.; Weissen-Plenz, G.; Fischer, R.J.; Mughal, Z.; Gregory, J.W.; et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet 2008, 1, 133–140. [Google Scholar]
- Ali, S.Y. Apatite crystal nodules in arthritic cartilage. Eur. J. Rheumatol. Inflamm 1978, 14, 115–119. [Google Scholar]
- Sampson, H.W.; Davis, R.W.; Dufner, D.C. Spondyloarthropathy in progressive ankylosis mice: Ultrastructural features of the intervertebral disk. Acta Anat 1991, 141, 36–41. [Google Scholar]
- Isakova, T. Fibroblast growth factor 23 and adverse clinical outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens 2012, 21, 334–340. [Google Scholar]
- Connor, J.M.; Evans, D.A. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J. Bone Joint Surg. Br 1982, 64, 76–83. [Google Scholar]
- Kaplan, F.S.; Shore, E.M.; Connor, J.M. Fibrodysplasia ossificans progressiva. In Connective Tissue and Its Heritable Disorders: Molecular, Genetic, and Medical Aspects; Royce, P.M., Steinmann, B., Eds.; John Wiley & Sons: New York, NY, USA, 2002; pp. 827–840. [Google Scholar]
- Gannon, F.H.; Kaplan, F.S.; Olmsted, E.; Finkel, G.C.; Zasloff, M.A.; Shore, E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Hum. Pathol 1997, 28, 339–343. [Google Scholar]
- Shafritz, A.B.; Shore, E.M.; Gannon, F.H.; Zasloff, M.A.; Taub, R.; Muenke, M.; Kaplan, F.S. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N. Engl. J. Med 1996, 335, 555–561. [Google Scholar]
- Kaplan, F.S.; Shore, E.M. Progressive osseous heteroplasia. J. Bone Miner. Res 2000, 15, 2084–2094. [Google Scholar]
- Mornet, E. Hypophosphatasia. Best Pract. Res. Clin. Rheumatol 2008, 22, 113–127. [Google Scholar]
- Whyte, M.P. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr. Rev 1994, 15, 439–461. [Google Scholar]
- Sambrook, P.; Cooper, C. Osteoporosis. Lancet 2006, 367, 2010–2018. [Google Scholar]
- Goronzy, J.J.; Weyand, C.M. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res. Ther 2009, 11, 249. [Google Scholar]
- Walsh, N.C.; Crotti, T.N.; Goldring, S.R.; Gravallese, E.M. Rheumatic diseases: The effects of inflammation on bone. Immunol. Rev 2005, 208, 228–251. [Google Scholar]
- Karouzakis, E.; Neidhart, M.; Gay, R.E.; Gay, S. Molecular and cellular basis of rheumatoid joint destruction. Immunol. Lett 2006, 106, 8–13. [Google Scholar]
- Mebarek, S.; Hamade, E.; Thouverey, C.; Bandorowicz-Pikula, J.; Pikula, S.; Magne, D.; Buchet, R. Ankylosing spondylitis, late osteoarthritis, vascular calcification, chondrocalcinosis and pseudo gout: Toward a possible drug therapy. Curr. Med. Chem 2011, 18, 2196–2203. [Google Scholar]
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001, 38, 938–942. [Google Scholar]
- Detrano, R.; Guerci, A.D.; Carr, J.J.; Bild, D.E.; Burke, G.; Folsom, A.R.; Liu, K.; Shea, S.; Szklo, M.; Bluemke, D.A.; et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med 2008, 358, 1336–1345. [Google Scholar]
- Ali, S.Y.; Sajdera, S.W.; Anderson, H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA 1970, 67, 1513–1520. [Google Scholar]
- Ali, S.Y.; Anderson, H.C.; Sajdera, S.W. Enzymic and electron-microscopic analysis of extracellular matrix vesicles associated with calcification in cartilage. Biochem. J 1971, 122, 56P–57P. [Google Scholar]
- Kaji, H. Pyrophosphate and mineralization (TNSALP, PC-1, ANK). Clin. Calcium 2007, 17, 1574–1579. [Google Scholar]
- Roberts, S.J.; Stewart, A.J.; Schmid, R.; Blindauer, C.A.; Bond, S.R.; Sadler, P.J.; Farquharson, C. Probing the substrate specificities of human PHOSPHO1 and PHOSPHO2. Biochim. Biophys. Acta 2005, 1752, 73–82. [Google Scholar]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J. Bone Miner. Res 2007, 22, 617–627. [Google Scholar]
- Hsu, H.H.; Anderson, H.C. A role for ATPase in the mechanisms of ATP-dependent Ca and phosphate deposition by isolated rachitic matrix vesicles. Int. J. Biochem. Cell. Biol 1995, 27, 1349–1356. [Google Scholar]
- Terkeltaub, R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal 2006, 2, 371–377. [Google Scholar]
- Wu, L.N.; Genge, B.R.; Kang, M.W.; Arsenault, A.L.; Wuthier, R.E. Changes in phospholipid extractability and composition accompany mineralization of chicken growth plate cartilage matrix vesicles. J. Biol. Chem 2002, 277, 5126–5133. [Google Scholar]
- Wuthier, R.E. Effect of phospholipids on the transformation of amorphous calcium phosphate to hydroxapatite in vitro. Calcif. Tissue Res 1975, 19, 197–210. [Google Scholar]
- Wuthier, R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta 1975, 409, 128–143. [Google Scholar]
- Wuthier, R.E.; Lipscomb, G.F. Matrix vesicles: Structure, composition, formation and function in calcification. Front. Biosci 2011, 16, 2812–2902. [Google Scholar]
- Golub, E.E. Biomineralization and matrix vesicles in biology and pathology. Semin. Immunopathol 2011, 33, 409–417. [Google Scholar]
- Duque, G. Bone and fat connection in aging bone. Curr. Opin. Rheumatol 2008, 20, 429–434. [Google Scholar]
- Kim, Y.; Ilich, J.Z. Implications of dietary α-linolenic acid in bone health. Nutrition 2011, 27, 1101–1107. [Google Scholar]
- Hur, S.J.; Park, Y. Effect of conjugated linoleic acid on bone formation and rheumatoid arthritis. Eur. J. Pharmacol 2007, 568, 16–24. [Google Scholar]
- Fernandes, G.; Bhattacharya, A.; Rahman, M.; Zaman, K.; Banu, J. Effects of n-3 fatty acids on autoimmunity and osteoporosis. Front. Biosci 2008, 13, 4015–4020. [Google Scholar]
- Salari, P.; Rezaie, A.; Larijani, B.; Abdollahi, M. A systematic review of the impact of n-3 fatty acids in bone health and osteoporosis. Med. Sci. Monit. 2008, 14, RA37–44. [Google Scholar]
- Poulsen, R.C.; Moughan, P.J.; Kruger, M.C. Long-chain polyunsaturated fatty acids and the regulation of bone metabolism. Exp. Biol. Med 2007, 232, 1275–1288. [Google Scholar]
- Kruger, M.C.; Coetzee, M.; Haag, M.; Weiler, H. Long-chain polyunsaturated fatty acids: Selected mechanisms of action on bone. Prog. Lipid Res 2010, 49, 438–449. [Google Scholar]
- Genuis, S.J.; Schwalfenberg, G.K. Picking a bone with contemporary osteoporosis management: Nutrient strategies to enhance skeletal integrity. Clin. Nutr 2007, 26, 193–207. [Google Scholar]
- Corwin, R.L.; Hartman, T.J.; Maczuga, S.A.; Graubard, B.I. Dietary saturated fat intake is inversely associated with bone density in humans: Analysis of NHANES III. J. Nutr 2006, 136, 159–165. [Google Scholar]
- Weinberg, J.M. Lipotoxicity. Kidney Int 2006, 70, 1560–1566. [Google Scholar]
- Unger, R.H.; Orci, L. Lipoapoptosis: Its mechanism and its diseases. Biochim. Biophys. Acta 2002, 1585, 202–212. [Google Scholar]
- Chapple, I.L. Potential mechanisms underpinning the nutritional modulation of periodontal inflammation. J. Am. Dent. Assoc 2009, 140, 178–184. [Google Scholar]
- Bab, I.; Smoum, R.; Bradshaw, H.; Mechoulam, R. Skeletal lipidomics: Regulation of bone metabolism by fatty acid amide family. Br. J. Pharmacol 2011, 163, 1441–1446. [Google Scholar]
- Wong, H.; Schotz, M.C. The lipase gene family. J. Lipid Res 2002, 43, 993–999. [Google Scholar]
- Aoki, J.; Inoue, A.; Makide, K.; Saiki, N.; Arai, H. Structure and function of extracellular phospholipase A1 belonging to the pancreatic lipase gene family. Biochimie 2007, 89, 197–204. [Google Scholar]
- Kudo, I.; Murakami, M. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat. 2002, 68–69, 3–58. [Google Scholar]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 biochemistry. Cardiovasc. Drugs Ther 2009, 23, 49–59. [Google Scholar]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta 2006, 1761, 1246–1259. [Google Scholar]
- Scott, K.F.; Sajinovic, M.; Hein, J.; Nixdorf, S.; Galettis, P.; Liauw, W.; de Souza, P.; Dong, Q.; Graham, G.G.; Russell, P.J. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie 2010, 92, 601–610. [Google Scholar]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res 2011, 50, 152–192. [Google Scholar]
- Fukami, K.; Inanobe, S.; Kanemaru, K.; Nakamura, Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res 2010, 49, 429–437. [Google Scholar]
- Bunney, T.D.; Katan, M. PLC regulation: Emerging pictures for molecular mechanisms. Trends Biochem. Sci 2011, 36, 88–96. [Google Scholar]
- Selvy, P.E.; Lavieri, R.R.; Lindsley, C.W.; Brown, H.A. Phospholipase D: Enzymology, functionality, and chemical modulation. Chem. Rev 2011, 111, 6064–6119. [Google Scholar]
- Peng, X.; Frohman, M.A. Mammalian phospholipase D physiological and pathological roles. Acta Physiol 2012, 204, 219–226. [Google Scholar]
- Kirsch, T. Physiological and pathological mineralization: A complex multifactorial process. Curr. Opin. Orthop 2007, 18, 425–427. [Google Scholar]
- Aoki, J.; Nagai, Y.; Hosono, H.; Inoue, K.; Arai, H. Structure and function of phosphatidylserine-specific phospholipase A1. Biochim. Biophys. Acta 2002, 1582, 26–32. [Google Scholar]
- Sonoda, H.; Aoki, J.; Hiramatsu, T.; Ishida, M.; Bandoh, K.; Nagai, Y.; Taguchi, R.; Inoue, K.; Arai, H. A novel phosphatidic acid-selective phospholipase A1 that produces lysophosphatidic acid. J. Biol. Chem 2002, 277, 34254–34263. [Google Scholar]
- Hiramatsu, T.; Sonoda, H.; Takanezawa, Y.; Morikawa, R.; Ishida, M.; Kasahara, K.; Sanai, Y.; Taguchi, R.; Aoki, J.; Arai, H. Biochemical and molecular characterization of two phosphatidic acid-selective phospholipase A1s, mPA-PLA1alpha and mPA-PLA1beta. J. Biol. Chem 2003, 278, 49438–49447. [Google Scholar]
- Carrière, F.; Withers-Martinez, C.; van Tilbeurgh, H.; Roussel, A.; Cambillau, C.; Verger, R. Structural basis for the substrate selectivity of pancreatic lipases and some related proteins. Biochim. Biophys. Acta 1998, 1376, 417–432. [Google Scholar]
- Hide, W.A.; Chan, L.; Li, W.H. Structure and evolution of the lipase superfamily. J. Lipid Res 1992, 33, 167–178. [Google Scholar]
- Pete, M.J.; Ross, A.H.; Exton, J.H. Purification and properties of phospholipase A1 from bovine brain. J. Biol. Chem 1994, 269, 19494–19500. [Google Scholar]
- Higgs, H.N.; Glomset, J.A. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. USA 1994, 91, 9574–9578. [Google Scholar]
- Higgs, H.N.; Han, M.H.; Johnson, G.E.; Glomset, J.A. Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J. Biol. Chem 1998, 273, 5468–5477. [Google Scholar]
- Tani, K.; Mizoguchi, T.; Iwamatsu, A.; Hatsuzawa, K.; Tagaya, M. p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J. Biol. Chem 1999, 274, 20505–20512. [Google Scholar]
- Nakajima, K.; Sonoda, H.; Mizoguchi, T.; Aoki, J.; Arai, H.; Nagahama, M.; Tagaya, M.; Tani, K. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem 2002, 277, 11329–11335. [Google Scholar]
- Murakami, M.; Taketomi, Y.; Sato, H.; Yamamoto, K. Secreted phospholipase A2 revisited. J. Biochem 2011, 150, 233–255. [Google Scholar]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev 2011, 111, 6130–6185. [Google Scholar]
- Menschikowski, M.; Hagelgans, A.; Siegert, G. Secretory phospholipase A2 of group IIA: Is it an offensive or a defensive player during atherosclerosis and other inflammatory diseases? Prostaglandins Other Lipid Mediat 2006, 79, 1–33. [Google Scholar]
- Rosenson, R.S.; Gelb, M.H. Secretory phospholipase A2: A multifaceted family of proatherogenic enzymes. Curr. Cardiol. Rep 2009, 11, 445–451. [Google Scholar]
- Nevalainen, T.J.; Graham, G.G.; Scott, K.F. Antibacterial actions of secreted phospholipases A2. Review. Biochim. Biophys. Acta 2008, 1781, 1–9. [Google Scholar]
- Lambeau, G.; Gelb, M.H. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem 2008, 77, 495–520. [Google Scholar]
- Boyanovsky, B.B.; Webb, N.R. Biology of secretory phospholipase A2. Cardiovasc. Drugs Ther 2009, 23, 61–72. [Google Scholar]
- Clark, J.D.; Lin, L.L.; Kriz, R.W.; Ramesha, C.S.; Sultzman, L.A.; Lin, A.Y.; Milona, N.; Knopf, J.L. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991, 65, 1043–1051. [Google Scholar]
- Kramer, R.M.; Roberts, E.F.; Manetta, J.; Putnam, J.E. The Ca2(+)-sensitive cytosolic phospholipase A2 is a 100-kDa protein in human monoblast U937 cells. J. Biol. Chem 1991, 266, 5268–5272. [Google Scholar]
- Ghosh, M.; Tucker, D.E.; Burchett, S.A.; Leslie, C.C. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res 2006, 45, 487–510. [Google Scholar]
- Leslie, C.C.; Gangelhoff, T.A.; Gelb, M.H. Localization and function of cytosolic phospholipase A2alpha at the Golgi. Biochimie 2010, 92, 620–626. [Google Scholar]
- Kita, Y.; Ohto, T.; Uozumi, N.; Shimizu, T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim. Biophys. Acta 2006, 1761, 1317–1322. [Google Scholar]
- Linkous, A.; Yazlovitskaya, E. Cytosolic phospholipase A2 as a mediator of disease pathogenesis. Cell. Microbiol 2010, 12, 1369–1377. [Google Scholar]
- Balsinde, J.; Pérez, R.; Balboa, M.A. Calcium-independent phospholipase A2 and apoptosis. Biochim. Biophys. Acta 2006, 1761, 1344–1350. [Google Scholar]
- Green, J.T.; Orr, S.K.; Bazinet, R.P. The emerging role of group VI calcium-independent phospholipase A2 in releasing docosahexaenoic acid from brain phospholipids. J. Lipid Res 2008, 49, 939–944. [Google Scholar]
- Hooks, S.B.; Cummings, B.S. Role of Ca2+-independent phospholipase A2 in cell growth and signaling. Biochem. Pharmacol 2008, 76, 1059–1067. [Google Scholar]
- Cedars, A.; Jenkins, C.M.; Mancuso, D.J.; Gross, R.W. Calcium-independent phospholipases in the heart: Mediators of cellular signaling, bioenergetics, and ischemia-induced electrophysiologic dysfunction. J. Cardiovasc. Pharmacol 2009, 53, 277–289. [Google Scholar]
- Lei, X.; Barbour, S.E.; Ramanadham, S. Group VIA Ca2+-independent phospholipase A2 (iPLA2beta) and its role in beta-cell programmed cell death. Biochimie 2010, 92, 627–637. [Google Scholar]
- Arai, H.; Koizumi, H.; Aoki, J.; Inoue, K. Platelet-activating factor acetylhydrolase (PAF-AH). J. Biochem 2002, 131, 635–640. [Google Scholar]
- Karasawa, K.; Harada, A.; Satoh, N.; Inoue, K.; Setaka, M. Plasma platelet activating factor-acetylhydrolase (PAF-AH). Prog. Lipid Res 2003, 42, 93–114. [Google Scholar]
- Mallat, Z.; Lambeau, G.; Tedgui, A. Lipoprotein-associated and secreted phospholipases A2 in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation 2010, 122, 2183–2200. [Google Scholar]
- McIntyre, T.M.; Prescott, S.M.; Stafforini, D.M. The emerging roles of PAF acetylhydrolase. J. Lipid Res 2009, 50, S255–S259. [Google Scholar]
- Stafforini, D.M. Biology of platelet-activating factor acetylhydrolase (PAF-AH, lipoprotein associated phospholipase A2). Cardiovasc. Drugs Ther 2009, 23, 73–83. [Google Scholar]
- Stafforini, D.M.; Elstad, M.R.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Human macrophages secret platelet-activating factor acetylhydrolase. J. Biol. Chem 1990, 265, 9682–9687. [Google Scholar]
- Stafforini, D.M.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Platelet-activating factor, a pleiotrophic mediator of physiological and pathological processes. Crit. Rev. Clin. Lab. Sci 2003, 40, 643–672. [Google Scholar]
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforini, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A.; et al. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553. [Google Scholar]
- Wilensky, R.L.; Macphee, C.H. Lipoprotein-associated phospholipase A2 and atherosclerosis. Curr. Opin. Lipidol 2009, 20, 415–420. [Google Scholar]
- Shayman, J.A.; Kelly, R.; Kollmeyer, J.; He, Y.; Abe, A. Group XV phospholipase A2, a lysosomal phospholipase A2. Prog. Lipid Res 2011, 50, 1–13. [Google Scholar]
- Duncan, R.E.; Sarkadi-Nagy, E.; Jaworski, K.; Ahmadian, M.; Sul, H.S. Identification and functional characterization of adipose-specific phospholipase A2 (AdPLA). J. Biol. Chem 2008, 283, 25428–25436. [Google Scholar]
- Jaworski, K.; Ahmadian, M.; Duncan, R.E.; Sarkadi-Nagy, E.; Varady, K.A.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; Kim, K.H.; et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med 2009, 15, 159–168. [Google Scholar]
- Yokota, Y.; Higashino, K.; Nakano, K.; Arita, H.; Hanasaki, K. Identification of group X secretory phospholipase A2 as a natural ligand for mouse phospholipase A2 receptor. FEBS Lett 2000, 478, 187–191. [Google Scholar]
- Ramanadham, S.; Yarasheski, K.E.; Silva, M.J.; Wohltmann, M.; Novack, D.V.; Christiansen, B.; Tu, X.; Zhang, S.; Lei, X.; Turk, J. Age-related changes in bone morphology are accelerated in group VIA phospholipase A2 (iPLA2beta)-null mice. Am. J. Pathol 2008, 172, 868–881. [Google Scholar]
- Bonventre, J. Cytosolic phospholipase A2alpha reigns supreme in arthritis and bone resorption. Trends Immunol 2004, 25, 116–119. [Google Scholar]
- Seilhamer, J.J.; Pruzanski, W.; Vadas, P.; Plant, S.; Miller, J.A.; Kloss, J.; Johnson, L.K. Cloning and recombinant expression of phospholipase A2 present in rheumatoid arthritic synovial fluid. J. Biol. Chem 1989, 264, 5335–5338. [Google Scholar]
- Masuda, S.; Murakami, M.; Komiyama, K.; Ishihara, M.; Ishikawa, Y.; Ishii, T.; Kudo, I. Various secretory phospholipase A2 enzymes are expressed in rheumatoid arthritis and augment prostaglandin production in cultured synovial cells. FEBS J 2005, 272, 655–672. [Google Scholar]
- Chang, J.; Gilman, S.C.; Lewis, A.J. Interleukin 1 activates phospholipase A2 in rabbit chondrocytes: A possible signal for IL 1 action. J. Immunol 1986, 136, 1283–1287. [Google Scholar]
- Jamal, O.S.; Conaghan, P.G.; Cunningham, A.M.; Brooks, P.M.; Munro, V.F.; Scott, K.F. Increased expression of human type IIa secretory phospholipase A2 antigen in arthritic synovium. Ann. Rheum. Dis 1998, 57, 550–558. [Google Scholar]
- Nevalainen, T.J.; Haapanen, T.J. Distribution of pancreatic (group I) and synovial-type (group II) phospholipases A2 in human tissues. Inflammation 1993, 17, 453–464. [Google Scholar]
- Nevalainen, T.J.; Märki, F.; Kortesuo, P.T.; Grütter, M.G.; di Marco, S.; Schmitz, A. Synovial type (group II) phospholipase A2 in cartilage. J. Rheumatol 1993, 20, 325–330. [Google Scholar]
- Nevalainen, T.J. Serum phospholipases A2 in inflammatory diseases. Clin. Chem 1993, 39, 2453–2459. [Google Scholar]
- Vignon, E.; Mathieu, P.; Louisot, P.; Vilamitjana, J.; Harmand, M.F.; Richard, M. Phospholipase A2 activity in human osteoarthritic cartilage. J. Rheumatol. Suppl 1989, 18, 35–38. [Google Scholar]
- Pruzanski, W.; Bogoch, E.; Katz, A.; Wloch, M.; Stefanski, E.; Grouix, B.; Sakotic, G.; Vadas, P. Induction of release of secretory nonpancreatic phospholipase A2 from human articular chondrocytes. J. Rheumatol 1995, 22, 2114–2119. [Google Scholar]
- Lyons-Giordano, B.; Davis, G.L.; Galbraith, W.; Pratta, M.A.; Arner, E.C. Interleukin-1 beta stimulates phospholipase A2 mRNA synthesis in rabbit articular chondrocytes. Biochem. Biophys. Res. Commun 1989, 164, 488–495. [Google Scholar]
- Nakano, T.; Ohara, O.; Teraoka, H.; Arita, H. Glucocorticoids suppress group II phospholipase A2 production by blocking mRNA synthesis and post-transcriptional expression. J. Biol. Chem 1990, 265, 12745–12748. [Google Scholar]
- Gilman, S.C. Activation of rabbit articular chondrocytes by recombinant human cytokines. J. Rheumatol 1987, 14, 1002–1007. [Google Scholar]
- Pfeilschifter, J.; Pignat, W.; Vosbeck, K.; Märki, F. Interleukin 1 and tumor necrosis factor synergistically stimulate prostaglandin synthesis and phospholipase A2 release from rat renal mesangial cells. Biochem. Biophys. Res. Commun 1989, 159, 385–394. [Google Scholar]
- Stevens, T.M.; Chin, J.E.; McGowan, M.; Giannaras, J.; Kerr, J.S. Phospholipase A2 (PLA2) activity in rabbit chondrocytes. Agents Actions 1989, 27, 385–387. [Google Scholar]
- Suffys, P.; Van Roy, F.; Fiers, W. Tumor necrosis factor and interleukin 1 activate phospholipase in rat chondrocytes. FEBS Lett 1988, 232, 24–28. [Google Scholar]
- Kudo, I.; Murakami, M.; Hara, S.; Inoue, K. Mammalian non-pancreatic phospholipases A2. Biochim. Biophys. Acta 1993, 1170, 217–231. [Google Scholar]
- Leistad, L.; Feuerherm, A.J.; Faxvaag, A.; Johansen, B. Multiple phospholipase A2 enzymes participate in the inflammatory process in osteoarthritic cartilage. Scand. J. Rheumatol 2011, 40, 308–316. [Google Scholar]
- Vadas, P.; Pruzanski, W.; Stefanski, E.; Ellies, L.G.; Aubin, J.E.; Sos, A.; Melcher, A. Extracellular phospholipase A2 secretion is a common effector pathway of interleukin-1 and tumour necrosis factor action. Immunol. Lett 1991, 28, 187–193. [Google Scholar]
- Bonewald, L.F.; Schwartz, Z.; Swain, L.D.; Boyan, B.D. Stimulation of matrix vesicle enzyme activity in osteoblast-like cells by 1,25(OH)2D3 and transforming growth factor beta (TGF beta). Bone Miner 1992, 17, 139–144. [Google Scholar]
- Schwartz, Z.; Dennis, R.; Bonewald, L.; Swain, L.; Gomez, R.; Boyan, B.D. Differential regulation of prostaglandin E2 synthesis and phospholipase A2 activity by 1,25-(OH)2D3 in three osteoblast-like cell lines (MC-3T3-E1, ROS 17/2.8, and MG-63). Bone 1992, 13, 51–58. [Google Scholar]
- Tokuda, H.; Oiso, Y.; Kozawa, O. Protein kinase C activation amplifies prostaglandin F2 alpha-induced prostaglandin E2 synthesis in osteoblast-like cells. J. Cell. Biochem 1992, 48, 262–268. [Google Scholar]
- Suzuki, N.; Matsunaga, T.; Kanaho, Y.; Nozawa, Y. The mechanism of bradykinin-induced arachidonic acid release in osteoblast-like MC3T3-E1 cells phospholipase A2 activation by bradykinin and its regulation by protein kinase C and calcium. Nihon Seikeigeka Gakkai Zasshi 1993, 67, 935–943. [Google Scholar]
- Suzuki, A.; Kozawa, O.; Shinoda, J.; Watanabe-Tomita, Y.; Saito, H.; Oiso, Y. Mechanism of thrombin-induced arachidonic acid release in osteoblast-like cells. Prostaglandins Leukot Essent Fatty Acids 1997, 56, 467–472. [Google Scholar]
- Miwa, M.; Kozawa, O.O.; Tokuda, H.; Uematsu, T. Involvement of arachidonic acid in chemical stress-induced interleukin-6 synthesis in osteoblast-like cells: Comparison with heat shock protein 27 induction. Prostaglandins Leukot Essent Fatty Acids 2000, 62, 189–193. [Google Scholar]
- Chu, S.T.; Cheng, H.H.; Huang, C.J.; Chang, H.C.; Chi, C.C.; Su, H.H.; Hsu, S.S.; Wang, J.L.; Chen, I.S.; Liu, S.I.; et al. Phospholipase A2-independent Ca2+ entry and subsequent apoptosis induced by melittin in human MG63 osteosarcoma cells. Life Sci 2007, 80, 364–369. [Google Scholar]
- Pruzanski, W.; Kennedy, B.P.; van den Bosch, H.; Stefanski, E.; Vadas, P. Microtubule depolymerization selectively down-regulates the synthesis of proinflammatory secretory nonpancreatic phospholipase A2. Lab. Invest 1997, 76, 171–178. [Google Scholar]
- Murakami, M.; Kuwata, H.; Amakasu, Y.; Shimbara, S.; Nakatani, Y.; Atsumi, G.; Kudo, I. Prostaglandin E2 amplifies cytosolic phospholipase A2- and cyclooxygenase-2-dependent delayed prostaglandin E2 generation in mouse osteoblastic cells. Enhancement by secretory phospholipase A2. J. Biol. Chem 1997, 272, 19891–19897. [Google Scholar]
- Kudo, I.; Murakami, M. Diverse functional coupling of prostanoid biosynthetic enzymes in various cell types. Adv. Exp. Med. Biol 1999, 469, 29–35. [Google Scholar]
- Higashi, S.; Ohishi, H.; Kudo, I. Augmented prostaglandin E2 generation resulting from increased activities of cytosolic and secretory phospholipase A2 and induction of cyclooxygenase-2 in interleukin-1 beta-stimulated rat calvarial cells during the mineralizing phase. Inflamm. Res 2000, 49, 102–111. [Google Scholar]
- Chen, Q.R.; Miyaura, C.; Higashi, S.; Murakami, M.; Kudo, I.; Saito, S.; Hiraide, T.; Shibasaki, Y.; Suda, T. Activation of cytosolic phospholipase A2 by platelet-derived growth factor is essential for cyclooxygenase-2-dependent prostaglandin E2 synthesis in mouse osteoblasts cultured with interleukin-1. J. Biol. Chem 1997, 272, 5952–5958. [Google Scholar]
- Miyahara, T.; Katoh, T.; Watanabe, M.; Mikami, Y.; Uchida, S.; Hosoe, M.; Sakuma, T.; Nemoto, N.; Takayama, K.; Komurasaki, T. Involvement of mitogen-activated protein kinases and protein kinase C in cadmium-induced prostaglandin E2 production in primary mouse osteoblastic cells. Toxicology 2004, 200, 159–167. [Google Scholar]
- Miyahara, T.; Tonoyama, H.; Watanabe, M.; Okajima, A.; Miyajima, S.; Sakuma, T.; Nemoto, N.; Takayama, K. Stimulative effect of cadmium on prostaglandin E2 production in primary mouse osteoblastic cells. Calcif. Tissue Int 2001, 68, 185–191. [Google Scholar]
- Leis, H.J.; Windischhofer, W. Inhibition of cyclooxygenases 1 and 2 by the phospholipase-blocker, arachidonyl trifluoromethyl ketone. Br. J. Pharmacol 2008, 155, 731–737. [Google Scholar]
- Miyaura, C.; Inada, M.; Matsumoto, C.; Ohshiba, T.; Uozumi, N.; Shimizu, T.; Ito, A. An essential role of cytosolic phospholipase A2alpha in prostaglandin E2-mediated bone resorption associated with inflammation. J. Exp. Med 2003, 197, 1303–1310. [Google Scholar]
- Hackett, J.A.; Allard-Chamard, H.; Sarrazin, P.; de Fatima Lucena, M.; Gallant, M.A.; Fortier, I.; Nader, M.; Parent, J.L.; Bkaily, G.; de Brum-Fernandes, A.J. Prostaglandin production by human osteoclasts in culture. J. Rheumatol 2006, 33, 1320–1328. [Google Scholar]
- Gregory, L.S.; Kelly, W.L.; Reid, R.C.; Fairlie, D.P.; Forwood, M.R. Inhibitors of cyclooxygenase-2 and secretory phospholipase A2 preserve bone architecture following ovariectomy in adult rats. Bone 2006, 39, 134–142. [Google Scholar]
- Kurihara, H.; Nakano, T.; Takasu, N.; Arita, H. Intracellular localization of group II phospholipase A2 in rat vascular smooth muscle cells and its possible relationship to eicosanoid formation. Biochim. Biophys. Acta 1991, 1082, 285–292. [Google Scholar]
- Nevalainen, T.J.; Grönroos, J.M.; Kallajoki, M. Expression of group II phospholipase A2 in the human gastrointestinal tract. Lab. Invest 1995, 72, 201–208. [Google Scholar]
- Romano, M.; Romano, E.; Björkerud, S.; Hurt-Camejo, E. Ultrastructural localization of secretory type II phospholipase A2 in atherosclerotic and nonatherosclerotic regions of human arteries. Arterioscler. Thromb. Vasc. Biol 1998, 18, 519–525. [Google Scholar]
- Sartipy, P.; Johansen, B.; Gâsvik, K.; Hurt-Camejo, E. Molecular basis for the association of group IIA phospholipase A(2) and decorin in human atherosclerotic lesions. Circ. Res 2000, 86, 707–714. [Google Scholar]
- Hurt-Camejo, E.; Camejo, G.; Peilot, H.; Oörni, K.; Kovanen, P. Phospholipase A(2) in vascular disease. Circ. Res 2001, 89, 298–304. [Google Scholar]
- Masuda, S.; Murakami, M.; Ishikawa, Y.; Ishii, T.; Kudo, I. Diverse cellular localizations of secretory phospholipase A2 enzymes in several human tissues. Biochim. Biophys. Acta 2005, 1736, 200–210. [Google Scholar]
- Pruzanski, W.; Keystone, E.C.; Sternby, B.; Bombardier, C.; Snow, K.M.; Vadas, P. Serum phospholipase A2 correlates with disease activity in rheumatoid arthritis. J. Rheumatol 1988, 15, 1351–1355. [Google Scholar]
- Pruzanski, W.; Vadas, P. Phospholipase A2—A mediator between proximal and distal effectors of inflammation. Immunol. Today 1991, 12, 143–146. [Google Scholar]
- Vignon, E.; Balblanc, J.C.; Mathieu, P.; Louisot, P.; Richard, M. Metalloprotease activity, phospholipase A2 activity and cytokine concentration in osteoarthritis synovial fluids. Osteoarthritis Cartilage 1993, 1, 115–120. [Google Scholar]
- Kortekangas, P.; Aro, H.T.; Nevalainen, T.J. Group II phospholipase A2 in synovial fluid and serum in acute arthritis. Scand. J. Rheumatol 1994, 23, 68–72. [Google Scholar]
- Pruzanski, W.; Albin-Cook, K.; Laxer, R.M.; MacMillan, J.; Stefanski, E.; Vadas, P.; Silverman, E.D. Phospholipase A2 in juvenile rheumatoid arthritis: Correlation to disease type and activity. J. Rheumatol 1994, 21, 1951–1954. [Google Scholar]
- Lin, M.K.; Farewell, V.; Vadas, P.; Bookman, A.A.; Keystone, E.C.; Pruzanski, W. Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. J. Rheumatol 1996, 23, 1162–1166. [Google Scholar]
- Michaels, R.M.; Reading, J.C.; Beezhold, D.H.; Ward, J.R. Serum phospholipase A2 activity in patients with rheumatoid arthritis before and after treatment with methotrexate, auranofin, or combination of the two. J. Rheumatol 1996, 23, 226–229. [Google Scholar]
- Bidgood, M.J.; Jamal, O.S.; Cunningham, A.M.; Brooks, P.M.; Scott, K.F. Type IIA secretory phospholipase A2 up-regulates cyclooxygenase-2 and amplifies cytokine-mediated prostaglandin production in human rheumatoid synoviocytes. J. Immunol 2000, 165, 2790–2797. [Google Scholar]
- Ellies, L.G.; Heersche, J.N.; Pruzanski, W.; Vadas, P.; Aubin, J.E. The role of phospholipase A2 in interleukin-1 alpha-mediated inhibition of mineralization of the osteoid formed by fetal rat calvaria cells in vitro. J. Dent. Res 1993, 72, 18–24. [Google Scholar]
- Hegen, M.; Sun, L.; Uozumi, N.; Kume, K.; Goad, M.E.; Nickerson-Nutter, C.L.; Shimizu, T.; Clark, J.D. Cytosolic phospholipase A2alpha-deficient mice are resistant to collagen-induced arthritis. J. Exp. Med 2003, 197, 1297–1302. [Google Scholar]
- Murakami, M.; Taketomi, Y.; Girard, C.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: Lessons from transgenic and knockout mice. Biochimie 2010, 92, 561–582. [Google Scholar]
- Bonventre, J.V.; Huang, Z.; Taheri, M.R.; O’Leary, E.; Li, E.; Moskowitz, M.A.; Sapirstein, A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 1997, 390, 622–625. [Google Scholar]
- Uozumi, N.; Kume, K.; Nagase, T.; Nakatani, N.; Ishii, S.; Tashiro, F.; Komagata, Y.; Maki, K.; Ikuta, K.; Ouchi, Y.; et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 1997, 390, 618–622. [Google Scholar]
- Sapirstein, A.; Bonventre, J.V. Specific physiological roles of cytosolic phospholipase A(2) as defined by gene knockouts. Biochim. Biophys. Acta 2000, 1488, 139–148. [Google Scholar]
- Bao, S.; Miller, D.J.; Ma, Z.; Wohltmann, M.; Eng, G.; Ramanadham, S.; Moley, K.; Turk, J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem 2004, 279, 38194–38200. [Google Scholar]
- Bao, S.; Jacobson, D.A.; Wohltmann, M.; Bohrer, A.; Jin, W.; Philipson, L.H.; Turk, J. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2beta in pancreatic beta-cells and in iPLA2beta-null mice. Am. J. Physiol. Endocrinol. Metab 2008, 294, E217–E229. [Google Scholar]
- Mancuso, D.J.; Kotzbauer, P.; Wozniak, D.F.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Han, X.; Yang, K.; Sun, G.; Malik, I.; et al. Genetic ablation of calcium-independent phospholipase A2 gamma leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J. Biol. Chem 2009, 284, 35632–35644. [Google Scholar]
- Mancuso, D.J.; Sims, H.F.; Han, X.; Jenkins, C.M.; Guan, S.P.; Yang, K.; Moon, S.H.; Pietka, T.; Abumrad, N.A.; Schlesinger, P.H.; et al. Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem 2007, 282, 34611–34622. [Google Scholar]
- Mancuso, D.J.; Sims, H.F.; Yang, K.; Kiebish, M.A.; Su, X.; Jenkins, C.M.; Guan, S.; Moon, S.H.; Pietka, T.; Nassir, F.; et al. Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J. Biol. Chem 2010, 285, 36495–36510. [Google Scholar]
- Boilard, E.; Lai, Y.; Larabee, K.; Balestrieri, B.; Ghomashchi, F.; Fujioka, D.; Gobezie, R.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; et al. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol. Med 2010, 2, 172–187. [Google Scholar]
- Adler, D.H.; Cogan, J.D.; Phillips, J.A.; Schnetz-Boutaud, N.; Milne, G.L.; Iverson, T.; Stein, J.A.; Brenner, D.A.; Morrow, J.D.; Boutaud, O.; et al. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J. Clin. Invest 2008, 118, 2121–2131. [Google Scholar]
- Uozumi, N.; Shimizu, T. Roles for cytosolic phospholipase A2alpha as revealed by gene-targeted mice. Prostaglandins Other Lipid Mediat. 2002, 68–69, 59–69. [Google Scholar]
- Balsinde, J.; Balboa, M.A.; Insel, P.A.; Dennis, E.A. Regulation and inhibition of phospholipase A2. Annu. Rev. Pharmacol. Toxicol 1999, 39, 175–189. [Google Scholar]
- Pruzanski, W.; Stefanski, E.; Vadas, P.; Ramamurthy, N.S. Inhibition of extracellular release of proinflammatory secretory phospholipase A2 (sPLA2) by sulfasalazine: A novel mechanism of anti-inflammatory activity. Biochem. Pharmacol 1997, 53, 1901–1907. [Google Scholar]
- Bradley, J.D.; Dmitrienko, A.A.; Kivitz, A.J.; Gluck, O.S.; Weaver, A.L.; Wiesenhutter, C.; Myers, S.L.; Sides, G.D. A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol 2005, 32, 417–423. [Google Scholar]
- Bryant, K.J.; Bidgood, M.J.; Lei, P.W.; Taberner, M.; Salom, C.; Kumar, V.; Lee, L.; Church, W.B.; Courtenay, B.; Smart, B.P.; et al. A bifunctional role for group IIA secreted phospholipase A2 in human rheumatoid fibroblast-like synoviocyte arachidonic acid metabolism. J. Biol. Chem 2011, 286, 2492–2503. [Google Scholar]
- Granata, F.; Petraroli, A.; Boilard, E.; Bezzine, S.; Bollinger, J.; Del Vecchio, L.; Gelb, M.H.; Lambeau, G.; Marone, G.; Triggiani, M. Activation of cytokine production by secreted phospholipase A2 in human lung macrophages expressing the M-type receptor. J. Immunol 2005, 174, 464–474. [Google Scholar]
- Rosenson, R.S.; Hislop, C.; Elliott, M.; Stasiv, Y.; Goulder, M.; Waters, D. Effects of varespladib methyl on biomarkers and major cardiovascular events in acute coronary syndrome patients. J. Am. Coll. Cardiol 2010, 56, 1079–1088. [Google Scholar]
- Divchev, D.; Schieffer, B. The secretory phospholipase A2 group IIA: A missing link between inflammation, activated renin-angiotensin system, and atherogenesis? Vasc. Health Risk Manag 2008, 4, 597–604. [Google Scholar]
- Tai, N.; Kuwabara, K.; Kobayashi, M.; Yamada, K.; Ono, T.; Seno, K.; Gahara, Y.; Ishizaki, J.; Hori, Y. Cytosolic phospholipase A2 alpha inhibitor, pyrroxyphene, displays anti-arthritic and anti-bone destructive action in a murine arthritis model. Inflamm. Res 2010, 59, 53–62. [Google Scholar]
- Kramer, R.M.; Hession, C.; Johansen, B.; Hayes, G.; McGray, P.; Chow, E.P.; Tizard, R.; Pepinsky, R.B. Structure and properties of a human non-pancreatic phospholipase A2. J. Biol. Chem 1989, 264, 5768–5775. [Google Scholar]
- Singer, A.G.; Ghomashchi, F.; Le Calvez, C.; Bollinger, J.; Bezzine, S.; Rouault, M.; Sadilek, M.; Nguyen, E.; Lazdunski, M.; Lambeau, G.; et al. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem 2002, 277, 48535–48549. [Google Scholar]
- Bezzine, S.; Bollinger, J.G.; Singer, A.G.; Veatch, S.L.; Keller, S.L.; Gelb, M.H. On the binding preference of human groups IIA and X phospholipases A2 for membranes with anionic phospholipids. J. Biol. Chem 2002, 277, 48523–48534. [Google Scholar]
- Mounier, C.M.; Ghomashchi, F.; Lindsay, M.R.; James, S.; Singer, A.G.; Parton, R.G.; Gelb, M.H. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A(2) occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A(2)-alpha. J. Biol. Chem 2004, 279, 25024–25038. [Google Scholar]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc. Natl. Acad. Sci. USA 1998, 95, 7951–7956. [Google Scholar]
- Han, S.K.; Kim, K.P.; Koduri, R.; Bittova, L.; Munoz, N.M.; Leff, A.R.; Wilton, D.C.; Gelb, M.H.; Cho, W. Roles of Trp31 in high membrane binding and proinflammatory activity of human group V phospholipase A2. J. Biol. Chem 1999, 274, 11881–11888. [Google Scholar]
- Hanasaki, K.; Ono, T.; Saiga, A.; Morioka, Y.; Ikeda, M.; Kawamoto, K.; Higashino, K.; Nakano, K.; Yamada, K.; Ishizaki, J.; et al. Purified group X secretory phospholipase A(2) induced prominent release of arachidonic acid from human myeloid leukemia cells. J. Biol. Chem 1999, 274, 34203–34211. [Google Scholar]
- Murakami, M.; Kambe, T.; Shimbara, S.; Higashino, K.; Hanasaki, K.; Arita, H.; Horiguchi, M.; Arita, M.; Arai, H.; Inoue, K.; et al. Different functional aspects of the group II subfamily (Types IIA and V) and type X secretory phospholipase A(2)s in regulating arachidonic acid release and prostaglandin generation. Implications of cyclooxygenase-2 induction and phospholipid scramblase-mediated cellular membrane perturbation. J. Biol. Chem 1999, 274, 31435–31444. [Google Scholar]
- Bezzine, S.; Koduri, R.S.; Valentin, E.; Murakami, M.; Kudo, I.; Ghomashchi, F.; Sadilek, M.; Lambeau, G.; Gelb, M.H. Exogenously added human group X secreted phospholipase A(2) but not the group IB, IIA, and V enzymes efficiently release arachidonic acid from adherent mammalian cells. J. Biol. Chem 2000, 275, 3179–3191. [Google Scholar]
- Kim, K.P.; Rafter, J.D.; Bittova, L.; Han, S.K.; Snitko, Y.; Munoz, N.M.; Leff, A.R.; Cho, W. Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J. Biol. Chem 2001, 276, 11126–11134. [Google Scholar]
- Murakami, M.; Koduri, R.S.; Enomoto, A.; Shimbara, S.; Seki, M.; Yoshihara, K.; Singer, A.; Valentin, E.; Ghomashchi, F.; Lambeau, G.; et al. Distinct arachidonate-releasing functions of mammalian secreted phospholipase A2s in human embryonic kidney 293 and rat mastocytoma RBL-2H3 cells through heparan sulfate shuttling and external plasma membrane mechanisms. J. Biol. Chem 2001, 276, 10083–10096. [Google Scholar]
- Kim, Y.J.; Kim, K.P.; Rhee, H.J.; Das, S.; Rafter, J.D.; Oh, Y.S.; Cho, W. Internalized group V secretory phospholipase A2 acts on the perinuclear membranes. J. Biol. Chem 2002, 277, 9358–9365. [Google Scholar]
- Muñoz, N.M.; Kim, Y.J.; Meliton, A.Y.; Kim, K.P.; Han, S.K.; Boetticher, E.; O’Leary, E.; Myou, S.; Zhu, X.; Bonventre, J.V.; et al. Human group V phospholipase A2 induces group IVA phospholipase A2-independent cysteinyl leukotriene synthesis in human eosinophils. J. Biol. Chem 2003, 278, 38813–38820. [Google Scholar]
- Murakami, M.; Shimbara, S.; Kambe, T.; Kuwata, H.; Winstead, M.V.; Tischfield, J.A.; Kudo, I. The functions of five distinct mammalian phospholipase A2S in regulating arachidonic acid release. Type IIa and type V secretory phospholipase A2S are functionally redundant and act in concert with cytosolic phospholipase A2. J. Biol. Chem 1998, 273, 14411–14423. [Google Scholar]
- Murakami, M.; Kambe, T.; Shimbara, S.; Yamamoto, S.; Kuwata, H.; Kudo, I. Functional association of type IIA secretory phospholipase A(2) with the glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan in the cyclooxygenase-2-mediated delayed prostanoid-biosynthetic pathway. J. Biol. Chem 1999, 274, 29927–29936. [Google Scholar]
- Murakami, M.; Kambe, T.; Shimbara, S.; Kudo, I. Functional coupling between various phospholipase A2s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J. Biol. Chem 1999, 274, 3103–3115. [Google Scholar]
- Balboa, M.A.; Shirai, Y.; Gaietta, G.; Ellisman, M.H.; Balsinde, J.; Dennis, E.A. Localization of group V phospholipase A2 in caveolin-enriched granules in activated P388D1 macrophage-like cells. J. Biol. Chem 2003, 278, 48059–48065. [Google Scholar]
- Lambeau, G.; Lazdunski, M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci 1999, 20, 162–170. [Google Scholar]
- Reynolds, L.J.; Hughes, L.L.; Louis, A.I.; Kramer, R.M.; Dennis, E.A. Metal ion and salt effects on the phospholipase A2, lysophospholipase, and transacylase activities of human cytosolic phospholipase A2. Biochim. Biophys. Acta 1993, 1167, 272–280. [Google Scholar]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar]
- Myers, L.K.; Kang, A.H.; Postlethwaite, A.E.; Rosloniec, E.F.; Morham, S.G.; Shlopov, B.V.; Goorha, S.; Ballou, L.R. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum 2000, 43, 2687–2693. [Google Scholar]
- Ochi, T.; Ohkubo, Y.; Mutoh, S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem. Pharmacol 2003, 66, 1055–1060. [Google Scholar]
- Trebino, C.E.; Stock, J.L.; Gibbons, C.P.; Naiman, B.M.; Wachtmann, T.S.; Umland, J.P.; Pandher, K.; Lapointe, J.M.; Saha, S.; Roach, M.L.; et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. USA 2003, 100, 9044–9049. [Google Scholar]
- Kamei, D.; Yamakawa, K.; Takegoshi, Y.; Mikami-Nakanishi, M.; Nakatani, Y.; Oh-Ishi, S.; Yasui, H.; Azuma, Y.; Hirasawa, N.; Ohuchi, K.; et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin e synthase-1. J. Biol. Chem 2004, 279, 33684–33695. [Google Scholar]
- Vane, J.R.; Botting, R.M. New insights into the mode of action of anti-inflammatory drugs. Inflamm. Res 1995, 44, 1–10. [Google Scholar]
- Altman, R.D.; Latta, L.L.; Keer, R.; Renfree, K.; Hornicek, F.J.; Banovac, K. Effect of nonsteroidal antiinflammatory drugs on fracture healing: A laboratory study in rats. J. Orthop. Trauma 1995, 9, 392–400. [Google Scholar]
- Törnkvist, H.; Lindholm, T.S.; Netz, P.; Strömberg, L.; Lindholm, T.C. Effect of ibuprofen and indomethacin on bone metabolism reflected in bone strength. Clin. Orthop. Relat. Res 1984, 187, 255–259. [Google Scholar]
- Ro, J.; Langeland, N.; Sander, J. Effect of indomethacin on collagen metabolism of rat fracture callus in vitro. Acta Orthop. Scand 1978, 49, 323–328. [Google Scholar]
- Keller, J.; Bünger, C.; Andreassen, T.T.; Bak, B.; Lucht, U. Bone repair inhibited by indomethacin. Effects on bone metabolism and strength of rabbit osteotomies. Acta Orthop. Scand 1987, 58, 379–383. [Google Scholar]
- Ritter, M.A.; Gioe, T.J. The effect of indomethacin on para-articular ectopic ossification following total hip arthroplasty. Clin. Orthop. Relat. Res 1982, 167, 113–117. [Google Scholar]
- Sodemann, B.; Persson, P.E.; Nilsson, O.S. Prevention of periarticular heterotopic ossification following total hip arthroplasty: Clinical experience with indomethacin and ibuprofen. Arch. Orthop. Trauma Surg 1988, 107, 329–333. [Google Scholar]
- Kjaersgaard-Andersen, P.; Ritter, M.A. Short-term treatment with nonsteroidal antiinflammatory medications to prevent heterotopic bone formation after total hip arthroplasty. A preliminary report. Clin. Orthop. Relat. Res 1992, 279, 157–162. [Google Scholar]
- Dimar, J.R.; Ante, W.A.; Zhang, Y.P.; Glassman, S.D. The effects of nonsteroidal anti-inflammatory drugs on posterior spinal fusions in the rat. Spine (Phila Pa 1976) 1996, 21, 1870–1876. [Google Scholar]
- Glassman, S.D.; Rose, S.M.; Dimar, J.R.; Puno, R.M.; Campbell, M.J.; Johnson, J.R. The effect of postoperative nonsteroidal anti-inflammatory drug administration on spinal fusion. Spine (Phila Pa 1976) 1998, 23, 834–838. [Google Scholar]
- Chang, J.K.; Li, C.J.; Wu, S.C.; Yeh, C.H.; Chen, C.H.; Fu, Y.C.; Wang, G.J.; Ho, M.L. Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem. Pharmacol 2007, 74, 1371–13782. [Google Scholar]
- Stafford, J.B.; Marnett, L.J. Prostaglandin E2 inhibits tumor necrosis factor-alpha RNA through PKA type I. Biochem. Biophys. Res. Commun 2008, 366, 104–109. [Google Scholar]
- Crofford, L.J.; Lipsky, P.E.; Brooks, P.; Abramson, S.B.; Simon, L.S.; van de Putte, L.B. Basic biology and clinical application of specific cyclooxygenase-2 inhibitors. Arthritis Rheum 2000, 43, 4–13. [Google Scholar]
- Martel-Pelletier, J.; Pelletier, J.P.; Fahmi, H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin. Arthritis Rheum 2003, 33, 155–167. [Google Scholar]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab 2010, 21, 294–301. [Google Scholar]
- Miller, S.B. Prostaglandins in health and disease: An overview. Semin. Arthritis Rheum 2006, 36, 37–49. [Google Scholar]
- Martel-Pelletier, J.; Lajeunesse, D.; Fahmi, H.; Tardif, G.; Pelletier, J.P. New thoughts on the pathophysiology of osteoarthritis: One more step toward new therapeutic targets. Curr. Rheumatol. Rep 2006, 8, 30–36. [Google Scholar]
- Zhang, X.; Schwarz, E.M.; Young, D.A.; Puzas, J.E.; Rosier, R.N.; O’Keefe, R.J. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J. Clin. Invest 2002, 109, 1405–1415. [Google Scholar]
- Atik, O.S.; Uslu, M.M.; Eksioglu, F.; Satana, T. Etiology of senile osteoporosis: A hypothesis. Clin. Orthop. Relat. Res 2006, 443, 25–27. [Google Scholar]
- Hikiji, H.; Takato, T.; Shimizu, T.; Ishii, S. The roles of prostanoids, leukotrienes, and platelet-activating factor in bone metabolism and disease. Prog. Lipid Res 2008, 47, 107–126. [Google Scholar]
- Lettesjö, H.; Nordström, E.; Ström, H.; Nilsson, B.; Glinghammar, B.; Dahlstedt, L.; Möller, E. Synovial fluid cytokines in patients with rheumatoid arthritis or other arthritic lesions. Scand. J. Immunol 1998, 48, 286–292. [Google Scholar]
- Hidaka, T.; Suzuki, K.; Kawakami, M.; Okada, M.; Kataharada, K.; Shinohara, T.; Takamizawa-Matsumoto, M.; Ohsuzu, F. Dynamic changes in cytokine levels in serum and synovial fluid following filtration leukocytapheresis therapy in patients with rheumatoid arthritis. J. Clin. Apher 2001, 16, 74–81. [Google Scholar]
- Hochberg, M.C. COX-2 selective inhibitors in the treatment of arthritis: A rheumatologist perspective. Curr. Top. Med. Chem 2005, 5, 443–448. [Google Scholar]
- Boyan, B.D.; Sylvia, V.L.; Liu, Y.; Sagun, R.; Cochran, D.L.; Lohmann, C.H.; Dean, D.D.; Schwartz, Z. Surface roughness mediates its effects on osteoblasts via protein kinase A and phospholipase A2. Biomaterials 1999, 20, 2305–2310. [Google Scholar]
- Kawaguchi, H.; Pilbeam, C.C.; Harrison, J.R.; Raisz, L.G. The role of prostaglandins in the regulation of bone metabolism. Clin. Orthop. Relat. Res. 1995, 36–46. [Google Scholar]
- Watkins, B.A.; Lippman, H.E.; Le Bouteiller, L.; Li, Y.; Seifert, M.F. Bioactive fatty acids: Role in bone biology and bone cell function. Prog. Lipid Res 2001, 40, 125–148. [Google Scholar]
- Knippenberg, M.; Helder, M.N.; de Blieck-Hogervorst, J.M.; Wuisman, P.I.; Klein-Nulend, J. Prostaglandins differentially affect osteogenic differentiation of human adipose tissue-derived mesenchymal stem cells. Tissue Eng 2007, 13, 2495–2503. [Google Scholar]
- Sato, I.; Suzuki, A.; Kakita, A.; Ono, Y.; Miura, Y.; Itoh, M.; Oiso, Y. Stimulatory effect of prostaglandin F(2alpha) on Na-dependent phosphate transport in osteoblast-like cells. Prostaglandins Leukotrienes Essent. Fatty Acids 2003, 68, 311–315. [Google Scholar]
- Montessuit, C.; Caverzasio, J.; Bonjour, J.P. Characterization of a Pi transport system in cartilage matrix vesicles. Potential role in the calcification process. J. Biol. Chem 1991, 266, 17791–17797. [Google Scholar]
- Khan, E.; Abu-Amer, Y. Activation of peroxisome proliferator-activated receptor-gamma inhibits differentiation of preosteoblasts. J. Lab. Clin. Med 2003, 142, 29–34. [Google Scholar]
- Koshihara, Y.; Kawamura, M. Prostaglandin D2 stimulates calcification of human osteoblastic cells. Biochem. Biophys. Res. Commun 1989, 159, 1206–1212. [Google Scholar]
- Baudry, A.; Bitard, J.; Mouillet-Richard, S.; Locker, M.; Poliard, A.; Launay, J.M.; Kellermann, O. Serotonergic 5-HT(2B) receptor controls tissue-nonspecific alkaline phosphatase activity in osteoblasts via eicosanoids and phosphatidylinositol-specific phospholipase C. J. Biol. Chem 2010, 285, 26066–26073. [Google Scholar]
- Tsushita, K.; Kozawa, O.; Tokuda, H.; Oiso, Y.; Saito, H. Proliferative effect of PGD2 on osteoblast-like cells; independent activation of pertussis toxin-sensitive GTP-binding protein from PGE2 or PGF2 alpha. Prostaglandins Leukotrienes Essent. Fatty Acids 1992, 45, 267–274. [Google Scholar]
- Makide, K.; Kitamura, H.; Sato, Y.; Okutani, M.; Aoki, J. Emerging lysophospholipid mediators, lysophosphatidylserine, lysophosphatidylthreonine, lysophosphatidylethanolamine and lysophosphatidylglycerol. Prostaglandins Other Lipid Mediat 2009, 89, 135–139. [Google Scholar]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun 2007, 362, 928–934. [Google Scholar]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand l-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J 2009, 23, 183–193. [Google Scholar]
- Henstridge, C.M. Off-target cannabinoid effects mediated by GPR55. Pharmacology 2012, 89, 179–187. [Google Scholar]
- Sharir, H.; Abood, M.E. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol. Ther 2010, 126, 301–313. [Google Scholar]
- Brown, A.J.; Daniels, D.A.; Kassim, M.; Brown, S.; Haslam, C.P.; Terrell, V.R.; Brown, J.; Nichols, P.L.; Staton, P.C.; Wise, A.; et al. Pharmacology of GPR55 in yeast and identification of GSK494581A as a mixed-activity glycine transporter subtype 1 inhibitor and GPR55 agonist. J. Pharmacol. Exp. Ther 2011, 337, 236–246. [Google Scholar]
- Oka, S.; Toshida, T.; Maruyama, K.; Nakajima, K.; Yamashita, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem 2009, 145, 13–20. [Google Scholar]
- Whyte, L.S.; Ryberg, E.; Sims, N.A.; Ridge, S.A.; Mackie, K.; Greasley, P.J.; Ross, R.A.; Rogers, M.J. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 16511–16516. [Google Scholar]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar]
- Blackburn, J.; Mansell, J.P. The emerging role of lysophosphatidic acid (LPA) in skeletal biology. Bone 2012, 50, 756–762. [Google Scholar]
- Hurst-Kennedy, J.; Boyan, B.D.; Schwartz, Z. Lysophosphatidic acid signaling promotes proliferation, differentiation, and cell survival in rat growth plate chondrocytes. Biochim. Biophys. Acta 2009, 1793, 836–846. [Google Scholar]
- Tigyi, G.; Miledi, R. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem 1992, 267, 21360–21367. [Google Scholar]
- Baker, D.L.; Umstot, E.S.; Desiderio, D.M.; Tigyi, G.J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci 2000, 905, 267–269. [Google Scholar]
- Sano, T.; Baker, D.; Virag, T.; Wada, A.; Yatomi, Y.; Kobayashi, T.; Igarashi, Y.; Tigyi, G. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J. Biol. Chem 2002, 277, 21197–21206. [Google Scholar]
- Aoki, J.; Taira, A.; Takanezawa, Y.; Kishi, Y.; Hama, K.; Kishimoto, T.; Mizuno, K.; Saku, K.; Taguchi, R.; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem 2002, 277, 48737–48744. [Google Scholar]
- Hosogaya, S.; Yatomi, Y.; Nakamura, K.; Ohkawa, R.; Okubo, S.; Yokota, H.; Ohta, M.; Yamazaki, H.; Koike, T.; Ozaki, Y. Measurement of plasma lysophosphatidic acid concentration in healthy subjects: Strong correlation with lysophospholipase D activity. Ann. Clin. Biochem 2008, 45, 364–368. [Google Scholar]
- Ishii, I.; Fukushima, N.; Ye, X.; Chun, J. Lysophospholipid receptors: Signaling and biology. Annu. Rev. Biochem 2004, 73, 321–354. [Google Scholar]
- Gerrard, J.M.; Robinson, P. Identification of the molecular species of lysophosphatidic acid produced when platelets are stimulated by thrombin. Biochim. Biophys. Acta 1989, 1001, 282–285. [Google Scholar]
- Xiao, Y.; Chen, Y.; Kennedy, A.W.; Belinson, J.; Xu, Y. Evaluation of plasma lysophospholipids for diagnostic significance using electrospray ionization mass spectrometry (ESI-MS) analyses. Ann. N. Y. Acad. Sci 2000, 905, 242–259. [Google Scholar]
- Baker, D.L.; Desiderio, D.M.; Miller, D.D.; Tolley, B.; Tigyi, G.J. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem 2001, 292, 287–295. [Google Scholar]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol 2004, 15, 477–489. [Google Scholar]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem 2010, 148, 13–24. [Google Scholar]
- Tokumura, A.; Fujimoto, H.; Yoshimoto, O.; Nishioka, Y.; Miyake, M.; Fukuzawa, K. Production of lysophosphatidic acid by lysophospholipase D in incubated plasma of spontaneously hypertensive rats and Wistar Kyoto rats. Life Sci 1999, 65, 245–253. [Google Scholar]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem 2002, 277, 39436–39442. [Google Scholar]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell. Biol 2002, 158, 227–233. [Google Scholar]
- Jonas, A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta 2000, 1529, 245–256. [Google Scholar]
- Sato, T.; Aoki, J.; Nagai, Y.; Dohmae, N.; Takio, K.; Doi, T.; Arai, H.; Inoue, K. Serine phospholipid-specific phospholipase A that is secreted from activated platelets. A new member of the lipase family. J. Biol. Chem 1997, 272, 2192–2198. [Google Scholar]
- Fourcade, O.; Simon, M.F.; Viodé, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournié, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927. [Google Scholar]
- Mauco, G.; Dajeans, P.; Chap, H.; Douste-Blazy, L. Subcellular localization of inositol lipids in blood platelets as deduced from the use of labelled precursors. Biochem. J 1987, 244, 757–761. [Google Scholar]
- Pages, C.; Simon, M.F.; Valet, P.; Saulnier-Blache, J.S. Lysophosphatidic acid synthesis and release. Prostaglandins Other Lipid Mediat 2001, 64, 1–10. [Google Scholar]
- Pasternack, S.M.; von Kügelgen, I.; Al Aboud, K.; Lee, Y.A.; Rüschendorf, F.; Voss, K.; Hillmer, A.M.; Molderings, G.J.; Franz, T.; Ramirez, A.; et al. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet 2008, 40, 329–334. [Google Scholar]
- Thumser, A.E.; Voysey, J.E.; Wilton, D.C. The binding of lysophospholipids to rat liver fatty acid-binding protein and albumin. Biochem. J 1994, 301, 801–806. [Google Scholar]
- Nochi, H.; Tomura, H.; Tobo, M.; Tanaka, N.; Sato, K.; Shinozaki, T.; Kobayashi, T.; Takagishi, K.; Ohta, H.; Okajima, F.; et al. Stimulatory role of lysophosphatidic acid in cyclooxygenase-2 induction by synovial fluid of patients with rheumatoid arthritis in fibroblast-like synovial cells. J. Immunol 2008, 181, 5111–5119. [Google Scholar]
- Ferry, G.; Tellier, E.; Try, A.; Grés, S.; Naime, I.; Simon, M.F.; Rodriguez, M.; Boucher, J.; Tack, I.; Gesta, S.; et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem 2003, 278, 18162–18169. [Google Scholar]
- Boucharaba, A.; Serre, C.M.; Grès, S.; Saulnier-Blache, J.S.; Bordet, J.C.; Guglielmi, J.; Clézardin, P.; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest 2004, 114, 1714–1725. [Google Scholar]
- Panupinthu, N.; Zhao, L.; Possmayer, F.; Ke, H.Z.; Sims, S.M.; Dixon, S.J. P2X7 nucleotide receptors mediate blebbing in osteoblasts through a pathway involving lysophosphatidic acid. J. Biol. Chem 2007, 282, 3403–3412. [Google Scholar]
- Panupinthu, N.; Rogers, J.T.; Zhao, L.; Solano-Flores, L.P.; Possmayer, F.; Sims, S.M.; Dixon, S.J. P2X7 receptors on osteoblasts couple to production of lysophosphatidic acid: A signaling axis promoting osteogenesis. J. Cell. Biol 2008, 181, 859–871. [Google Scholar]
- Gaits, F.; Fourcade, O.; Le Balle, F.; Gueguen, G.; Gaigé, B.; Gassama-Diagne, A.; Fauvel, J.; Salles, J.P.; Mauco, G.; Simon, M.F.; et al. Lysophosphatidic acid as a phospholipid mediator: Pathways of synthesis. FEBS Lett 1997, 410, 54–58. [Google Scholar]
- Chun, J.; Hla, T.; Lynch, K.R.; Spiegel, S.; Moolenaar, W.H. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol. Rev 2010, 62, 579–587. [Google Scholar]
- Grey, A.; Banovic, T.; Naot, D.; Hill, B.; Callon, K.; Reid, I.; Cornish, J. Lysophosphatidic acid is an osteoblast mitogen whose proliferative actions involve G(i) proteins and protein kinase C, but not P42/44 mitogen-activated protein kinases. Endocrinology 2001, 142, 1098–1106. [Google Scholar]
- Caverzasio, J.; Palmer, G.; Suzuki, A.; Bonjour, J.P. Evidence for the involvement of two pathways in activation of extracellular signal-regulated kinase (Erk) and cell proliferation by Gi and Gq protein-coupled receptors in osteoblast-like cells. J. Bone Miner. Res 2000, 15, 1697–1706. [Google Scholar]
- Dziak, R.; Yang, B.M.; Leung, B.W.; Li, S.; Marzec, N.; Margarone, J.; Bobek, L. Effects of sphingosine-1-phosphate and lysophosphatidic acid on human osteoblastic cells. Prostaglandins Leukotrienes Essent. Fatty Acids 2003, 68, 239–249. [Google Scholar]
- Gidley, J.; Openshaw, S.; Pring, E.T.; Sale, S.; Mansell, J.P. Lysophosphatidic acid cooperates with 1alpha,25(OH)2D3 in stimulating human MG63 osteoblast maturation. Prostaglandins Other Lipid Mediat 2006, 80, 46–61. [Google Scholar]
- Kim, M.K.; Lee, H.Y.; Park, K.S.; Shin, E.H.; Jo, S.H.; Yun, J.; Lee, S.W.; Yoo, Y.H.; Lee, Y.S.; Baek, S.H.; et al. Lysophosphatidic acid stimulates cell proliferation in rat chondrocytes. Biochem. Pharmacol 2005, 70, 1764–1771. [Google Scholar]
- Grey, A.; Chen, Q.; Callon, K.; Xu, X.; Reid, I.R.; Cornish, J. The phospholipids sphingosine-1-phosphate and lysophosphatidic acid prevent apoptosis in osteoblastic cells via a signaling pathway involving G(i) proteins and phosphatidylinositol-3 kinase. Endocrinology 2002, 143, 4755–4763. [Google Scholar]
- Lyons, J.M.; Karin, N.J. A role for G protein-coupled lysophospholipid receptors in sphingolipid-induced Ca2+ signaling in MC3T3-E1 osteoblastic cells. J. Bone Miner. Res 2001, 16, 2035–2042. [Google Scholar]
- Masiello, L.M.; Fotos, J.S.; Galileo, D.S.; Karin, N.J. Lysophosphatidic acid induces chemotaxis in MC3T3-E1 osteoblastic cells. Bone 2006, 39, 72–82. [Google Scholar]
- Checovich, W.J.; Mosher, D.F. Lysophosphatidic acid enhances fibronectin binding to adherent cells. Arterioscler. Thromb 1993, 13, 1662–1667. [Google Scholar]
- Zhang, Q.; Checovich, W.J.; Peters, D.M.; Albrecht, R.M.; Mosher, D.F. Modulation of cell surface fibronectin assembly sites by lysophosphatidic acid. J. Cell. Biol 1994, 127, 1447–1459. [Google Scholar]
- Zhang, Q.; Magnusson, M.K.; Mosher, D.F. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell 1997, 8, 1415–1425. [Google Scholar]
- Karagiosis, S.A.; Karin, N.J. Lysophosphatidic acid induces osteocyte dendrite outgrowth. Biochem. Biophys. Res. Commun 2007, 357, 194–199. [Google Scholar]
- Liu, Y.B.; Kharode, Y.; Bodine, P.V.; Yaworsky, P.J.; Robinson, J.A.; Billiard, J. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J. Cell. Biochem 2010, 109, 794–800. [Google Scholar]
- Contos, J.J.; Fukushima, N.; Weiner, J.A.; Kaushal, D.; Chun, J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. USA 2000, 97, 13384–13389. [Google Scholar]
- Gennero, I.; Laurencin-Dalicieux, S.; Conte-Auriol, F.; Briand-Mésange, F.; Laurencin, D.; Rue, J.; Beton, N.; Malet, N.; Mus, M.; Tokumura, A.; et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone 2011, 49, 395–403. [Google Scholar]
- Lapierre, D.M.; Tanabe, N.; Pereverzev, A.; Spencer, M.; Shugg, R.P.; Dixon, S.J.; Sims, S.M. Lysophosphatidic acid signals through multiple receptors in osteoclasts to elevate cytosolic calcium concentration, evoke retraction, and promote cell survival. J. Biol. Chem 2010, 285, 25792–25801. [Google Scholar]
- Itoh, R.; Miura, S.; Takimoto, A.; Kondo, S.; Sano, H.; Hiraki, Y. Stimulatory actions of lysophosphatidic acid on mouse ATDC5 chondroprogenitor cells. J. Bone Miner. Metab 2010, 28, 659–671. [Google Scholar] [Green Version]
- Hale, J.E.; Wuthier, R.E. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J. Biol. Chem 1987, 262, 1916–1925. [Google Scholar]
- Peress, N.S.; Anderson, H.C.; Sajdera, S.W. The lipids of matrix vesicles from bovine fetal epiphyseal cartilage. Calcif. Tissue Res 1974, 14, 275–281. [Google Scholar]
- Wuthier, R.E. Lipids of matrix vesicles. Fed. Proc 1976, 35, 117–121. [Google Scholar]
- Genge, B.R.; Wu, L.N.; Wuthier, R.E. Separation and quantification of chicken and bovine growth plate cartilage matrix vesicle lipids by high-performance liquid chromatography using evaporative light scattering detection. Anal. Biochem 2003, 322, 104–115. [Google Scholar]
- Say, J.C.; Ciuffi, K.; Furriel, R.P.; Ciancaglini, P.; Leone, F.A. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta 1991, 1074, 256–262. [Google Scholar]
- Pizauro, J.M.; Ciancaglini, P.; Leone, F.A. Phosphotransferase activity associated with rat osseous plate alkaline phosphatase: A possible role in biomineralization. Int. J. Biochem 1992, 24, 1391–1396. [Google Scholar]
- Zhang, L.; Balcerzak, M.; Radisson, J.; Thouverey, C.; Pikula, S.; Azzar, G.; Buchet, R. Phosphodiesterase activity of alkaline phosphatase in ATP-initiated Ca(2+) and phosphate deposition in isolated chicken matrix vesicles. J. Biol. Chem 2005, 280, 37289–37296. [Google Scholar]
- Schwartz, Z.; Boyan, B. The effects of vitamin D metabolites on phospholipase A2 activity of growth zone and resting zone cartilage cells in vitro. Endocrinology 1988, 122, 2191–2198. [Google Scholar]
- Schwartz, Z.; Schlader, D.L.; Swain, L.D.; Boyan, B.D. Direct effects of 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 on growth zone and resting zone chondrocyte membrane alkaline phosphatase and phospholipase-A2 specific activities. Endocrinology 1988, 123, 2878–2884. [Google Scholar]
- Nishizuka, Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature 1984, 308, 693–698. [Google Scholar]
- Vines, C.M. Phospholipase C. Adv. Exp. Med. Biol 2012, 740, 235–254. [Google Scholar]
- Li, H.; Zhang, L.; Yin, D.; Zhang, Y.; Miao, J. Targeting phosphatidylcholine-specific phospholipase C for atherogenesis therapy. Trends Cardiovasc. Med 2010, 20, 172–176. [Google Scholar]
- Spadaro, F.; Ramoni, C.; Mezzanzanica, D.; Miotti, S.; Alberti, P.; Cecchetti, S.; Iorio, E.; Dolo, V.; Canevari, S.; Podo, F. Phosphatidylcholine-specific phospholipase C activation in epithelial ovarian cancer cells. Cancer Res 2008, 68, 6541–6549. [Google Scholar]
- Tsai, C.C.; Kai, J.I.; Huang, W.C.; Wang, C.Y.; Wang, Y.; Chen, C.L.; Fang, Y.T.; Lin, Y.S.; Anderson, R.; Chen, S.H.; et al. Glycogen synthase kinase-3beta facilitates IFN-gamma-induced STAT1 activation by regulating Src homology-2 domain-containing phosphatase 2. J. Immunol 2009, 183, 856–864. [Google Scholar]
- Horstmeyer, A.; Licht, C.; Scherr, G.; Eckes, B.; Krieg, T. Signalling and regulation of collagen I synthesis by ET-1 and TGF-beta1. FEBS J 2005, 272, 6297–6309. [Google Scholar]
- Weber, C.; Erl, W.; Pietsch, A.; Danesch, U.; Weber, P.C. Docosahexaenoic acid selectively attenuates induction of vascular cell adhesion molecule-1 and subsequent monocytic cell adhesion to human endothelial cells stimulated by tumor necrosis factor-alpha. Arterioscler. Thromb. Vasc. Biol 1995, 15, 622–628. [Google Scholar]
- Zhang, L.; Zhao, J.; Su, L.; Huang, B.; Wang, L.; Su, H.; Zhang, Y.; Zhang, S.; Miao, J. D609 inhibits progression of preexisting atheroma and promotes lesion stability in apolipoprotein e−/− mice: A role of phosphatidylcholine-specific phospholipase in atherosclerosis. Arterioscler. Thromb. Vasc. Biol 2010, 30, 411–418. [Google Scholar]
- Rapuano, B.E.; Bockman, R.S. Protein kinase C-independent activation of a novel nonspecific phospholipase C pathway by phorbol myristate acetate releases arachidonic acid for prostaglandin synthesis in MC3T3-E1 osteoblasts. Prostaglandins 1997, 53, 163–186. [Google Scholar]
- Ishimi, Y.; Miyaura, C.; Jin, C.H.; Akatsu, T.; Abe, E.; Nakamura, Y.; Yamaguchi, A.; Yoshiki, S.; Matsuda, T.; Hirano, T. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol 1990, 145, 3297–3303. [Google Scholar]
- Nijweide, P.J.; Burger, E.H.; Feyen, J.H. Cells of bone: Proliferation, differentiation, and hormonal regulation. Physiol. Rev 1986, 66, 855–886. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Tokuda, H.; Kaida, T.; Uematsu, T. Protein kinase C activation by interleukin (IL)-1 limits IL-1-induced IL-6 synthesis in osteoblast-like cells: Involvement of phosphatidylcholine-specific phospholipase C. J. Cell. Biochem 1997, 67, 103–111. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Kaida, T.; Tokuda, H.; Uematsu, T. Tumor necrosis factor-alpha autoregulates interleukin-6 synthesis via activation of protein kinase C. Function of sphingosine 1-phosphate and phosphatidylcholine-specific phospholipase C. J. Biol. Chem 1997, 272, 25099–25104. [Google Scholar]
- Sakai, T.; Sugiyama, T.; Banno, Y.; Kato, Y.; Nozawa, Y. Involvement of phosphatidylcholine hydrolysis by phospholipase C in prostaglandin F2alpha-induced 1,2-diacylglycerol formation in osteoblast-like MC3T3-E1 cells. J. Bone Miner. Metab 2004, 22, 198–206. [Google Scholar]
- Ma, Y.; Fu, D.; Liu, Z. Effect of lead on apoptosis in cultured rat primary osteoblasts. Toxicol. Ind. Health 2012, 28, 136–146. [Google Scholar]
- Xia, L.; Zhang, D.; Wang, C.; Wei, F.; Hu, Y. PC-PLC is involved in osteoclastogenesis induced by TNF-α through upregulating IP3R1 expression. FEBS Lett 2012, 586, 3341–3348. [Google Scholar]
- Hofbauer, L.C.; Schrader, J.; Niebergall, U.; Viereck, V.; Burchert, A.; Hörsch, D.; Preissner, K.T.; Schoppet, M. Interleukin-4 differentially regulates osteoprotegerin expression and induces calcification in vascular smooth muscle cells. Thromb. Haemost 2006, 95, 708–714. [Google Scholar]
- Whyte, M.P.; Landt, M.; Ryan, L.M.; Mulivor, R.A.; Henthorn, P.S.; Fedde, K.N.; Mahuren, J.D.; Coburn, S.P. Alkaline phosphatase: Placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5′-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J. Clin. Invest 1995, 95, 1440–1445. [Google Scholar]
- Fedde, K.N.; Lane, C.C.; Whyte, M.P. Alkaline phosphatase is an ectoenzyme that acts on micromolar concentrations of natural substrates at physiologic pH in human osteosarcoma (SAOS-2) cells. Arch. Biochem. Biophys 1988, 264, 400–409. [Google Scholar]
- Berridge, M.J. Inositol trisphosphate and calcium signaling. Ann. N. Y. Acad. Sci 1995, 766, 31–43. [Google Scholar]
- Yin, H.L.; Janmey, P.A. Phosphoinositide regulation of the actin cytoskeleton. Annu. Rev. Physiol 2003, 65, 761–789. [Google Scholar]
- Di Paolo, G.; de Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar]
- Suh, B.C.; Hille, B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol 2005, 15, 370–378. [Google Scholar]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev 2010, 20, 87–90. [Google Scholar]
- Kashiwada, M.; Lu, P.; Rothman, P.B. PIP3 pathway in regulatory T cells and autoimmunity. Immunol. Res 2007, 39, 194–224. [Google Scholar]
- Suh, P.G.; Ryu, S.H.; Moon, K.H.; Suh, H.W.; Rhee, S.G. Cloning and sequence of multiple forms of phospholipase C. Cell 1988, 54, 161–169. [Google Scholar]
- Kim, J.K.; Lim, S.; Kim, J.; Kim, S.; Kim, J.H.; Ryu, S.H.; Suh, P.G. Subtype-specific roles of phospholipase C-β via differential interactions with PDZ domain proteins. Adv. Enzyme Regul 2011, 51, 138–151. [Google Scholar]
- Cocco, L.; Follo, M.Y.; Faenza, I.; Fiume, R.; Ramazzotti, G.; Weber, G.; Martelli, A.M.; Manzoli, F.A. Physiology and pathology of nuclear phospholipase C β1. Adv. Enzyme Regul 2011, 51, 2–12. [Google Scholar]
- Faccio, R.; Cremasco, V. PLCgamma2: Where bone and immune cells find their common ground. Ann. N. Y. Acad. Sci 2010, 1192, 124–130. [Google Scholar]
- Choi, J.H.; Ryu, S.H.; Suh, P.G. On/off-regulation of phospholipase C-gamma 1-mediated signal transduction. Adv. Enzyme Regul 2007, 47, 104–116. [Google Scholar]
- Kelley, G.G.; Reks, S.E.; Ondrako, J.M.; Smrcka, A.V. Phospholipase C(epsilon): A novel Ras effector. EMBO J 2001, 20, 743–754. [Google Scholar]
- Lopez, I.; Mak, E.C.; Ding, J.; Hamm, H.E.; Lomasney, J.W. A novel bifunctional phospholipase c that is regulated by Galpha 12 and stimulates the Ras/mitogen-activated protein kinase pathway. J. Biol. Chem 2001, 276, 2758–2765. [Google Scholar]
- Smrcka, A.V.; Brown, J.H.; Holz, G.G. Role of phospholipase Cɛ in physiological phosphoinositide signaling networks. Cell. Signal 2012, 24, 1333–1343. [Google Scholar]
- Song, C.; Hu, C.D.; Masago, M.; Kariyai, K.; Yamawaki-Kataoka, Y.; Shibatohge, M.; Wu, D.; Satoh, T.; Kataoka, T. Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J. Biol. Chem 2001, 276, 2752–2757. [Google Scholar]
- Saunders, C.M.; Larman, M.G.; Parrington, J.; Cox, L.J.; Royse, J.; Blayney, L.M.; Swann, K.; Lai, F.A. PLC zeta: A sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 2002, 129, 3533–3544. [Google Scholar]
- Hwang, J.I.; Oh, Y.S.; Shin, K.J.; Kim, H.; Ryu, S.H.; Suh, P.G. Molecular cloning and characterization of a novel phospholipase C, PLC-eta. Biochem. J 2005, 389, 181–186. [Google Scholar]
- Nakahara, M.; Shimozawa, M.; Nakamura, Y.; Irino, Y.; Morita, M.; Kudo, Y.; Fukami, K. A novel phospholipase C, PLC(eta)2, is a neuron-specific isozyme. J. Biol. Chem 2005, 280, 29128–29134. [Google Scholar]
- Zhou, Y.; Wing, M.R.; Sondek, J.; Harden, T.K. Molecular cloning and characterization of PLC-eta2. Biochem. J 2005, 391, 667–676. [Google Scholar]
- Suh, P.G.; Park, J.I.; Manzoli, L.; Cocco, L.; Peak, J.C.; Katan, M.; Fukami, K.; Kataoka, T.; Yun, S.; Ryu, S.H. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep 2008, 41, 415–434. [Google Scholar]
- Harden, T.K.; Sondek, J. Regulation of phospholipase C isozymes by ras superfamily GTPases. Annu. Rev. Pharmacol. Toxicol 2006, 46, 355–379. [Google Scholar]
- Rhee, S.G.; Suh, P.G.; Ryu, S.H.; Lee, S.Y. Studies of inositol phospholipid-specific phospholipase C. Science 1989, 244, 546–550. [Google Scholar]
- Katan, M. Families of phosphoinositide-specific phospholipase C: Structure and function. Biochim. Biophys. Acta 1998, 1436, 5–17. [Google Scholar]
- Sorli, S.C.; Bunney, T.D.; Sugden, P.H.; Paterson, H.F.; Katan, M. Signaling properties and expression in normal and tumor tissues of two phospholipase C epsilon splice variants. Oncogene 2005, 24, 90–100. [Google Scholar]
- Kim, C.G.; Park, D.; Rhee, S.G. The role of carboxyl-terminal basic amino acids in Gqalpha-dependent activation, particulate association, and nuclear localization of phospholipase C-beta1. J. Biol. Chem 1996, 271, 21187–21192. [Google Scholar]
- Manzoli, L.; Martelli, A.M.; Billi, A.M.; Faenza, I.; Fiume, R.; Cocco, L. Nuclear phospholipase C: Involvement in signal transduction. Prog. Lipid Res 2005, 44, 185–206. [Google Scholar]
- Martelli, A.M.; Fiume, R.; Faenza, I.; Tabellini, G.; Evangelista, C.; Bortul, R.; Follo, M.Y.; Falà, F.; Cocco, L. Nuclear phosphoinositide specific phospholipase C (PI-PLC)-beta 1: A central intermediary in nuclear lipid-dependent signal transduction. Histol. Histopathol 2005, 20, 1251–1260. [Google Scholar]
- Bae, Y.S.; Cantley, L.G.; Chen, C.S.; Kim, S.R.; Kwon, K.S.; Rhee, S.G. Activation of phospholipase C-gamma by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem 1998, 273, 4465–4469. [Google Scholar]
- Huang, P.S.; Davis, L.; Huber, H.; Goodhart, P.J.; Wegrzyn, R.E.; Oliff, A.; Heimbrook, D.C. An SH3 domain is required for the mitogenic activity of microinjected phospholipase C-gamma 1. FEBS Lett 1995, 358, 287–292. [Google Scholar]
- Smith, M.R.; Ryu, S.H.; Suh, P.G.; Rhee, S.G.; Kung, H.F. S-phase induction and transformation of quiescent NIH 3T3 cells by microinjection of phospholipase C. Proc. Natl. Acad. Sci. USA 1989, 86, 3659–3663. [Google Scholar]
- Wang, Z.; Glück, S.; Zhang, L.; Moran, M.F. Requirement for phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol. Cell. Biol 1998, 18, 590–597. [Google Scholar]
- Cuvillier, O. Sphingosine in apoptosis signaling. Biochim. Biophys. Acta 2002, 1585, 153–162. [Google Scholar]
- Park, D.; Jhon, D.Y.; Kriz, R.; Knopf, J.; Rhee, S.G. Cloning, sequencing, expression, and Gq-independent activation of phospholipase C-beta 2. J. Biol. Chem 1992, 267, 16048–16055. [Google Scholar]
- Homma, Y.; Takenawa, T.; Emori, Y.; Sorimachi, H.; Suzuki, K. Tissue- and cell type-specific expression of mRNAs for four types of inositol phospholipid-specific phospholipase C. Biochem. Biophys. Res. Commun 1989, 164, 406–412. [Google Scholar]
- Sun, L.; Mao, G.; Kunapuli, S.P.; Dhanasekaran, D.N.; Rao, A.K. Alternative splice variants of phospholipase C-beta2 are expressed in platelets: Effect on Galphaq-dependent activation and localization. Platelets 2007, 18, 217–223. [Google Scholar]
- Jhon, D.Y.; Lee, H.H.; Park, D.; Lee, C.W.; Lee, K.H.; Yoo, O.J.; Rhee, S.G. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-beta 3. J. Biol. Chem 1993, 268, 6654–6661. [Google Scholar]
- Adamski, F.M.; Timms, K.M.; Shieh, B.H. A unique isoform of phospholipase Cbeta4 highly expressed in the cerebellum and eye. Biochim. Biophys. Acta 1999, 1444, 55–60. [Google Scholar]
- Mizuguchi, M.; Yamada, M.; Kim, S.U.; Rhee, S.G. Phospholipase C isozymes in neurons and glial cells in culture: An immunocytochemical and immunochemical study. Brain Res 1991, 548, 35–40. [Google Scholar]
- Lin, F.G.; Cheng, H.F.; Lee, I.F.; Kao, H.J.; Loh, S.H.; Lee, W.H. Downregulation of phospholipase C delta3 by cAMP and calcium. Biochem. Biophys. Res. Commun 2001, 286, 274–280. [Google Scholar]
- Lee, S.B.; Rhee, S.G. Molecular cloning, splice variants, expression, and purification of phospholipase C-delta 4. J. Biol. Chem 1996, 271, 25–31. [Google Scholar]
- Nameroff, M.; Trotter, J.A.; Keller, J.M.; Munar, E. Inhibition of cellular differentiation by phospholipase C.I. Effects of the enzyme on myogenesis and chondrogenesis in vitro. J. Cell. Biol 1973, 58, 107–118. [Google Scholar]
- Das, P.; Schurman, D.J.; Smith, R.L. Nitric oxide and G proteins mediate the response of bovine articular chondrocytes to fluid-induced shear. J. Orthop. Res 1997, 15, 87–93. [Google Scholar]
- D’Andrea, P.; Calabrese, A.; Capozzi, I.; Grandolfo, M.; Tonon, R.; Vittur, F. Intercellular Ca2+ waves in mechanically stimulated articular chondrocytes. Biorheology 2000, 37, 75–83. [Google Scholar]
- Nong, L.; Yin, G.; Ren, K.; Tang, J.; Fan, W. Periodic mechanical stress enhances rat chondrocyte area expansion and migration through Src-PLCgamma1-ERK1/2 signaling. Eur. J. Cell. Biol 2010, 89, 705–711. [Google Scholar]
- Rhee, S.G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem 2001, 70, 281–312. [Google Scholar]
- Bleasdale, J.E.; Bundy, G.L.; Bunting, S.; Fitzpatrick, F.A.; Huff, R.M.; Sun, F.F.; Pike, J.E. Inhibition of phospholipase C dependent processes by U-73, 122. Adv. Prostaglandin Thromboxane Leukotriene Res 1989, 19, 590–593. [Google Scholar]
- Poiraudeau, S.; Lieberherr, M.; Kergosie, N.; Corvol, M.T. Different mechanisms are involved in intracellular calcium increase by insulin-like growth factors 1 and 2 in articular chondrocytes: Voltage-gated calcium channels, and/or phospholipase C coupled to a pertussis-sensitive G-protein. J. Cell. Biochem 1997, 64, 414–422. [Google Scholar]
- Grandolfo, M.; Calabrese, A.; D’Andrea, P. Mechanism of mechanically induced intercellular calcium waves in rabbit articular chondrocytes and in HIG-82 synovial cells. J. Bone Miner. Res 1998, 13, 443–453. [Google Scholar]
- Capozzi, I.; Tonon, R.; D’andrea, P. Ca2+-sensitive phosphoinositide hydrolysis is activated in synovial cells but not in articular chondrocytes. Biochem. J 1999, 344, 545–553. [Google Scholar]
- Boyan, B.D.; Sylvia, V.L.; Dean, D.D.; Schwartz, Z. Membrane mediated signaling mechanisms are used differentially by metabolites of vitamin D(3) in musculoskeletal cells. Steroids 2002, 67, 421–427. [Google Scholar]
- Boyan, B.D.; Sylvia, V.L.; Dean, D.D.; Del Toro, F.; Schwartz, Z. Differential regulation of growth plate chondrocytes by 1alpha,25-(OH)2D3 and 24R,25-(OH)2D3 involves cell-maturation-specific membrane-receptor-activated phospholipid metabolism. Crit. Rev. Oral Biol. Med 2002, 13, 143–154. [Google Scholar]
- Schwartz, Z.; Graham, E.J.; Wang, L.; Lossdörfer, S.; Gay, I.; Johnson-Pais, T.L.; Carnes, D.L.; Sylvia, V.L.; Boyan, B.D. Phospholipase A2 activating protein (PLAA) is required for 1alpha,25(OH)2D3 signaling in growth plate chondrocytes. J. Cell. Physiol 2005, 203, 54–70. [Google Scholar]
- Boyan, B.D.; Wang, L.; Wong, K.L.; Jo, H.; Schwartz, Z. Plasma membrane requirements for 1alpha,25(OH)2D3 dependent PKC signaling in chondrocytes and osteoblasts. Steroids 2006, 71, 286–290. [Google Scholar]
- Sylvia, V.L.; Schwartz, Z.; Curry, D.B.; Chang, Z.; Dean, D.D.; Boyan, B.D. 1,25(OH)2D3 regulates protein kinase C activity through two phospholipid-dependent pathways involving phospholipase A2 and phospholipase C in growth zone chondrocytes. J. Bone Miner. Res 1998, 13, 559–569. [Google Scholar]
- Schwartz, Z.; Sylvia, V.L.; Luna, M.H.; DeVeau, P.; Whetstone, R.; Dean, D.D.; Boyan, B.D. The effect of 24R,25-(OH)(2)D(3) on protein kinase C activity in chondrocytes is mediated by phospholipase D whereas the effect of 1alpha,25-(OH)(2)D(3) is mediated by phospholipase C. Steroids 2001, 66, 683–694. [Google Scholar]
- Schwartz, Z.; Ehland, H.; Sylvia, V.L.; Larsson, D.; Hardin, R.R.; Bingham, V.; Lopez, D.; Dean, D.D.; Boyan, B.D. 1alpha,25-dihydroxyvitamin D(3) and 24R,25-dihydroxyvitamin D(3) modulate growth plate chondrocyte physiology via protein kinase C-dependent phosphorylation of extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase. Endocrinology 2002, 143, 2775–2786. [Google Scholar]
- Schwartz, Z.; Shaked, D.; Hardin, R.R.; Gruwell, S.; Dean, D.D.; Sylvia, V.L.; Boyan, B.D. 1alpha,25(OH)2D3 causes a rapid increase in phosphatidylinositol-specific PLC-beta activity via phospholipase A2-dependent production of lysophospholipid. Steroids 2003, 68, 423–437. [Google Scholar]
- Ekstein, J.; Nasatzky, E.; Boyan, B.D.; Ornoy, A.; Schwartz, Z. Growth-plate chondrocytes respond to 17beta-estradiol with sex-specific increases in IP3 and intracellular calcium ion signalling via a capacitative entry mechanism. Steroids 2005, 70, 775–786. [Google Scholar]
- Sylvia, V.L.; Boyan, B.D.; Dean, D.D.; Schwartz, Z. The membrane effects of 17beta-estradiol on chondrocyte phenotypic expression are mediated by activation of protein kinase C through phospholipase C and G-proteins. J. Steroid Biochem. Mol. Biol 2000, 73, 211–224. [Google Scholar]
- Sylvia, V.L.; Walton, J.; Lopez, D.; Dean, D.D.; Boyan, B.D.; Schwartz, Z. 17 beta-estradiol-BSA conjugates and 17 beta-estradiol regulate growth plate chondrocytes by common membrane associated mechanisms involving PKC dependent and independent signal transduction. J. Cell. Biochem 2001, 81, 413–429. [Google Scholar]
- Hurst-Kennedy, J.; Zhong, M.; Gupta, V.; Boyan, B.D.; Schwartz, Z. 24R,25-Dihydroxyvitamin D3, lysophosphatidic acid, and p53: A signaling axis in the inhibition of phosphate-induced chondrocyte apoptosis. J. Steroid Biochem. Mol. Biol 2010, 122, 264–271. [Google Scholar]
- Evans, J.F.; Shen, C.L.; Pollack, S.; Aloia, J.F.; Yeh, J.K. Adrenocorticotropin evokes transient elevations in intracellular free calcium ([Ca2+]i) and increases basal [Ca2+]i in resting chondrocytes through a phospholipase C-dependent mechanism. Endocrinology 2005, 146, 3123–3132. [Google Scholar]
- Riggs, B.L.; Khosla, S.; Melton, L.J. Sex steroids and the construction and conservation of the adult skeleton. Endocr. Rev 2002, 23, 279–302. [Google Scholar]
- ElBaradie, K.; Wang, Y.; Boyan, B.D.; Schwartz, Z. Rapid membrane responses to dihydrotestosterone are sex dependent in growth plate chondrocytes. J. Steroid Biochem. Mol. Biol 2012, 132, 15–23. [Google Scholar]
- Chao, P.Z.; Hsieh, M.S.; Cheng, C.W.; Lin, Y.F.; Chen, C.H. Regulation of MMP-3 expression and secretion by the chemokine eotaxin-1 in human chondrocytes. J. Biomed. Sci 2011, 18, 86. [Google Scholar]
- Harada, D.; Yamanaka, Y.; Ueda, K.; Nishimura, R.; Morishima, T.; Seino, Y.; Tanaka, H. Sustained phosphorylation of mutated FGFR3 is a crucial feature of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCgamma-activated STAT1. Bone 2007, 41, 273–281. [Google Scholar]
- Suzuki, Y.; Hruska, K.A.; Reid, I.; Alvarez, U.M.; Avioli, L.V. Characterization of phospholipase C activity of the plasma membrane and cytosol of an osteoblast-like cell line. Am. J. Med. Sci 1989, 297, 135–144. [Google Scholar]
- Hoberg, M.; Gratz, H.H.; Noll, M.; Jones, D.B. Mechanosensitivity of human osteosarcoma cells and phospholipase C beta2 expression. Biochem. Biophys. Res. Commun 2005, 333, 142–149. [Google Scholar]
- Bai, X.C.; Lu, D.; Bai, J.; Zheng, H.; Ke, Z.Y.; Li, X.M.; Luo, S.Q. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun 2004, 314, 197–207. [Google Scholar]
- Godwin, S.L.; Soltoff, S.P. Calcium-sensing receptor-mediated activation of phospholipase C-gamma1 is downstream of phospholipase C-beta and protein kinase C in MC3T3-E1 osteoblasts. Bone 2002, 30, 559–566. [Google Scholar]
- Takuwa, Y.; Ohue, Y.; Takuwa, N.; Yamashita, K. Endothelin-1 activates phospholipase C and mobilizes Ca2+ from extra- and intracellular pools in osteoblastic cells. Am. J. Physiol 1989, 257, E797–E803. [Google Scholar]
- Takuwa, Y.; Masaki, T.; Yamashita, K. The effects of the endothelin family peptides on cultured osteoblastic cells from rat calvariae. Biochem. Biophys. Res. Commun 1990, 170, 998–1005. [Google Scholar]
- Lam, H.C.; Lee, J.K.; Lai, K.H. Detection and characterization of endothelin in transformed human osteoblast cell culture medium. Endocrine 2000, 12, 77–80. [Google Scholar]
- Masukawa, H.; Miura, Y.; Sato, I.; Oiso, Y.; Suzuki, A. Stimulatory effect of endothelin-1 on Na-dependent phosphate transport and its signaling mechanism in osteoblast-like cells. J. Cell. Biochem 2001, 83, 47–55. [Google Scholar]
- Zach, D.; Windischhofer, W.; Leis, H.J. Endothelin- and sarafotoxin-induced receptor-mediated calcium mobilization in a clonal murine osteoblast-like cell line, MC3T3-E1/B. Bone 2001, 28, 595–602. [Google Scholar]
- Suzuki, A.; Palmer, G.; Bonjour, J.P.; Caverzasio, J. Stimulation of sodium-dependent phosphate transport and signaling mechanisms induced by basic fibroblast growth factor in MC3T3-E1 osteoblast-like cells. J. Bone Miner. Res 2000, 15, 95–102. [Google Scholar]
- Tang, C.H.; Yang, R.S.; Chen, Y.F.; Fu, W.M. Basic fibroblast growth factor stimulates fibronectin expression through phospholipase C gamma, protein kinase C alpha, c-Src, NF-kappaB, and p300 pathway in osteoblasts. J. Cell. Physiol 2007, 211, 45–55. [Google Scholar]
- Zhen, X.; Bonjour, J.P.; Caverzasio, J. Platelet-derived growth factor stimulates sodium-dependent Pi transport in osteoblastic cells via phospholipase Cgamma and phosphatidylinositol 3′-kinase. J. Bone Miner. Res 1997, 12, 36–44. [Google Scholar]
- Godwin, S.L.; Soltoff, S.P. Extracellular calcium and platelet-derived growth factor promote receptor-mediated chemotaxis in osteoblasts through different signaling pathways. J. Biol. Chem 1997, 272, 11307–11312. [Google Scholar]
- Moenning, A.; Jäger, R.; Egert, A.; Kress, W.; Wardelmann, E.; Schorle, H. Sustained platelet-derived growth factor receptor alpha signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-gamma pathway. Mol. Cell. Biol 2009, 29, 881–891. [Google Scholar]
- Swarthout, J.T.; Doggett, T.A.; Lemker, J.L.; Partridge, N.C. Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C-dependent. J. Biol. Chem 2001, 276, 7586–7592. [Google Scholar]
- Hömme, M.; Schmitt, C.P.; Himmele, R.; Hoffmann, G.F.; Mehls, O.; Schaefer, F. Vitamin D and dexamethasone inversely regulate parathyroid hormone-induced regulator of G protein signaling-2 expression in osteoblast-like cells. Endocrinology 2003, 144, 2496–2504. [Google Scholar]
- Hilal, G.; Massicotte, F.; Martel-Pelletier, J.; Fernandes, J.C.; Pelletier, J.P.; Lajeunesse, D. Endogenous prostaglandin E2 and insulin-like growth factor 1 can modulate the levels of parathyroid hormone receptor in human osteoarthritic osteoblasts. J. Bone Miner. Res 2001, 16, 713–721. [Google Scholar]
- Reeve, J.; Zanelli, J.M. Parathyroid hormone and bone. Clin. Sci 1986, 71, 231–238. [Google Scholar]
- Wu, Y.; Kumar, R. Parathyroid hormone regulates transforming growth factor beta1 and beta2 synthesis in osteoblasts via divergent signaling pathways. J. Bone Miner. Res 2000, 15, 879–884. [Google Scholar]
- Erclik, M.S.; Mitchell, J. The role of protein kinase C-delta in PTH stimulation of IGF-binding protein-5 mRNA in UMR-106–01 cells. Am. J. Physiol. Endocrinol. Metab 2002, 282, E534–E541. [Google Scholar]
- Cheung, R.; Erclik, M.S.; Mitchell, J. Increased expression of G11alpha in osteoblastic cells enhances parathyroid hormone activation of phospholipase C and AP-1 regulation of matrix metalloproteinase-13 mRNA. J. Cell. Physiol 2005, 204, 336–343. [Google Scholar]
- Imamura, Y.; Kozawa, O.; Suzuki, A.; Watanabe, Y.; Saito, H.; Oiso, Y. Mechanism of phospholipase D activation induced by prostaglandin D2 in osteoblast-like cells: Function of Ca2+/calmodulin. Cell. Signal 1995, 7, 45–51. [Google Scholar]
- Babich, M.; King, K.L.; Nissenson, R.A. Thrombin stimulates inositol phosphate production and intracellular free calcium by a pertussis toxin-insensitive mechanism in osteosarcoma cells. Endocrinology 1990, 126, 948–954. [Google Scholar]
- Kawase, T.; Suzuki, A. Initial responses of a clonal osteoblast-like cell line, MOB 3–4, to phosphatidic acid in vitro. Bone Miner 1990, 10, 61–70. [Google Scholar]
- Tokuda, H.; Kozawa, O.; Niwa, M.; Matsuno, H.; Kato, K.; Uematsu, T. Mechanism of prostaglandin E2-stimulated heat shock protein 27 induction in osteoblast-like MC3T3-E1 cells. J. Endocrinol 2002, 172, 271–281. [Google Scholar]
- Kondo, A.; Togari, A. Activation of osteoblastic functions by a mediator of pain, bradykinin. Biochem. Pharmacol 2004, 68, 1423–1431. [Google Scholar]
- Toriyama, K.; Morita, I.; Seyama, Y.; Yamashita, S.; Murota, S. Prostaglandin E2 evokes intracellular calcium rise in mouse osteoblastic cell line, MC3T3E-1. Eicosanoids 1990, 3, 157–160. [Google Scholar]
- Tomura, H.; Wang, J.Q.; Liu, J.P.; Komachi, M.; Damirin, A.; Mogi, C.; Tobo, M.; Nochi, H.; Tamoto, K.; Im, D.S.; et al. Cyclooxygenase-2 expression and prostaglandin E2 production in response to acidic pH through OGR1 in a human osteoblastic cell line. J. Bone Miner. Res 2008, 23, 1129–1139. [Google Scholar]
- Hakeda, Y.; Shiokawa, M.; Mano, H.; Kameda, T.; Raisz, L.G.; Kumegawa, M. Prostaglandin F2alpha stimulates tyrosine phosphorylation and mitogen-activated protein kinase in osteoblastic MC3T3-E1 cells via protein kinase C activation. Endocrinology 1997, 138, 1821–1828. [Google Scholar]
- Quarles, L.D.; Haupt, D.M.; Davidai, G.; Middleton, J.P. Prostaglandin F2 alpha-induced mitogenesis in MC3T3-E1 osteoblasts: Role of protein kinase-C-mediated tyrosine phosphorylation. Endocrinology 1993, 132, 1505–1513. [Google Scholar]
- Hatakeyama, D.; Kozawa, O.; Otsuka, T.; Shibata, T.; Uematsu, T. Zinc suppresses IL-6 synthesis by prostaglandin F2alpha in osteoblasts: Inhibition of phospholipase C and phospholipase D. J. Cell. Biochem 2002, 85, 621–628. [Google Scholar]
- Kozawa, O.; Tokuda, H.; Suzuki, A.; Kotoyori, J.; Ito, Y.; Oiso, Y. Effect of glucocorticoid on prostaglandin F2 alpha-induced prostaglandin E2 synthesis in osteoblast-like cells: Inhibition of phosphoinositide hydrolysis by phospholipase C as well as phospholipase A2. Eur. J. Endocrinol 1994, 131, 510–515. [Google Scholar]
- Kozawa, O.; Tokuda, H.; Matsuno, H.; Uematsu, T. Activation of mitogen-activated protein kinase is involved in sphingosine 1-phosphate-stimulated interleukin-6 synthesis in osteoblasts. FEBS Lett 1997, 418, 149–151. [Google Scholar]
- Kozawa, O.; Kawamura, H.; Matsuno, H.; Uematsu, T. p38 MAP kinase is involved in the signalling of sphingosine in osteoblasts: Sphingosine inhibits prostaglandin F(2alpha)-induced phosphoinositide hydrolysis. Cell. Signal 2000, 12, 447–450. [Google Scholar]
- Le Mellay, V.; Grosse, B.; Lieberherr, M. Phospholipase C beta and membrane action of calcitriol and estradiol. J. Biol. Chem 1997, 272, 11902–11907. [Google Scholar]
- Le Mellay, V.; Lasmoles, F.; Lieberherr, M. Galpha(q/11) and gbetagamma proteins and membrane signaling of calcitriol and estradiol. J. Cell. Biochem 1999, 75, 138–146. [Google Scholar]
- Schwartz, Z.; Lohmann, C.H.; Vocke, A.K.; Sylvia, V.L.; Cochran, D.L.; Dean, D.D.; Boyan, B.D. Osteoblast response to titanium surface roughness and 1alpha,25-(OH)(2)D(3) is mediated through the mitogen-activated protein kinase (MAPK) pathway. J. Biomed. Mater. Res 2001, 56, 417–426. [Google Scholar]
- Marmiroli, S.; Ognibene, A.; Bavelloni, A.; Cinti, C.; Cocco, L.; Maraldi, N.M. Interleukin 1 alpha stimulates nuclear phospholipase C in human osteosarcoma SaOS-2 cells. J. Biol. Chem 1994, 269, 13–16. [Google Scholar]
- Zini, N.; Sabatelli, P.; Faenza, I.; Ognibene, A.; Maraldi, N.M. Interleukin-1 alpha induces variations of the intranuclear amount of phosphatidylinositol 4,5-bisphosphate and phospholipase C beta 1 in human osteosarcoma Saos-2 cells. Histochem. J 1996, 28, 495–504. [Google Scholar]
- Rodan, S.B.; Wesolowski, G.; Chin, J.; Limjuco, G.A.; Schmidt, J.A.; Rodan, G.A. IL-1 binds to high affinity receptors on human osteosarcoma cells and potentiates prostaglandin E2 stimulation of cAMP production. J. Immunol 1990, 145, 1231–1237. [Google Scholar]
- Martelli, A.M.; Billi, A.M.; Manzoli, L.; Faenza, I.; Aluigi, M.; Falconi, M.; De Pol, A.; Gilmour, R.S.; Cocco, L. Insulin selectively stimulates nuclear phosphoinositide-specific phospholipase C (PI-PLC) beta1 activity through a mitogen-activated protein (MAP) kinase-dependent serine phosphorylation. FEBS Lett 2000, 486, 230–236. [Google Scholar]
- Piecyk, M.; Anderson, P. Signal transduction in rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol 2001, 15, 789–803. [Google Scholar]
- Lax, A.J.; Grigoriadis, A.E. Pasteurella multocida toxin: The mitogenic toxin that stimulates signalling cascades to regulate growth and differentiation. Int. J. Med. Microbiol 2001, 291, 261–268. [Google Scholar]
- Chang, H.T.; Hsu, S.S.; Chou, C.T.; Cheng, J.S.; Wang, J.L.; Lin, K.L.; Fang, Y.C.; Chen, W.C.; Chien, J.M.; Lu, T.; et al. Effect of thymol on Ca2+ homeostasis and viability in MG63 human osteosarcoma cells. Pharmacology 2011, 88, 201–212. [Google Scholar]
- Shin, M.K.; Jang, Y.H.; Yoo, H.J.; Kang, D.W.; Park, M.H.; Kim, M.K.; Song, J.H.; Kim, S.D.; Min, G.; You, H.K.; et al. N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow. J. Biol. Chem 2011, 286, 17133–17143. [Google Scholar]
- Aki, Y.; Kondo, A.; Nakamura, H.; Togari, A. Lysophosphatidic acid-stimulated interleukin-6 and -8 synthesis through LPA1 receptors on human osteoblasts. Arch. Oral Biol 2008, 53, 207–213. [Google Scholar]
- Katz, S.; Boland, R.; Santillán, G. Modulation of ERK 1/2 and p38 MAPK signaling pathways by ATP in osteoblasts: Involvement of mechanical stress-activated calcium influx, PKC and Src activation. Int. J. Biochem. Cell. Biol 2006, 38, 2082–2091. [Google Scholar]
- Nishii, N.; Nejime, N.; Yamauchi, C.; Yanai, N.; Shinozuka, K.; Nakabayashi, T. Effects of ATP on the intracellular calcium level in the osteoblastic TBR31–2 cell line. Biol. Pharm. Bull 2009, 32, 18–23. [Google Scholar]
- Nakamura, I.; Lipfert, L.; Rodan, G.A.; Duong, L.T. Convergence of alpha(v)beta(3) integrin- and macrophage colony stimulating factor-mediated signals on phospholipase Cgamma in prefusion osteoclasts. J. Cell. Biol 2001, 152, 361–373. [Google Scholar]
- Epple, H.; Cremasco, V.; Zhang, K.; Mao, D.; Longmore, G.D.; Faccio, R. Phospholipase Cgamma2 modulates integrin signaling in the osteoclast by affecting the localization and activation of Src kinase. Mol. Cell. Biol 2008, 28, 3610–3622. [Google Scholar]
- Lucht, U. Effects of calcitonin on osteoclasts in vivo. An ultrastructural and histochemical study. Zeitschrift Zellforsch. Mikrosk. Anat 1973, 145, 75–87. [Google Scholar]
- Baron, R.; Neff, L.; Brown, W.; Louvard, D.; Courtoy, P.J. Selective internalization of the apical plasma membrane and rapid redistribution of lysosomal enzymes and mannose 6-phosphate receptors during osteoclast inactivation by calcitonin. J. Cell. Sci 1990, 97, 439–447. [Google Scholar]
- Henriksen, K.; Bay-Jensen, A.C.; Christiansen, C.; Karsdal, M.A. Oral salmon calcitonin— Pharmacology in osteoporosis. Expert Opin. Biol. Ther 2010, 10, 1617–1629. [Google Scholar]
- Stenbeck, G.; Lawrence, K.M.; Albert, A.P. Hormone-stimulated modulation of endocytic trafficking in osteoclasts. Front. Endocrinol 2012, 3, 103. [Google Scholar]
- Silver, I.A.; Murrills, R.J.; Etherington, D.J. Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp. Cell. Res 1988, 175, 266–276. [Google Scholar]
- Malgaroli, A.; Meldolesi, J.; Zallone, A.Z.; Teti, A. Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. J. Biol. Chem 1989, 264, 14342–14347. [Google Scholar]
- Miyauchi, A.; Hruska, K.A.; Greenfield, E.M.; Duncan, R.; Alvarez, J.; Barattolo, R.; Colucci, S.; Zambonin-Zallone, A.; Teitelbaum, S.L.; Teti, A. Osteoclast cytosolic calcium, regulated by voltage-gated calcium channels and extracellular calcium, controls podosome assembly and bone resorption. J. Cell. Biol 1990, 111, 2543–2552. [Google Scholar]
- Zaidi, M.; Moonga, B.S.; Adebanjo, O.A. Novel mechanisms of calcium handling by the osteoclast: A review-hypothesis. Proc. Assoc. Am. Physicians 1999, 111, 319–327. [Google Scholar]
- Lorget, F.; Kamel, S.; Mentaverri, R.; Wattel, A.; Naassila, M.; Maamer, M.; Brazier, M. High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem. Biophys. Res. Commun 2000, 268, 899–903. [Google Scholar]
- Chamoux, E.; Bisson, M.; Payet, M.D.; Roux, S. TRPV-5 mediates a receptor activator of NF-kappaB (RANK) ligand-induced increase in cytosolic Ca2+ in human osteoclasts and down-regulates bone resorption. J. Biol. Chem 2010, 285, 25354–25362. [Google Scholar]
- Kajiya, H.; Okamoto, F.; Nemoto, T.; Kimachi, K.; Toh-Goto, K.; Nakayana, S.; Okabe, K. RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations. Cell Calcium 2010, 48, 260–269. [Google Scholar]
- Mentaverri, R.; Yano, S.; Chattopadhyay, N.; Petit, L.; Kifor, O.; Kamel, S.; Terwilliger, E.F.; Brazier, M.; Brown, E.M. The calcium sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J 2006, 20, 2562–2564. [Google Scholar]
- Hurtel-Lemaire, A.S.; Mentaverri, R.; Caudrillier, A.; Cournarie, F.; Wattel, A.; Kamel, S.; Terwilliger, E.F.; Brown, E.M.; Brazier, M. The calcium-sensing receptor is involved in strontium ranelate-induced osteoclast apoptosis. New insights into the associated signaling pathways. J. Biol. Chem 2009, 284, 575–584. [Google Scholar]
- Seuwen, K.; Boddeke, H.G.; Migliaccio, S.; Perez, M.; Taranta, A.; Teti, A. A novel calcium sensor stimulating inositol phosphate formation and [Ca2+]i signaling expressed by GCT23 osteoclast-like cells. Proc. Assoc. Am. Physicians 1999, 111, 70–81. [Google Scholar]
- Yoon, S.H.; Lee, Y.; Kim, H.J.; Lee, Z.H.; Hyung, S.W.; Lee, S.W.; Kim, H.H. Lyn inhibits osteoclast differentiation by interfering with PLCgamma1-mediated Ca2+ signaling. FEBS Lett 2009, 583, 1164–1170. [Google Scholar]
- Sakai, H.; Moriura, Y.; Notomi, T.; Kawawaki, J.; Ohnishi, K.; Kuno, M. Phospholipase C-dependent Ca2+-sensing pathways leading to endocytosis and inhibition of the plasma membrane vacuolar H+-ATPase in osteoclasts. Am. J. Physiol. Cell. Physiol 2010, 299, C570–C578. [Google Scholar]
- Kim, T.; Kim, K.; Lee, S.H.; So, H.S.; Lee, J.; Kim, N.; Choi, Y. Identification of LRRc17 as a negative regulator of receptor activator of NF-kappaB ligand (RANKL)-induced osteoclast differentiation. J. Biol. Chem 2009, 284, 15308–15316. [Google Scholar]
- Bennett, B.D.; Alvarez, U.; Hruska, K.A. Receptor-operated osteoclast calcium sensing. Endocrinology 2001, 142, 1968–1974. [Google Scholar]
- Weidema, A.F.; Dixon, S.J.; Sims, S.M. Activation of P2Y but not P2X(4) nucleotide receptors causes elevation of [Ca2+]i in mammalian osteoclasts. Am. J. Physiol. Cell. Physiol 2001, 280, C1531–C1539. [Google Scholar]
- Komarova, S.V.; Pilkington, M.F.; Weidema, A.F.; Dixon, S.J.; Sims, S.M. RANK ligand-induced elevation of cytosolic Ca2+ accelerates nuclear translocation of nuclear factor kappa B in osteoclasts. J. Biol. Chem 2003, 278, 8286–8293. [Google Scholar]
- Komarova, S.V.; Pereverzev, A.; Shum, J.W.; Sims, S.M.; Dixon, S.J. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc. Natl. Acad. Sci. USA 2005, 102, 2643–2648. [Google Scholar]
- Yang, S.; Li, Y.P. RGS12 is essential for RANKL-evoked signaling for terminal differentiation of osteoclasts in vitro. J. Bone Miner. Res 2007, 22, 45–54. [Google Scholar]
- Mao, D.; Epple, H.; Uthgenannt, B.; Novack, D.V.; Faccio, R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J. Clin. Invest 2006, 116, 2869–2879. [Google Scholar]
- Stein, N.C.; Kreutzmann, C.; Zimmermann, S.P.; Niebergall, U.; Hellmeyer, L.; Goettsch, C.; Schoppet, M.; Hofbauer, L.C. Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteoprotegerin by human endothelial cells through the STAT6 pathway. J. Bone Miner. Res 2008, 23, 750–758. [Google Scholar]
- Shinohara, M.; Koga, T.; Okamoto, K.; Sakaguchi, S.; Arai, K.; Yasuda, H.; Takai, T.; Kodama, T.; Morio, T.; Geha, R.S.; et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008, 132, 794–806. [Google Scholar]
- Taguchi, Y.; Gohda, J.; Koga, T.; Takayanagi, H.; Inoue, J. A unique domain in RANK is required for Gab2 and PLCgamma2 binding to establish osteoclastogenic signals. Genes Cells 2009, 14, 1331–1345. [Google Scholar]
- Kim, K.; Kim, J.H.; Moon, J.B.; Lee, J.; Kwak, H.B.; Park, Y.W.; Kim, N. The transmembrane adaptor protein, linker for activation of T cells (LAT), regulates RANKL-induced osteoclast differentiation. Mol. Cells 2012, 33, 401–406. [Google Scholar]
- Kim, M.S.; Yang, Y.M.; Son, A.; Tian, Y.S.; Lee, S.I.; Kang, S.W.; Muallem, S.; Shin, D.M. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J. Biol. Chem 2010, 285, 6913–6921. [Google Scholar]
- Lietman, S.A.; Yin, L.; Levine, M.A. SH3BP2 mutations potentiate osteoclastogenesis via PLCγ. J. Orthop. Res 2010, 28, 1425–1430. [Google Scholar]
- Kawamoto, T.; Fan, C.; Gaivin, R.J.; Levine, M.A.; Lietman, S.A. Decreased SH3BP2 inhibits osteoclast differentiation and function. J. Orthop. Res 2011, 29, 1521–1527. [Google Scholar]
- Guo, J.; Liu, M.; Yang, D.; Bouxsein, M.L.; Thomas, C.C.; Schipani, E.; Bringhurst, F.R.; Kronenberg, H.M. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology 2010, 151, 3502–3513. [Google Scholar]
- Liu, B.Y.; Wu, P.W.; Bringhurst, F.R.; Wang, J.T. Estrogen inhibition of PTH-stimulated osteoclast formation and attachment in vitro: Involvement of both PKA and PKC. Endocrinology 2002, 143, 627–635. [Google Scholar]
- Baines, R.J.; Brown, C.; Ng, L.L.; Boarder, M.R. Angiotensin II-stimulated phospholipase C responses of two vascular smooth muscle-derived cell lines. Role of cyclic GMP. Hypertension 1996, 28, 772–778. [Google Scholar]
- Boss, V.; Abbott, K.L.; Wang, X.F.; Pavlath, G.K.; Murphy, T.J. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J. Biol. Chem 1998, 273, 19664–19671. [Google Scholar]
- Paulhe, F.; Bogyo, A.; Chap, H.; Perret, B.; Racaud-Sultan, C. Vascular smooth muscle cell spreading onto fibrinogen is regulated by calpains and phospholipase C. Biochem. Biophys. Res. Commun 2001, 288, 875–881. [Google Scholar]
- Kelly, J.; Brazil, D.; Clyne, C.; McHale, N.G.; Gierschik, P.; Keenan, A.K. Evidence for the presence of G-proteins, adenylyl cyclase and phospholipase C activities in lymphatic smooth muscle cell membranes. Cell. Signal 1996, 8, 425–432. [Google Scholar]
- Grillone, L.R.; Clark, M.A.; Godfrey, R.W.; Stassen, F.; Crooke, S.T. Vasopressin induces V1 receptors to activate phosphatidylinositol- and phosphatidylcholine-specific phospholipase C and stimulates the release of arachidonic acid by at least two pathways in the smooth muscle cell line, A-10. J. Biol. Chem 1988, 263, 2658–2663. [Google Scholar]
- Liu, B.; Itoh, H.; Louie, O.; Kubota, K.; Kent, K.C. The role of phospholipase C and phosphatidylinositol 3-kinase in vascular smooth muscle cell migration and proliferation. J. Surg. Res 2004, 120, 256–265. [Google Scholar]
- Roztocil, E.; Nicholl, S.M.; Davies, M.G. Sphingosine-1-phosphate-induced oxygen free radical generation in smooth muscle cell migration requires Galpha12/13 protein-mediated phospholipase C activation. J. Vasc. Surg 2007, 46, 1253–1259. [Google Scholar]
- Shibukawa, Y.; Suzuki, T. Ca2+ signaling mediated by IP3-dependent Ca2+ releasing and store-operated Ca2+ channels in rat odontoblasts. J. Bone Miner. Res 2003, 18, 30–38. [Google Scholar]
- Sooampon, S.; Manokawinchoke, J.; Pavasant, P. Transient receptor potential vanilloid-1 regulates osteoprotegerin/RANKL homeostasis in human periodontal ligament cells. J. Periodontal Res. 2012. [Google Scholar] [CrossRef]
- Osathanon, T.; Nowwarote, N.; Pavasant, P. Basic fibroblast growth factor inhibits mineralization but induces neuronal differentiation by human dental pulp stem cells through a FGFR and PLCγ signaling pathway. J. Cell. Biochem 2011, 112, 1807–1816. [Google Scholar]
- Nakamura, Y.; Fukami, K. Roles of phospholipase C isozymes in organogenesis and embryonic development. Physiology 2009, 24, 332–341. [Google Scholar]
- Liao, H.J.; Kume, T.; McKay, C.; Xu, M.J.; Ihle, J.N.; Carpenter, G. Absence of erythrogenesis and vasculogenesis in Plcg1-deficient mice. J. Biol. Chem 2002, 277, 9335–9341. [Google Scholar]
- Nakamura, Y.; Hamada, Y.; Fujiwara, T.; Enomoto, H.; Hiroe, T.; Tanaka, S.; Nose, M.; Nakahara, M.; Yoshida, N.; Takenawa, T.; et al. Phospholipase C-delta1 and -delta3 are essential in the trophoblast for placental development. Mol. Cell. Biol 2005, 25, 10979–10988. [Google Scholar]
- Bhattacharya, R.; Kwon, J.; Li, X.; Wang, E.; Patra, S.; Bida, J.P.; Bajzer, Z.; Claesson-Welsh, L.; Mukhopadhyay, D. Distinct role of PLCbeta3 in VEGF-mediated directional migration and vascular sprouting. J. Cell. Sci 2009, 122, 1025–1034. [Google Scholar]
- Shirane, M.; Sawa, H.; Kobayashi, Y.; Nakano, T.; Kitajima, K.; Shinkai, Y.; Nagashima, K.; Negishi, I. Deficiency of phospholipase C-gamma1 impairs renal development and hematopoiesis. Development 2001, 128, 5173–5180. [Google Scholar]
- Hashimoto, A.; Takeda, K.; Inaba, M.; Sekimata, M.; Kaisho, T.; Ikehara, S.; Homma, Y.; Akira, S.; Kurosaki, T. Cutting edge: Essential role of phospholipase C-gamma 2 in B cell development and function. J. Immunol 2000, 165, 1738–1742. [Google Scholar]
- Tassi, I.; Presti, R.; Kim, S.; Yokoyama, W.M.; Gilfillan, S.; Colonna, M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. J. Immunol 2005, 175, 749–754. [Google Scholar]
- Tassi, I.; Cella, M.; Castro, I.; Gilfillan, S.; Khan, W.N.; Colonna, M. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur. J. Immunol 2009, 39, 1369–1378. [Google Scholar]
- Cremasco, V.; Graham, D.B.; Novack, D.V.; Swat, W.; Faccio, R. Vav/Phospholipase Cgamma2-mediated control of a neutrophil-dependent murine model of rheumatoid arthritis. Arthritis. Rheum 2008, 58, 2712–2722. [Google Scholar]
- Cremasco, V.; Benasciutti, E.; Cella, M.; Kisseleva, M.; Croke, M.; Faccio, R. Phospholipase C gamma 2 is critical for development of a murine model of inflammatory arthritis by affecting actin dynamics in dendritic cells. PLoS One 2010, 5, e8909. [Google Scholar]
- Graham, D.B.; Robertson, C.M.; Bautista, J.; Mascarenhas, F.; Diacovo, M.J.; Montgrain, V.; Lam, S.K.; Cremasco, V.; Dunne, W.M.; Faccio, R.; et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J. Clin. Invest 2007, 117, 3445–3452. [Google Scholar]
- Zhang, K.; Kim, S.; Cremasco, V.; Hirbe, A.C.; Collins, L.; Piwnica-Worms, D.; Novack, D.V.; Weilbaecher, K.; Faccio, R. CD8+ T cells regulate bone tumor burden independent of osteoclast resorption. Cancer Res 2011, 71, 4799–4808. [Google Scholar]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem 2004, 279, 26475–26480. [Google Scholar]
- Chen, Y.; Wang, X.; Di, L.; Fu, G.; Bai, L.; Liu, J.; Feng, X.; McDonald, J.M.; Michalek, S.; He, Y.; et al. Phospholipase Cgamma2 mediates RANKL-stimulated lymph node organogenesis and osteoclastogenesis. J. Biol. Chem 2008, 283, 29593–29601. [Google Scholar]
- Kertész, Z.; Gyori, D.; Körmendi, S.; Fekete, T.; Kis-Tóth, K.; Jakus, Z.; Schett, G.; Rajnavölgyi, E.; Dobó-Nagy, C.; Mócsai, A. Phospholipase Cγ2 is required for basal but not oestrogen deficiency-induced bone resorption. Eur. J. Clin. Invest 2012, 42, 49–60. [Google Scholar]
- Kanematsu, T.; Takeya, H.; Watanabe, Y.; Ozaki, S.; Yoshida, M.; Koga, T.; Iwanaga, S.; Hirata, M. Putative inositol 1,4,5-trisphosphate binding proteins in rat brain cytosol. J. Biol. Chem 1992, 267, 6518–6525. [Google Scholar]
- Kanematsu, T.; Yoshimura, K.; Hidaka, K.; Takeuchi, H.; Katan, M.; Hirata, M. Domain organization of p130, PLC-related catalytically inactive protein, and structural basis for the lack of enzyme activity. Eur. J. Biochem 2000, 267, 2731–2737. [Google Scholar]
- Yoshimura, K.; Takeuchi, H.; Sato, O.; Hidaka, K.; Doira, N.; Terunuma, M.; Harada, K.; Ogawa, Y.; Ito, Y.; Kanematsu, T.; et al. Interaction of p130 with, and consequent inhibition of, the catalytic subunit of protein phosphatase 1alpha. J. Biol. Chem 2001, 276, 17908–17913. [Google Scholar]
- Kanematsu, T.; Yasunaga, A.; Mizoguchi, Y.; Kuratani, A.; Kittler, J.T.; Jovanovic, J.N.; Takenaka, K.; Nakayama, K.I.; Fukami, K.; Takenawa, T.; et al. Modulation of GABA(A) receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. J. Biol. Chem 2006, 281, 22180–22189. [Google Scholar]
- Fujii, M.; Kanematsu, T.; Ishibashi, H.; Fukami, K.; Takenawa, T.; Nakayama, K.I.; Moss, S.J.; Nabekura, J.; Hirata, M. Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of gamma-aminobutyric acid type A receptors. J. Biol. Chem 2010, 285, 4837–4846. [Google Scholar]
- Takeuchi, H.; Kanematsu, T.; Misumi, Y.; Yaakob, H.B.; Yagisawa, H.; Ikehara, Y.; Watanabe, Y.; Tan, Z.; Shears, S.B.; Hirata, M. Localization of a high-affinity inositol 1,4,5-trisphosphate/inositol 1,4,5,6-tetrakisphosphate binding domain to the pleckstrin homology module of a new 130 kDa protein: Characterization of the determinants of structural specificity. Biochem. J 1996, 318, 561–568. [Google Scholar]
- Mizokami, A.; Kanematsu, T.; Ishibashi, H.; Yamaguchi, T.; Tanida, I.; Takenaka, K.; Nakayama, K.I.; Fukami, K.; Takenawa, T.; Kominami, E.; et al. Phospholipase C-related inactive protein is involved in trafficking of gamma2 subunit-containing GABA(A) receptors to the cell surface. J. Neurosci 2007, 27, 1692–1701. [Google Scholar]
- Matsuda, M.; Tsutsumi, K.; Kanematsu, T.; Fukami, K.; Terada, Y.; Takenawa, T.; Nakayama, K.I.; Hirata, M. Involvement of phospholipase C-related inactive protein in the mouse reproductive system through the regulation of gonadotropin levels. Biol. Reprod 2009, 81, 681–689. [Google Scholar]
- Tsutsumi, K.; Matsuda, M.; Kotani, M.; Mizokami, A.; Murakami, A.; Takahashi, I.; Terada, Y.; Kanematsu, T.; Fukami, K.; Takenawa, T.; et al. Involvement of PRIP, phospholipase C-related, but catalytically inactive protein, in bone formation. J. Biol. Chem 2011, 286, 31032–31042. [Google Scholar]
- Gao, J.; Takeuchi, H.; Zhang, Z.; Fukuda, M.; Hirata, M. Phospholipase C-related but catalytically inactive protein (PRIP) modulates synaptosomal-associated protein 25 (SNAP-25) phosphorylation and exocytosis. J. Biol. Chem 2012, 287, 10565–10578. [Google Scholar]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett 2000, 476, 55–57. [Google Scholar]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar]
- Khavandgar, Z.; Poirier, C.; Clarke, C.J.; Li, J.; Wang, N.; McKee, M.D.; Hannun, Y.A.; Murshed, M. A cell-autonomous requirement for neutral sphingomyelinase 2 in bone mineralization. J. Cell. Biol 2011, 194, 277–289. [Google Scholar]
- Gilbert, S.J.; Blain, E.J.; Jones, P.; Duance, V.C.; Mason, D.J. Exogenous sphingomyelinase increases collagen and sulphated glycosaminoglycan production by primary articular chondrocytes: An in vitro study. Arthritis. Res. Ther 2006, 8, 89. [Google Scholar]
- Sabatini, M.; Rolland, G.; Léonce, S.; Thomas, M.; Lesur, C.; Pérez, V.; de Nanteuil, G.; Bonnet, J. Effects of ceramide on apoptosis, proteoglycan degradation, and matrix metalloproteinase expression in rabbit articular cartilage. Biochem. Biophys. Res. Commun 2000, 267, 438–444. [Google Scholar]
- Ehlert, K.; Frosch, M.; Fehse, N.; Zander, A.; Roth, J.; Vormoor, J. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 2007, 5, 15:1–15:7. [Google Scholar]
- Gilbert, S.J.; Blain, E.J.; Duance, V.C.; Mason, D.J. Sphingomyelinase decreases type II collagen expression in bovine articular cartilage chondrocytes via the ERK signaling pathway. Arthritis. Rheum 2008, 58, 209–220. [Google Scholar]
- Kitajima, I.; Soejima, Y.; Takasaki, I.; Beppu, H.; Tokioka, T.; Maruyama, I. Ceramide-induced nuclear translocation of NF-kappa B is a potential mediator of the apoptotic response to TNF-alpha in murine clonal osteoblasts. Bone 1996, 19, 263–270. [Google Scholar]
- Takeshita, A.; Watanabe, A.; Takada, Y.; Hanazawa, S. Selective stimulation by ceramide of the expression of the alpha isoform of retinoic acid and retinoid X receptors in osteoblastic cells. A role of sphingosine 1-phosphate-mediated AP-1 in the ligand-dependent transcriptional activity of these receptors. J. Biol. Chem 2000, 275, 32220–32226. [Google Scholar]
- Kozawa, O.; Hatakeyama, D.; Tokuda, H.; Oiso, Y.; Matsuno, H.; Uematsu, T. Sphingomyelinase amplifies BMP-4-induced osteocalcin synthesis in osteoblasts: Role of ceramide. Cell. Signal 2002, 14, 999–1004. [Google Scholar]
- Klein, B.Y.; Kerem, Z.; Rojansky, N. LDL induces Saos2 osteoblasts death via Akt pathways responsive to a neutral sphingomyelinase inhibitor. J. Cell. Biochem 2006, 98, 661–671. [Google Scholar]
- Hill, P.A.; Tumber, A. Ceramide-induced cell death/survival in murine osteoblasts. J. Endocrinol 2010, 206, 225–233. [Google Scholar]
- Tokuda, H.; Kozawa, O.; Harada, A.; Uematsu, T. Extracellular sphingomyelinase induces interleukin-6 synthesis in osteoblasts. J. Cell. Biochem 1999, 72, 262–268. [Google Scholar]
- Takeshita, A.; Shinoda, H.; Nakabayashi, Y.; Takano, A.; Matsumoto, K.; Suetsugu, M.; Miyazawa, K.; Tanaka, S.; Endo, H.; Ueyama, Y.; et al. Sphingosine 1-phosphate acts as a signal molecule in ceramide signal transduction of TNF-alpha-induced activator protein-1 in osteoblastic cell line MC3T3-E1 cells. J. Oral Sci 2005, 47, 43–51. [Google Scholar]
- Spiegel, S.; Merrill, A.H. Sphingolipid metabolism and cell growth regulation. FASEB J 1996, 10, 1388–1397. [Google Scholar]
- Bao, X.; Ogawa, T.; Se, S.; Akiyama, M.; Bahtiar, A.; Takeya, T.; Ishida-Kitagawa, N. Acid sphingomyelinase regulates osteoclastogenesis by modulating sphingosine kinases downstream of RANKL signaling. Biochem. Biophys. Res. Commun 2011, 405, 533–537. [Google Scholar]
- Takeda, H.; Ozaki, K.; Yasuda, H.; Ishida, M.; Kitano, S.; Hanazawa, S. Sphingomyelinase and ceramide inhibit formation of F-actin ring in and bone resorption by rabbit mature osteoclasts. FEBS Lett 1998, 422, 255–258. [Google Scholar]
- Aubin, I.; Adams, C.P.; Opsahl, S.; Septier, D.; Bishop, C.E.; Auge, N.; Salvayre, R.; Negre-Salvayre, A.; Goldberg, M.; Guénet, J.L.; et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat. Genet 2005, 37, 803–805. [Google Scholar]
- Stoffel, W.; Jenke, B.; Blöck, B.; Zumbansen, M.; Koebke, J. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc. Natl. Acad. Sci. USA 2005, 102, 4554–4559. [Google Scholar]
- Stoffel, W.; Jenke, B.; Holz, B.; Binczek, E.; Günter, R.H.; Knifka, J.; Koebke, J.; Niehoff, A. Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am. J. Pathol 2007, 171, 153–161. [Google Scholar]
- Thouverey, C.; Malinowska, A.; Balcerzak, M.; Strzelecka-Kiliszek, A.; Buchet, R.; Dadlez, M.; Pikula, S. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J. Proteomics 2011, 74, 1123–1134. [Google Scholar]
- Balcerzak, M.; Malinowska, A.; Thouverey, C.; Sekrecka, A.; Dadlez, M.; Buchet, R.; Pikula, S. Proteome analysis of matrix vesicles isolated from femurs of chicken embryo. Proteomics 2008, 8, 192–205. [Google Scholar]
- Ponting, C.P.; Kerr, I.D. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: Identification of duplicated repeats and potential active site residues. Protein Sci 1996, 5, 914–922. [Google Scholar]
- Lavieri, R.; Scott, S.A.; Lewis, J.A.; Selvy, P.E.; Armstrong, M.D.; Alex Brown, H.; Lindsley, C.W. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II. Identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett 2009, 19, 2240–2243. [Google Scholar]
- Stace, C.L.; Ktistakis, N.T. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 2006, 1761, 913–926. [Google Scholar]
- Exton, J.H. Phosphatidylcholine breakdown and signal transduction. Biochim. Biophys. Acta 1994, 1212, 26–42. [Google Scholar]
- Exton, J.H. Phospholipase D: Enzymology, mechanisms of regulation, and function. Physiol. Rev 1997, 77, 303–320. [Google Scholar]
- Kanoh, H.; Sakane, F.; Imai, S.; Wada, I. Diacylglycerol kinase and phosphatidic acid phosphatase—Enzymes metabolizing lipid second messengers. Cell. Signal 1993, 5, 495–503. [Google Scholar]
- Martin, T.W. Formation of diacylglycerol by a phospholipase d-phosphatidate phosphatase pathway specific for phosphatidylcholine in endothelial cells. Biochim. Biophys. Acta 1988, 962, 282–296. [Google Scholar]
- Mackay, H.J.; Twelves, C.J. Targeting the protein kinase C family: Are we there yet? Nat. Rev. Cancer 2007, 7, 554–562. [Google Scholar]
- Khan, W.A.; Blobe, G.C.; Hannun, Y.A. Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell. Signal 1995, 7, 171–184. [Google Scholar]
- Ghosh, S.; Strum, J.C.; Sciorra, V.A.; Daniel, L.; Bell, R.M. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-o-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J. Biol. Chem 1996, 271, 8472–8480. [Google Scholar]
- Rizzo, M.A.; Shome, K.; Watkins, S.C.; Romero, G. The recruitment of Raf-1 to membranes is mediated by direct interaction with phosphatidic acid and is independent of association with Ras. J. Biol. Chem 2000, 275, 23911–23918. [Google Scholar]
- Fang, Y.; Vilella-Bach, M.; Bachmann, R.; Flanigan, A.; Chen, J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science 2001, 294, 1942–1945. [Google Scholar]
- Exton, J.H. Phospholipase d-structure, regulation and function. Rev. Physiol. Biochem. Pharmacol 2002, 144, 1–94. [Google Scholar]
- Steed, P.M.; Clark, K.L.; Boyar, W.C.; Lasala, D.J. Characterization of human PLD2 and the analysis of PLD isoform splice variants. FASEB J 1998, 12, 1309–1317. [Google Scholar]
- Cao, J.X.; Koop, B.F.; Upton, C. A human homolog of the vaccinia virus HindIII K4L gene is a member of the phospholipase D superfamily. Virus Res 1997, 48, 11–18. [Google Scholar]
- Choi, S.Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell. Biol 2006, 8, 1255–1262. [Google Scholar]
- McDermott, M.; Wakelam, M.J.; Morris, A.J. Phospholipase D. Biochem. Cell. Biol 2004, 82, 225–253. [Google Scholar]
- Watanabe-Tomita, Y.; Suzuki, A.; Shinoda, J.; Oiso, Y.; Kozawa, O. Arachidonic acid release induced by extracellular ATP in osteoblasts: Role of phospholipase D. Prostaglandins Leukotrienes Essent. Fatty Acids 1997, 57, 335–339. [Google Scholar]
- Hughes, W.E.; Parker, P.J. Endosomal localization of phospholipase D 1a and 1b is defined by the C-termini of the proteins, and is independent of activity. Biochem. J 2001, 356, 727–736. [Google Scholar]
- Lee, S.; Park, J.B.; Kim, J.H.; Kim, Y.; Shin, K.J.; Lee, J.S.; Ha, S.H.; Suh, P.G.; Ryu, S.H. Actin directly interacts with phospholipase D, inhibiting its activity. J. Biol. Chem 2001, 276, 28252–28260. [Google Scholar]
- Sugars, J.M.; Cellek, S.; Manifava, M.; Coadwell, J.; Ktistakis, N.T. Fatty acylation of phospholipase D1 on cysteine residues 240 and 241 determines localization on intracellular membranes. J. Biol. Chem 1999, 274, 30023–30027. [Google Scholar]
- Xie, Z.; Ho, W.T.; Exton, J.H. Functional implications of post-translational modifications of phospholipases D1 and D2. Biochim. Biophys. Acta 2002, 1580, 9–21. [Google Scholar]
- Jang, J.H.; Lee, C.S.; Hwang, D.; Ryu, S.H. Understanding of the roles of phospholipase D and phosphatidic acid through their binding partners. Prog. Lipid Res 2012, 51, 71–81. [Google Scholar]
- Rizzo, M.; Romero, G. Pharmacological importance of phospholipase D and phosphatidic acid in the regulation of the mitogen-activated protein kinase cascade. Pharmacol. Ther 2002, 94, 35–50. [Google Scholar]
- Szumiło, M.; Rahden-Staroń, I. Phospholipase D in mammalian cells: Structure, properties, physiological and pathological role. Postepy Hig. Med. Dosw 2006, 60, 421–430. [Google Scholar]
- Park, M.K.; Her, Y.M.; Cho, M.L.; Oh, H.J.; Park, E.M.; Kwok, S.K.; Ju, J.H.; Park, K.S.; Min, D.S.; Kim, H.Y.; et al. IL-15 promotes osteoclastogenesis via the PLD pathway in rheumatoid arthritis. Immunol. Lett 2011, 139, 42–51. [Google Scholar]
- Jenkins, G.M.; Frohman, M.A. Phospholipase D: A lipid centric review. Cell. Mol. Life Sci 2005, 62, 2305–2316. [Google Scholar]
- Conquer, J.A.; Jones, S.A.; Cruz, T.F. Effect of interleukin 1, lipopolysaccharide and phorbol esters on phospholipase D activity in chondrocytes. Osteoarthritis Cartilage 1994, 2, 269–273. [Google Scholar]
- Sylvia, V.L.; Schwartz, Z.; Del Toro, F.; DeVeau, P.; Whetstone, R.; Hardin, R.R.; Dean, D.D.; Boyan, B.D. Regulation of phospholipase D (PLD) in growth plate chondrocytes by 24R,25-(OH)2D3 is dependent on cell maturation state (resting zone cells) and is specific to the PLD2 isoform. Biochim. Biophys. Acta 2001, 1499, 209–221. [Google Scholar]
- Dean, D.D.; Muniz, O.E.; Berman, I.; Pita, J.C.; Carreno, M.R.; Woessner, J.F.; Howell, D.S. Localization of collagenase in the growth plate of rachitic rats. J. Clin. Invest 1985, 76, 716–722. [Google Scholar]
- Dean, D.D.; Muniz, O.E.; Howell, D.S. Association of collagenase and tissue inhibitor of metalloproteinases (TIMP) with hypertrophic cell enlargement in the growth plate. Matrix 1989, 9, 366–375. [Google Scholar]
- Boyan, B.D.; Dean, D.D.; Sylvia, V.L.; Schwartz, Z. Steroid hormone action in musculoskeletal cells involves membrane receptor and nuclear receptor mechanisms. Connect. Tissue Res 2003, 44, 130–135. [Google Scholar]
- Langston, G.G.; Swain, L.D.; Schwartz, Z.; Del Toro, F.; Gomez, R.; Boyan, B.D. Effect of 1,25(OH)2D3 and 24,25(OH)2D3 on calcium ion fluxes in costochondral chondrocyte cultures. Calcif. Tissue Int 1990, 47, 230–236. [Google Scholar]
- Schwartz, Z.; Schlader, D.L.; Ramirez, V.; Kennedy, M.B.; Boyan, B.D. Effects of vitamin D metabolites on collagen production and cell proliferation of growth zone and resting zone cartilage cells in vitro. J. Bone Miner. Res 1989, 4, 199–207. [Google Scholar]
- Boyan, B.D.; Schwartz, Z.; Swain, L.D.; Carnes, D.L.; Zislis, T. Differential expression of phenotype by resting zone and growth region costochondral chondrocytes in vitro. Bone 1988, 9, 185–194. [Google Scholar]
- Helm, S.; Sylvia, V.L.; Harmon, T.; Dean, D.D.; Boyan, B.D.; Schwartz, Z. 24,25-(OH)2D3 regulates protein kinase C through two distinct phospholipid-dependent mechanisms. J. Cell. Physiol 1996, 169, 509–521. [Google Scholar]
- Boyan, B.D.; Sylvia, V.L.; McKinney, N.; Schwartz, Z. Membrane actions of vitamin D metabolites 1alpha,25(OH)2D3 and 24R,25(OH)2D3 are retained in growth plate cartilage cells from vitamin D receptor knockout mice. J. Cell. Biochem 2003, 90, 1207–1223. [Google Scholar]
- Boyan, B.D.; Jennings, E.G.; Wang, L.; Schwartz, Z. Mechanisms regulating differential activation of membrane-mediated signaling by 1alpha,25(OH)2D3 and 24R,25(OH)2D3. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 309–315. [Google Scholar]
- Boyan, B.D.; Sylvia, V.L.; Dean, D.D.; Pedrozo, H.; Del Toro, F.; Nemere, I.; Posner, G.H.; Schwartz, Z. 1,25-(OH)2D3 modulates growth plate chondrocytes via membrane receptor-mediated protein kinase C by a mechanism that involves changes in phospholipid metabolism and the action of arachidonic acid and PGE2. Steroids 1999, 64, 129–136. [Google Scholar]
- Schwartz, Z.; Sylvia, V.L.; Del Toro, F.; Hardin, R.R.; Dean, D.D.; Boyan, B.D. 24R,25-(OH)(2)D(3) mediates its membrane receptor-dependent effects on protein kinase C and alkaline phosphatase via phospholipase A(2) and cyclooxygenase-1 but not cyclooxygenase-2 in growth plate chondrocytes. J. Cell. Physiol 2000, 182, 390–401. [Google Scholar]
- Sylvia, V.L.; Schwartz, Z.; Holmes, S.C.; Dean, D.D.; Boyan, B.D. 24,25-(OH)2D3 regulation of matrix vesicle protein kinase C occurs both during biosynthesis and in the extracellular matrix. Calcif. Tissue Int 1997, 61, 313–321. [Google Scholar]
- Dean, D.D.; Boyan, B.D.; Schwart, Z.; Muniz, O.E.; Carreno, M.R.; Maeda, S.; Howell, D.S. Effect of 1alpha,25-dihydroxyvitamin D3 and 24R,25-dihydroxyvitamin D3 on metalloproteinase activity and cell maturation in growth plate cartilage in vivo. Endocrine 2001, 14, 311–323. [Google Scholar]
- Kusner, D.J.; Barton, J.A.; Qin, C.; Wang, X.; Iyer, S.S. Evolutionary conservation of physical and functional interactions between phospholipase D and actin. Arch. Biochem. Biophys 2003, 412, 231–241. [Google Scholar]
- Blackwood, R.A.; Smolen, J.E.; Transue, A.; Hessler, R.J.; Harsh, D.M.; Brower, R.C.; French, S. Phospholipase D activity facilitates Ca2+-induced aggregation and fusion of complex liposomes. Am. J. Physiol 1997, 272, C1279–C1285. [Google Scholar]
- Burger, K.N. Greasing membrane fusion and fission machineries. Traffic 2000, 1, 605–613. [Google Scholar]
- Burger, K.N.; Demel, R.A.; Schmid, S.L.; de Kruijff, B. Dynamin is membrane-active: Lipid insertion is induced by phosphoinositides and phosphatidic acid. Biochemistry 2000, 39, 12485–12493. [Google Scholar]
- Bader, M.F.; Vitale, N. Phospholipase D in calcium-regulated exocytosis: Lessons from chromaffin cells. Biochim. Biophys. Acta 2009, 1791, 936–941. [Google Scholar]
- Laulagnier, K.; Grand, D.; Dujardin, A.; Hamdi, S.; Vincent-Schneider, H.; Lankar, D.; Salles, J.P.; Bonnerot, C.; Perret, B.; Record, M. PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett 2004, 572, 11–14. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Watanabe, Y.; Shinoda, J.; Oiso, Y. Effect of platelet-derived growth factor on phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like cells. Endocrinology 1995, 136, 4473–4478. [Google Scholar]
- Shinoda, J.; Suzuki, A.; Oiso, Y.; Kozawa, O. Thromboxane A2-stimulated phospholipase D in osteoblast-like cells: Possible involvement of PKC. Am. J. Physiol 1995, 269, E524–E529. [Google Scholar]
- Tokuda, H.; Suzuki, A.; Watanabe-Tomita, Y.; Shinoda, J.; Imamura, Y.; Oiso, Y.; Igata, A.; Kozawa, O. Function of Ca2+ in phosphatidylcholine-hydrolyzing phospholipase D activation in osteoblast-like cells. Bone 1996, 19, 347–352. [Google Scholar]
- Oiso, Y.; Suzuki, A.; Kozawa, O. Effect of prostaglandin E2 on phospholipase D activity in osteoblast-like MC3T3-E1 cells. J. Bone Miner. Res 1995, 10, 1185–1190. [Google Scholar]
- Suzuki, A.; Kozawa, O.; Shinoda, J.; Watanabe, Y.; Saito, H.; Oiso, Y. Thrombin induces proliferation of osteoblast-like cells through phosphatidylcholine hydrolysis. J. Cell. Physiol 1996, 168, 209–216. [Google Scholar]
- Sugiyama, T.; Sakai, T.; Nozawa, Y.; Oka, N. Prostaglandin F2 alpha-stimulated phospholipase D activation in osteoblast-like MC3T3-E1 cells: Involvement in sustained 1,2-diacylglycerol production. Biochem. J. 1994, 298( Pt. 2), 479–484. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Kotoyori, J.; Tokuda, H.; Watanabe, Y.; Ito, Y.; Oiso, Y. Prostaglandin F2 alpha activates phospholipase D independently from activation of protein kinase C in osteoblast-like cells. J. Cell. Biochem 1994, 55, 373–379. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Shinoda, J.; Oiso, Y. Effect of retinoic acid on prostaglandin F2 alpha-induced phospholipase D activity in osteoblast-like cells. Prostaglandins Leukotrienes Essent. Fatty Acids 1996, 55, 151–154. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Watanabe, Y.; Shinoda, J.; Oiso, Y. Function of Ca2+ influx in phospholipase D activation induced by prostaglandin F2 alpha in osteoblast-like cells: Involvement of tyrosine kinase. Prostaglandins Leukotrienes Essent. Fatty Acids 1995, 52, 319–323. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Oiso, Y. Tyrosine kinase regulates phospholipase D activation at a point downstream from protein kinase C in osteoblast-like cells. J. Cell. Biochem 1995, 57, 251–255. [Google Scholar]
- Suzuki, A.; Oiso, Y.; Kozawa, O. Effect of endothelin-1 on phospholipase D activity in osteoblast-like cells. Mol. Cell. Endocrinol 1994, 105, 193–196. [Google Scholar]
- Kozawa, O.; Suzuki, A.; Shinoda, J.; Ozaki, N.; Oiso, Y.; Uematsu, T. Involvement of phospholipase D activation in endothelin-1-induced release of arachidonic acid in osteoblast-like cells. J. Cell. Biochem 1997, 64, 376–381. [Google Scholar]
- Suzuki, A.; Shinoda, J.; Kanda, S.; Oiso, Y.; Kozawa, O. Basic fibroblast growth factor stimulates phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like cells. J. Cell. Biochem 1996, 63, 491–499. [Google Scholar]
- Bourgoin, S.G.; Harbour, D.; Poubelle, P.E. Role of protein kinase C alpha, Arf, and cytoplasmic calcium transients in phospholipase D activation by sodium fluoride in osteoblast-like cells. J. Bone Miner. Res 1996, 11, 1655–1665. [Google Scholar]
- Kaneki, H.; Yokozawa, J.; Fujieda, M.; Mizuochi, S.; Ishikawa, C.; Ide, H. Phorbol ester-induced production of prostaglandin E2 from phosphatidylcholine through the activation of phospholipase D in UMR-106 cells. Bone 1998, 23, 213–222. [Google Scholar]
- Singh, A.T.; Gilchrist, A.; Voyno-Yasenetskaya, T.; Radeff-Huang, J.M.; Stern, P.H. G alpha12/G alpha13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology 2005, 146, 2171–2175. [Google Scholar]
- Radeff, J.M.; Singh, A.T.; Stern, P.H. Role of protein kinase A, phospholipase C and phospholipase D in parathyroid hormone receptor regulation of protein kinase Calpha and interleukin-6 in UMR-106 osteoblastic cells. Cell. Signal 2004, 16, 105–114. [Google Scholar]
- Singh, A.T.; Frohman, M.A.; Stern, P.H. Parathyroid hormone stimulates phosphatidylethanolamine hydrolysis by phospholipase D in osteoblastic cells. Lipids 2005, 40, 1135–1140. [Google Scholar]
- Igarashi, K.; Hirafuji, M.; Adachi, H.; Shinoda, H.; Mitani, H. Role of endogenous PGE2 in osteoblastic functions of a clonal osteoblast-like cell, MC3T3-E1. Prostaglandins Leukotrienes Essent. Fatty Acids 1994, 50, 169–172. [Google Scholar]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest 1999, 104, 439–446. [Google Scholar]
- Tashjian, A.H.; Levine, L. Epidermal growth factor stimulates prostaglandin production and bone resorption in cultured mouse calvaria. Biochem. Biophys. Res. Commun 1978, 85, 966–975. [Google Scholar]
- Carpio, L.C.; Dziak, R. Activation of phospholipase D signaling pathway by epidermal growth factor in osteoblastic cells. J. Bone Miner. Res 1998, 13, 1707–1713. [Google Scholar]
- Kim, M.J.; Choi, M.U.; Kim, C.W. Activation of phospholipase D1 by surface roughness of titanium in MG63 osteoblast-like cell. Biomaterials 2006, 27, 5502–5511. [Google Scholar]
- Fang, M.; Olivares-Navarrete, R.; Wieland, M.; Cochran, D.L.; Boyan, B.D.; Schwartz, Z. The role of phospholipase D in osteoblast response to titanium surface microstructure. J. Biomed. Mater. Res. A 2010, 93, 897–909. [Google Scholar]
- Sumikawa, K.; Okochi, T.; Adachi, K. Differences in phosphatidate hydrolytic activity of human alkaline phosphatase isozymes. Biochim. Biophys. Acta 1990, 1046, 27–31. [Google Scholar]
- Al Husaini, H.; Wheatley-Price, P.; Clemons, M.; Shepherd, F.A. Prevention and management of bone metastases in lung cancer: A review. J. Thorac. Oncol 2009, 4, 251–259. [Google Scholar]
- Hirsh, V.; Major, P.P.; Lipton, A.; Cook, R.J.; Langer, C.J.; Smith, M.R.; Brown, J.E.; Coleman, R.E. Zoledronic acid and survival in patients with metastatic bone disease from lung cancer and elevated markers of osteoclast activity. J. Thorac. Oncol 2008, 3, 228–236. [Google Scholar]
- Bendre, M.S.; Margulies, A.G.; Walser, B.; Akel, N.S.; Bhattacharrya, S.; Skinner, R.A.; Swain, F.; Ramani, V.; Mohammad, K.S.; Wessner, L.L.; et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res 2005, 65, 11001–11009. [Google Scholar]
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem 2009, 284, 6038–6042. [Google Scholar]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res 2008, 14, 6735–6741. [Google Scholar]
- MacManus, C.F.; Pettigrew, J.; Seaton, A.; Wilson, C.; Maxwell, P.J.; Berlingeri, S.; Purcell, C.; McGurk, M.; Johnston, P.G.; Waugh, D.J. Interleukin-8 signaling promotes translational regulation of cyclin D in androgen-independent prostate cancer cells. Mol. Cancer Res 2007, 5, 737–748. [Google Scholar]
- Hsu, Y.L.; Hung, J.Y.; Ko, Y.C.; Hung, C.H.; Huang, M.S.; Kuo, P.L. Phospholipase D signaling pathway is involved in lung cancer-derived IL-8 increased osteoclastogenesis. Carcinogenesis 2010, 31, 587–596. [Google Scholar]
- Gravallese, E.M.; Manning, C.; Tsay, A.; Naito, A.; Pan, C.; Amento, E.; Goldring, S.R. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis. Rheum 2000, 43, 250–258. [Google Scholar]
- Kong, Y.Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar]
- Ogata, Y.; Kukita, A.; Kukita, T.; Komine, M.; Miyahara, A.; Miyazaki, S.; Kohashi, O. A novel role of IL-15 in the development of osteoclasts: Inability to replace its activity with IL-2. J. Immunol 1999, 162, 2754–2760. [Google Scholar]
- Miranda-Carús, M.E.; Benito-Miguel, M.; Balsa, A.; Cobo-Ibáñez, T.; Pérez de Ayala, C.; Pascual-Salcedo, D.; Martín-Mola, E. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis. Rheum 2006, 54, 1151–1164. [Google Scholar]
- Elvers, M.; Stegner, D.; Hagedorn, I.; Kleinschnitz, C.; Braun, A.; Kuijpers, M.E.; Boesl, M.; Chen, Q.; Heemskerk, J.W.; Stoll, G.; et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. 2010, 3, ra1. [Google Scholar]
- Norton, L.J.; Zhang, Q.; Saqib, K.M.; Schrewe, H.; Macura, K.; Anderson, K.E.; Lindsley, C.W.; Brown, H.A.; Rudge, S.A.; Wakelam, M.J. PLD1 rather than PLD2 regulates phorbol-ester-, adhesion-dependent and Fc gamma-receptor-stimulated ROS production in neutrophils. J. Cell. Sci 2011, 124, 1973–1983. [Google Scholar]
- Balcerzak, M.; Pikula, S.; Buchet, R. Phosphorylation-dependent phospholipase D activity of matrix vesicles. FEBS Lett 2006, 580, 5676–5680. [Google Scholar]
- Tsujioka, H.; Takami, N.; Misumi, Y.; Ikehara, Y. Intracellular cleavage of glycosylphosphatidylinositol by phospholipase D induces activation of protein kinase Calpha. Biochem. J 1999, 342, 449–455. [Google Scholar]
- LeBoeuf, R.C.; Caldwell, M.; Guo, Y.; Metz, C.; Davitz, M.A.; Olson, L.K.; Deeg, M.A. Mouse glycosylphosphatidylinositol-specific phospholipase D (Gpld1) characterization. Mamm. Genome 1998, 9, 710–714. [Google Scholar]
- Scallon, B.J.; Fung, W.J.; Tsang, T.C.; Li, S.; Kado-Fong, H.; Huang, K.S.; Kochan, J.P. Primary structure and functional activity of a phosphatidylinositol-glycan-specific phospholipase D. Science 1991, 252, 446–448. [Google Scholar]
- Tang, J.H.; Gu, S.L.; Zhang, X.J. Preliminary study of the gene structure of human glycosylphosphatidylinositol specific phospholipase D. Hunan Yi Ke Da Xue Xue Bao 2001, 26, 95–97. [Google Scholar]
- Brunner, G.; Metz, C.N.; Nguyen, H.; Gabrilove, J.; Patel, S.R.; Davitz, M.A.; Rifkin, D.B.; Wilson, E.L. An endogenous glycosylphosphatidylinositol-specific phospholipase D releases basic fibroblast growth factor-heparan sulfate proteoglycan complexes from human bone marrow cultures. Blood 1994, 83, 2115–2125. [Google Scholar]
- Gregory, P.; Kraemer, E.; Zürcher, G.; Gentinetta, R.; Rohrbach, V.; Brodbeck, U.; Andres, A.C.; Ziemiecki, A.; Bütikofer, P. GPI-specific phospholipase D (GPI-PLD) is expressed during mouse development and is localized to the extracellular matrix of the developing mouse skeleton. Bone 2005, 37, 139–147. [Google Scholar]
- Larson, D.M.; Kennedy, M.A.; Bowen, R.F.; Verchere, C.B.; Deeg, M.A. Glycosylphosphatidylinositol-specific phospholipase D immunoreactivity is present in islet amyloid in type 2 diabetes. J. Pathol 2004, 203, 961–967. [Google Scholar]
- Rhode, H.; Lopatta, E.; Schulze, M.; Pascual, C.; Schulze, H.P.; Schubert, K.; Schubert, H.; Reinhart, K.; Horn, A. Glycosylphosphatidylinositol-specific phospholipase D in blood serum: Is the liver the only source of the enzyme? Clin. Chim. Acta 1999, 281, 127–145. [Google Scholar]
- Xiaotong, H.; Hannocks, M.J.; Hampson, I.; Brunner, G. GPI-specific phospholipase D mRNA expression in tumor cells of different malignancy. Clin. Exp. Metastasis 2002, 19, 291–299. [Google Scholar]
- Knudson, C.B.; Knudson, W. Cartilage proteoglycans. Semin. Cell Dev. Biol 2001, 12, 69–78. [Google Scholar]
- Paine-Saunders, S.; Viviano, B.L.; Zupicich, J.; Skarnes, W.C.; Saunders, S. Glypican-3 controls cellular responses to Bmp4 in limb patterning and skeletal development. Dev. Biol 2000, 225, 179–187. [Google Scholar]
- Pilia, G.; Hughes-Benzie, R.M.; MacKenzie, A.; Baybayan, P.; Chen, E.Y.; Huber, R.; Neri, G.; Cao, A.; Forabosco, A.; Schlessinger, D. Mutations in GPC3, a glypican gene, cause the Simpson-Golabi-Behmel overgrowth syndrome. Nat. Genet 1996, 12, 241–247. [Google Scholar]
- Schwab, W.; Gavlik, J.M.; Beichler, T.; Funk, R.H.; Albrecht, S.; Magdolen, V.; Luther, T.; Kasper, M.; Shakibaei, M. Expression of the urokinase-type plasminogen activator receptor in human articular chondrocytes: Association with caveolin and beta 1-integrin. Histochem. Cell. Biol 2001, 115, 317–323. [Google Scholar]
- Daci, E.; Everts, V.; Torrekens, S.; Van Herck, E.; Tigchelaar-Gutterr, W.; Bouillon, R.; Carmeliet, G. Increased bone formation in mice lacking plasminogen activators. J. Bone Miner. Res 2003, 18, 1167–1176. [Google Scholar]
- Xing, Y.; Gu, Y.; Xu, L.C.; Siedlecki, C.A.; Donahue, H.J.; You, J. Effects of membrane cholesterol depletion and GPI-anchored protein reduction on osteoblastic mechanotransduction. J. Cell. Physiol 2011, 226, 2350–2359. [Google Scholar]
- Goding, J.W.; Terkeltaub, R.; Maurice, M.; Deterre, P.; Sali, A.; Belli, S.I. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev 1998, 161, 11–26. [Google Scholar]
- Van Meeteren, L.A.; Ruurs, P.; Christodoulou, E.; Goding, J.W.; Takakusa, H.; Kikuchi, K.; Perrakis, A.; Nagano, T.; Moolenaar, W.H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem 2005, 280, 21155–21161. [Google Scholar]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res 2007, 46, 145–160. [Google Scholar]
- Tania, M.; Khan, M.A.; Zhang, H.; Li, J.; Song, Y. Autotaxin: A protein with two faces. Biochem. Biophys. Res. Commun 2010, 401, 493–497. [Google Scholar]
- Giganti, A.; Rodriguez, M.; Fould, B.; Moulharat, N.; Cogé, F.; Chomarat, P.; Galizzi, J.P.; Valet, P.; Saulnier-Blache, J.S.; Boutin, J.A.; et al. Murine and human autotaxin alpha, beta, and gamma isoforms: Gene organization, tissue distribution, and biochemical characterization. J. Biol. Chem 2008, 283, 7776–7789. [Google Scholar]
- Fukushima, N. LPA in neural cell development. J. Cell. Biochem 2004, 92, 993–1003. [Google Scholar]
- Hu, P.F.; Chen, Y.; Cai, P.F.; Jiang, L.F.; Wu, L.D. Sphingosine-1-phosphate: A potential therapeutic target for rheumatoid arthritis. Mol. Biol. Rep 2011, 38, 4225–4230. [Google Scholar]
- Clair, T.; Aoki, J.; Koh, E.; Bandle, R.W.; Nam, S.W.; Ptaszynska, M.M.; Mills, G.B.; Schiffmann, E.; Liotta, L.A.; Stracke, M.L. Autotaxin hydrolyzes sphingosylphosphorylcholine to produce the regulator of migration, sphingosine-1-phosphate. Cancer Res 2003, 63, 5446–5453. [Google Scholar]
- Bourgoin, S.G.; Zhao, C. Autotaxin and lysophospholipids in rheumatoid arthritis. Curr. Opin. Investig. Drugs 2010, 11, 515–526. [Google Scholar]
- Lai, W.Q.; Irwan, A.W.; Goh, H.H.; Howe, H.S.; Yu, D.T.; Valle-Oñate, R.; McInnes, I.B.; Melendez, A.J.; Leung, B.P. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J. Immunol 2008, 181, 8010–8017. [Google Scholar]
- Kitano, M.; Hla, T.; Sekiguchi, M.; Kawahito, Y.; Yoshimura, R.; Miyazawa, K.; Iwasaki, T.; Sano, H.; Saba, J.D.; Tam, Y.Y. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: Regulation of synovial proliferation and inflammatory gene expression. Arthritis. Rheum 2006, 54, 742–753. [Google Scholar]
- Deutschman, D.H.; Carstens, J.S.; Klepper, R.L.; Smith, W.S.; Page, M.T.; Young, T.R.; Gleason, L.A.; Nakajima, N.; Sabbadini, R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J 2003, 146, 62–68. [Google Scholar]
- Pyne, S.; Pyne, N. Sphingosine 1-phosphate signalling via the endothelial differentiation gene family of G-protein-coupled receptors. Pharmacol. Ther 2000, 88, 115–131. [Google Scholar]
- Takeshita, H.; Kitano, M.; Iwasaki, T.; Kitano, S.; Tsunemi, S.; Sato, C.; Sekiguchi, M.; Azuma, N.; Miyazawa, K.; Hla, T.; et al. Sphingosine 1-phosphate (S1P)/S1P receptor 1 signaling regulates receptor activator of NF-κB ligand (RANKL) expression in rheumatoid arthritis. Biochem. Biophys. Res. Commun 2012, 419, 154–159. [Google Scholar]
- Bächner, D.; Ahrens, M.; Schröder, D.; Hoffmann, A.; Lauber, J.; Betat, N.; Steinert, P.; Flohé, L.; Gross, G. Bmp-2 downstream targets in mesenchymal development identified by subtractive cloning from recombinant mesenchymal progenitors (C3H10T1/2). Dev. Dyn 1998, 213, 398–411. [Google Scholar]
- Bächner, D.; Ahrens, M.; Betat, N.; Schröder, D.; Gross, G. Developmental expression analysis of murine autotaxin (ATX). Mech. Dev 1999, 84, 121–125. [Google Scholar]
- Garciadiego-Cázares, D.; Rosales, C.; Katoh, M.; Chimal-Monroy, J. Coordination of chondrocyte differentiation and joint formation by alpha5beta1 integrin in the developing appendicular skeleton. Development 2004, 131, 4735–4742. [Google Scholar]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar]
- Peyruchaud, O.; Leblanc, R.; David, M. Pleiotropic activity of lysophosphatidic acid in bone metastasis. Biochim. Biophys. Acta 2013, 1831, 99–104. [Google Scholar]
- Karagiosis, S.A.; Chrisler, W.B.; Bollinger, N.; Karin, N.J. Lysophosphatidic acid-induced ERK activation and chemotaxis in MC3T3-E1 preosteoblasts are independent of EGF receptor transactivation. J. Cell. Physiol 2009, 219, 716–723. [Google Scholar]
- David, M.; Wannecq, E.; Descotes, F.; Jansen, S.; Deux, B.; Ribeiro, J.; Serre, C.M.; Grès, S.; Bendriss-Vermare, N.; Bollen, M.; et al. Cancer cell expression of autotaxin controls bone metastasis formation in mouse through lysophosphatidic acid-dependent activation of osteoclasts. PLoS One 2010, 5, e9741. [Google Scholar]
- Murakami-Murofushi, K.; Uchiyama, A.; Fujiwara, Y.; Kobayashi, T.; Kobayashi, S.; Mukai, M.; Murofushi, H.; Tigyi, G. Biological functions of a novel lipid mediator, cyclic phosphatidic acid. Biochim. Biophys. Acta 2002, 1582, 1–7. [Google Scholar]
- Mansell, J.P.; Nowghani, M.; Pabbruwe, M.; Paterson, I.C.; Smith, A.J.; Blom, A.W. Lysophosphatidic acid and calcitriol co-operate to promote human osteoblastogenesis: Requirement of albumin-bound LPA. Prostaglandins Other Lipid Mediat 2011, 95, 45–52. [Google Scholar]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Rojas, M.; End, C.; Vallant, C.; Dong, A.; Lynch, K.; Chun, J.; Morris, A.J.; et al. Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ. Res 2008, 103, 662–670. [Google Scholar]
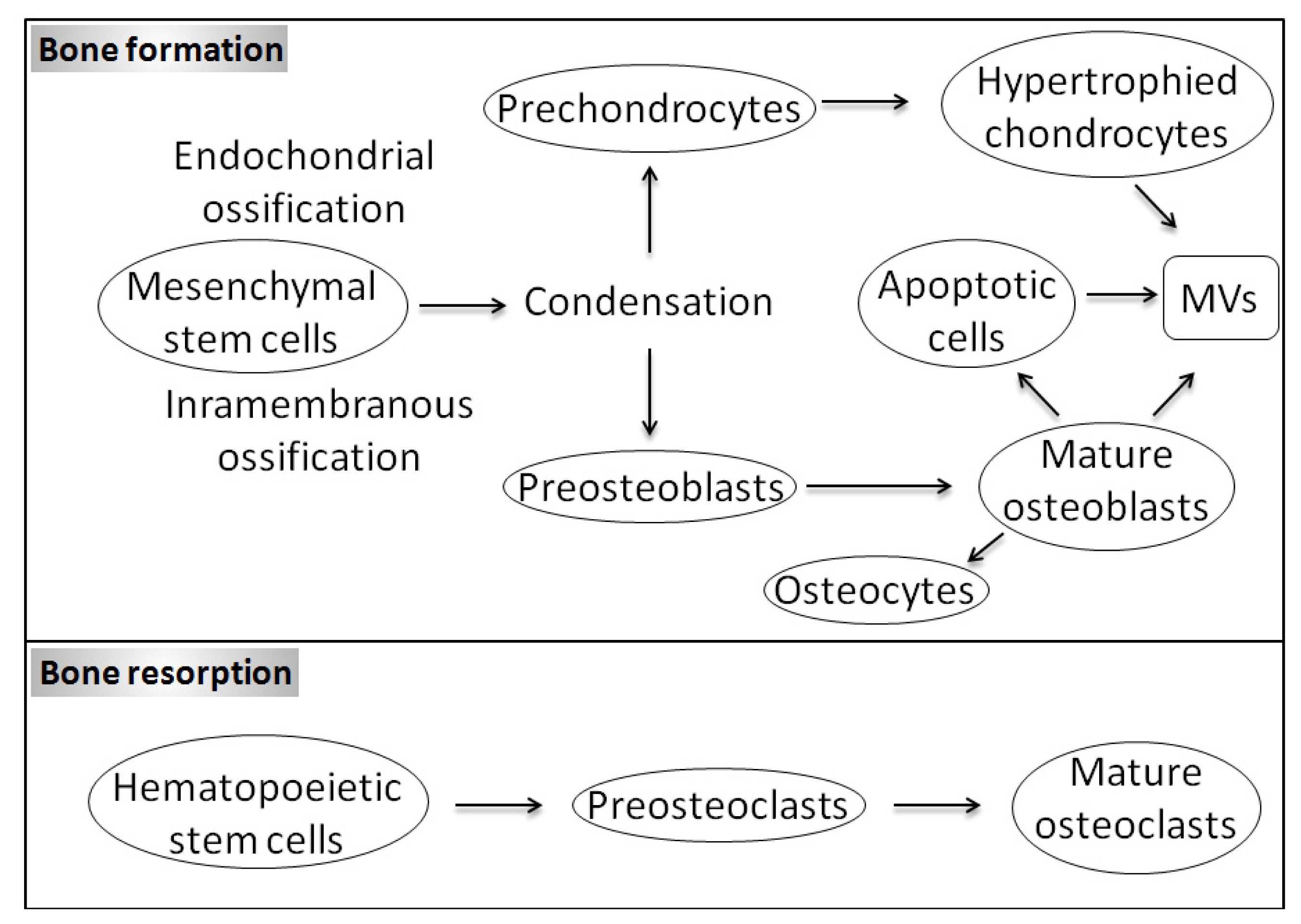
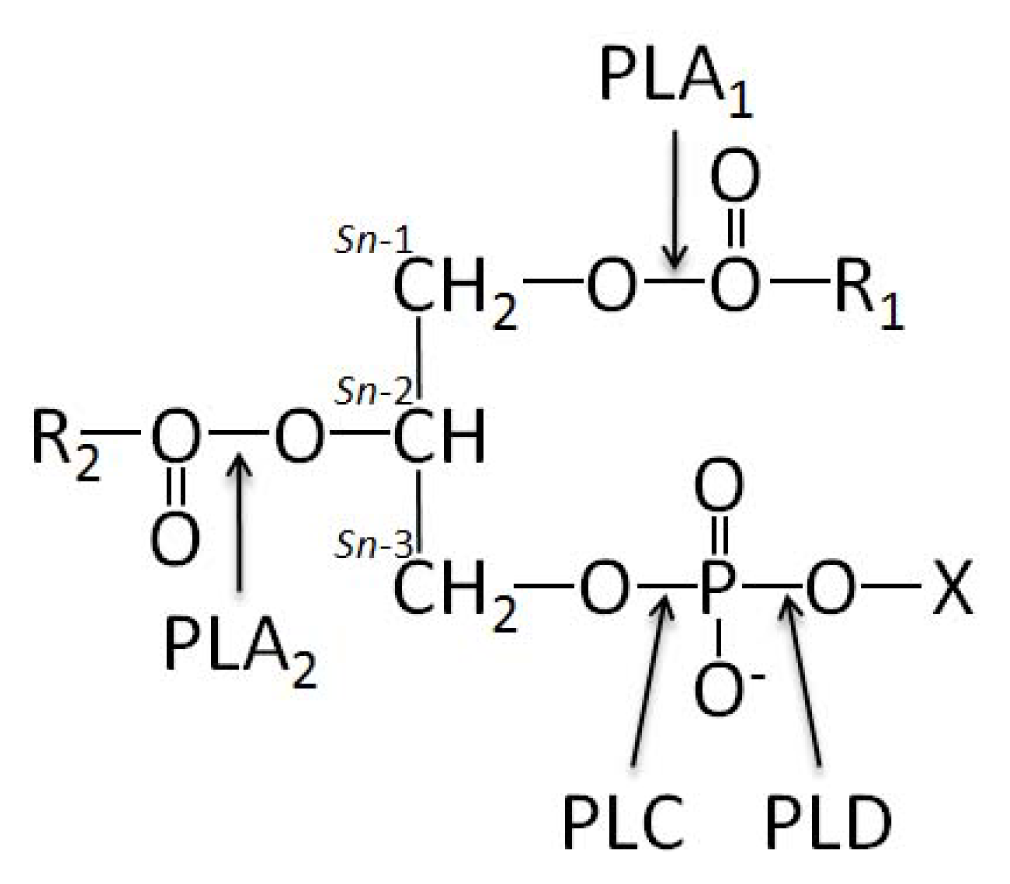
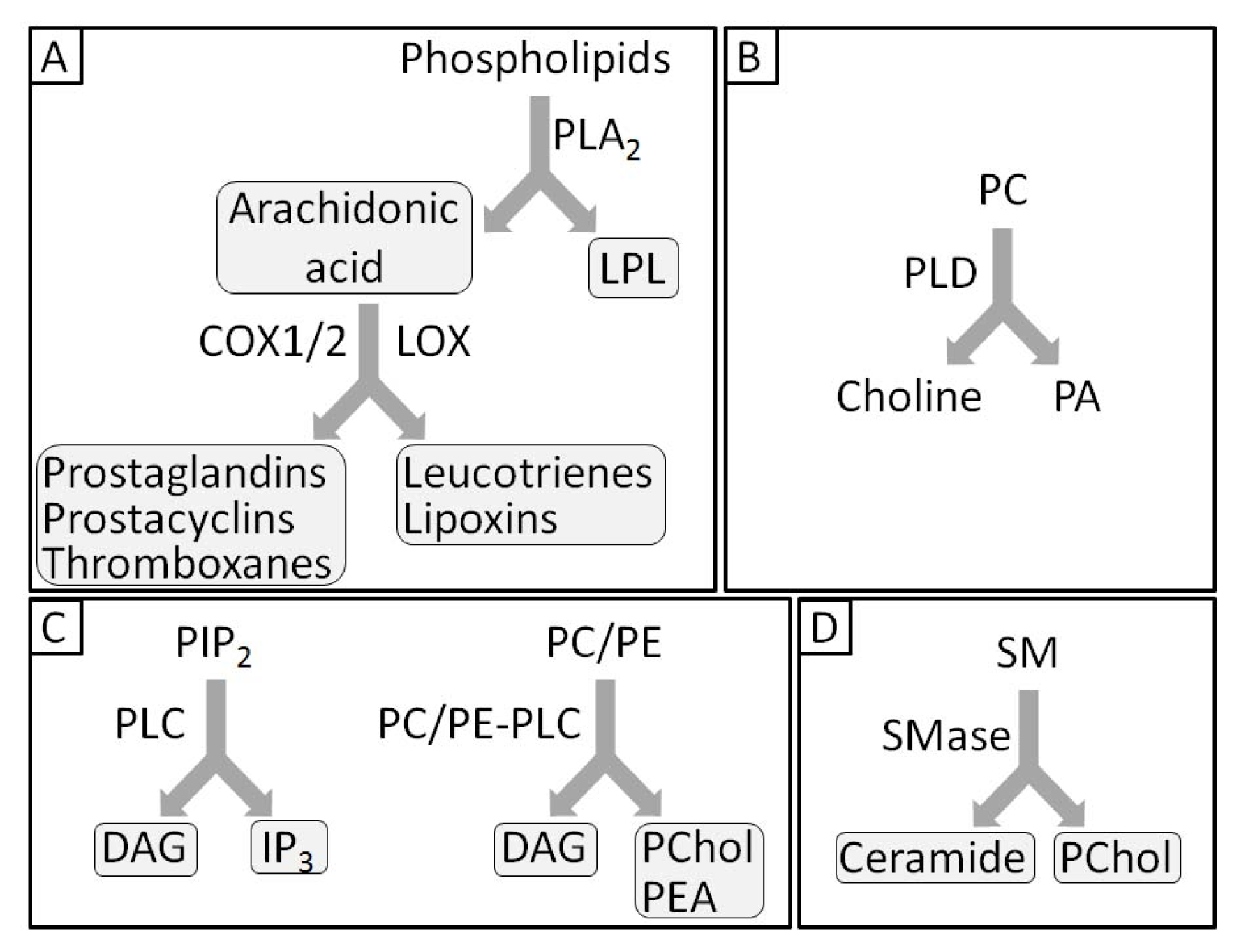
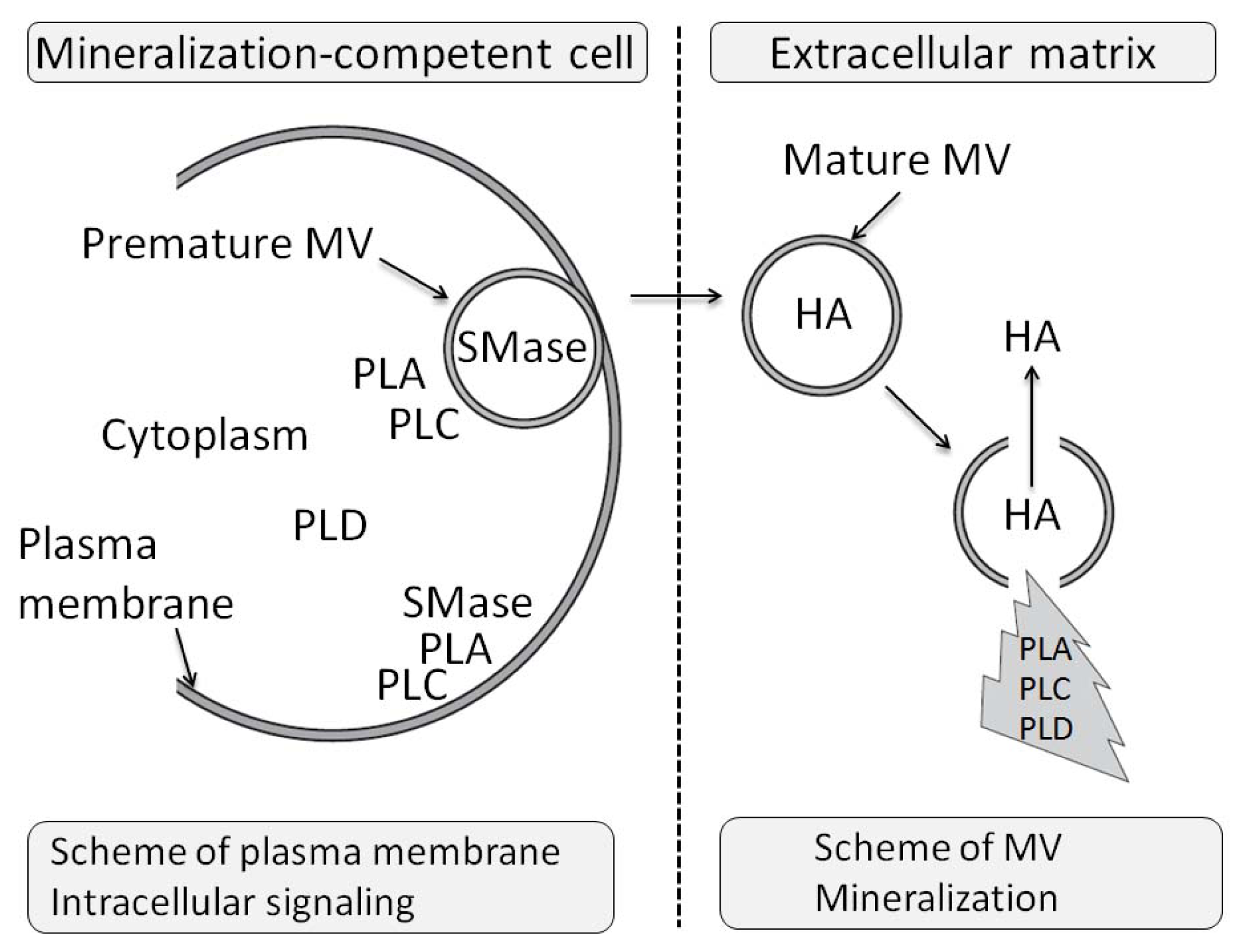
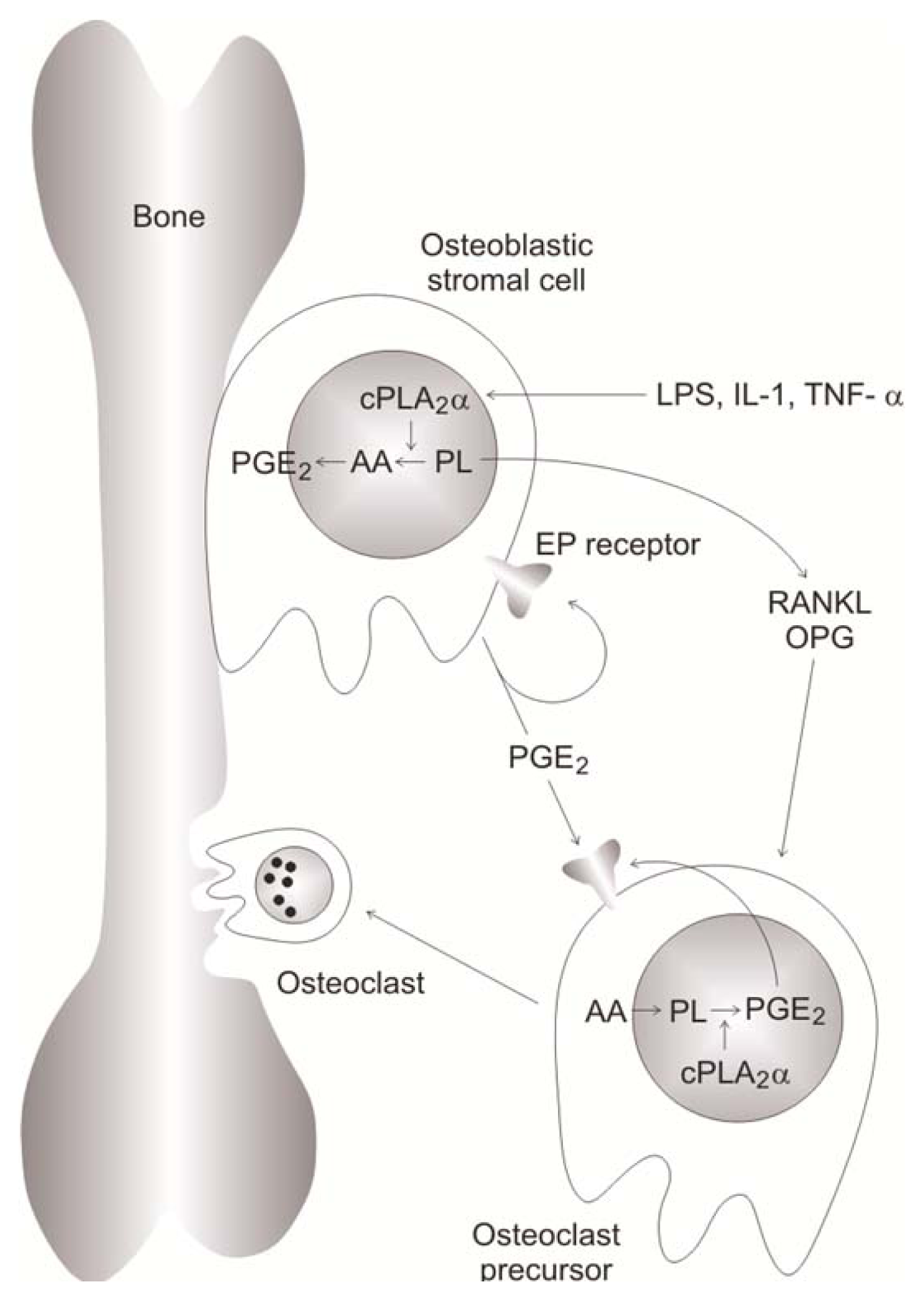
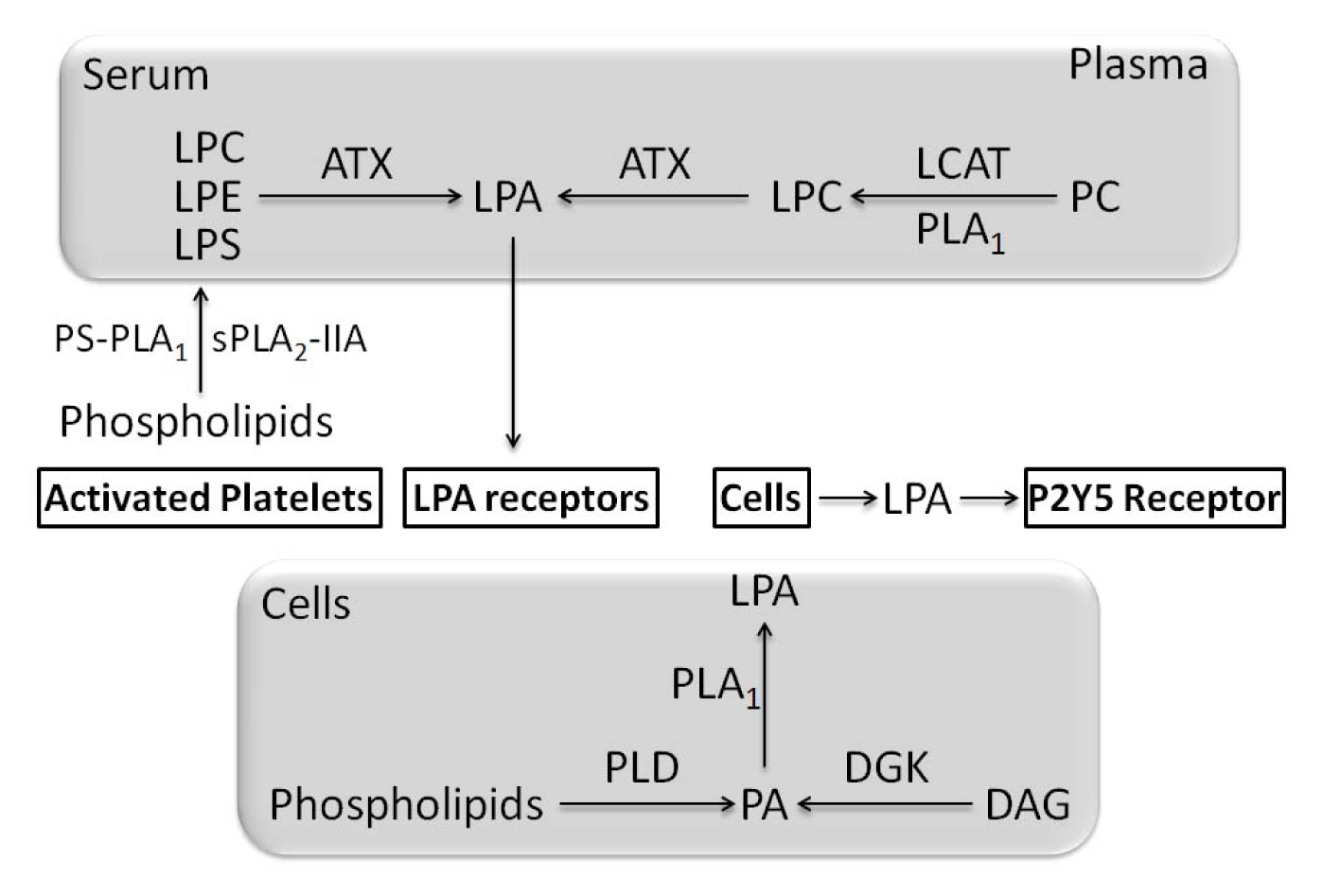
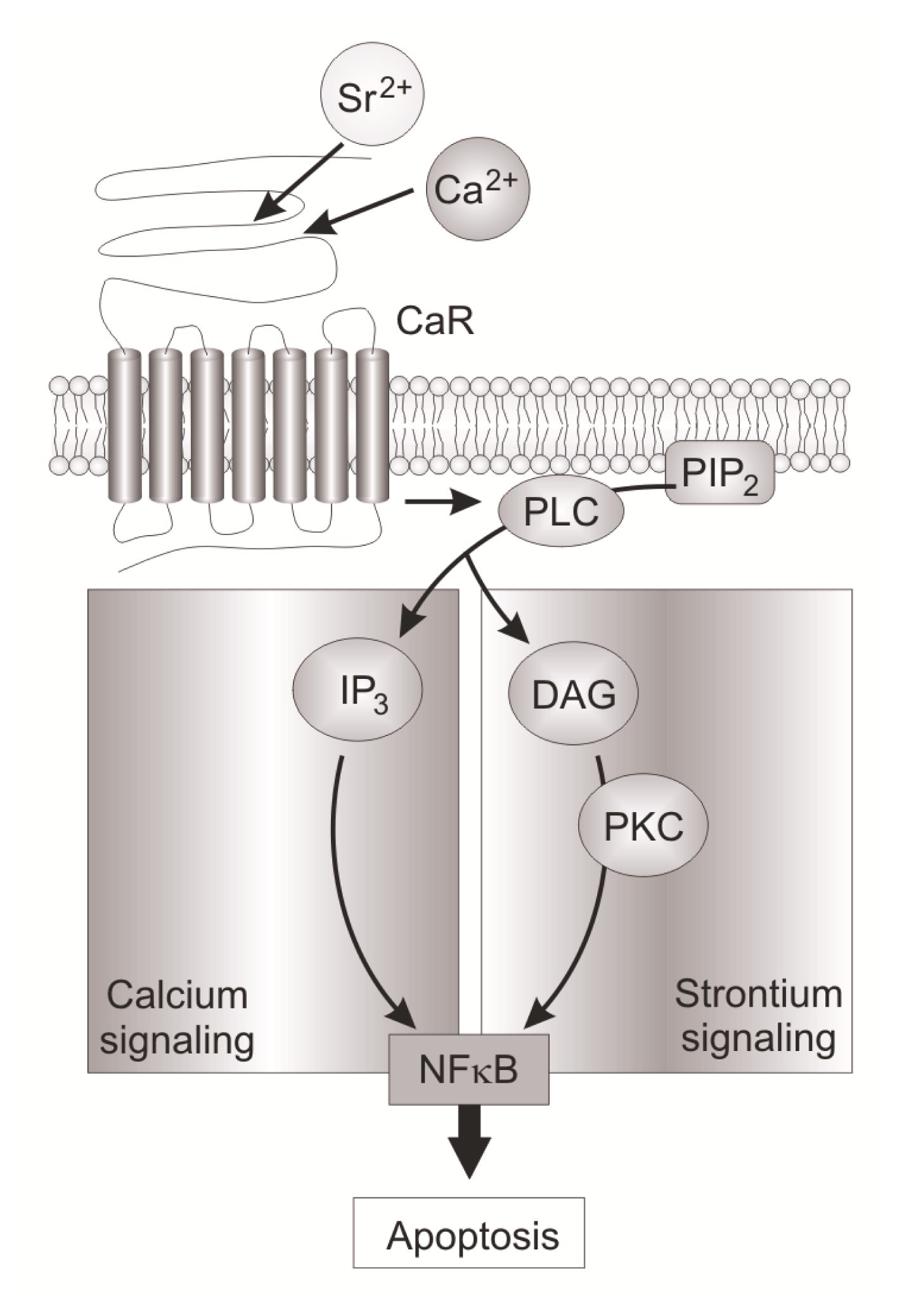
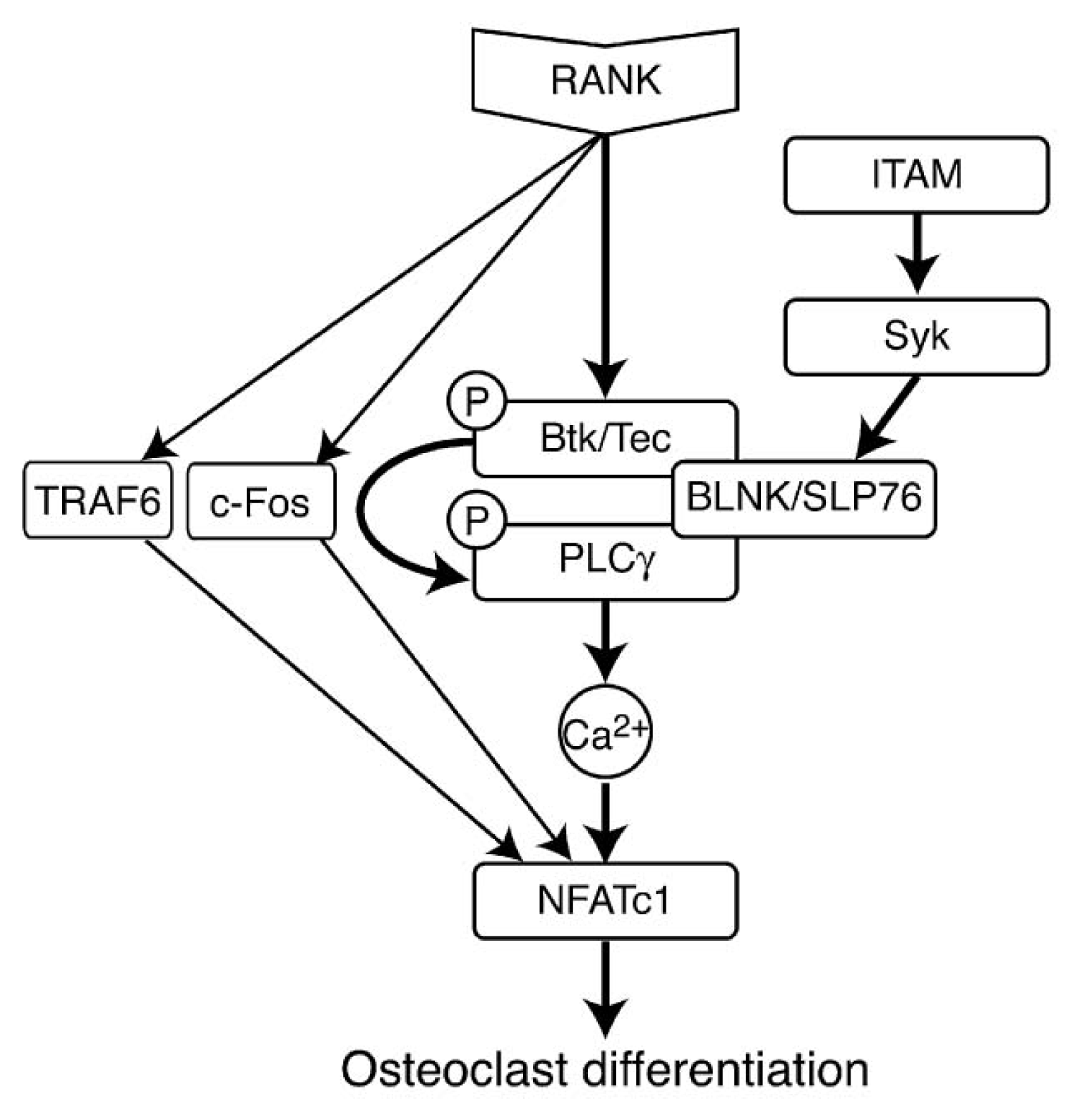

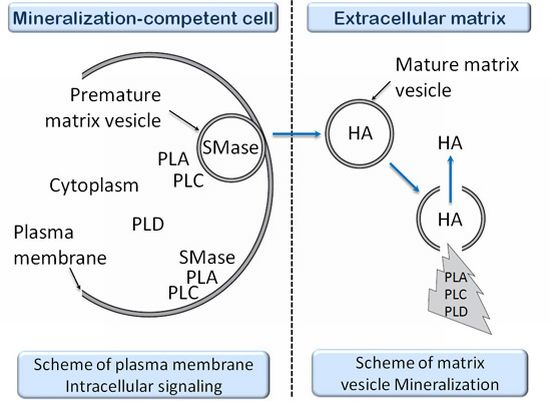{kind=link}
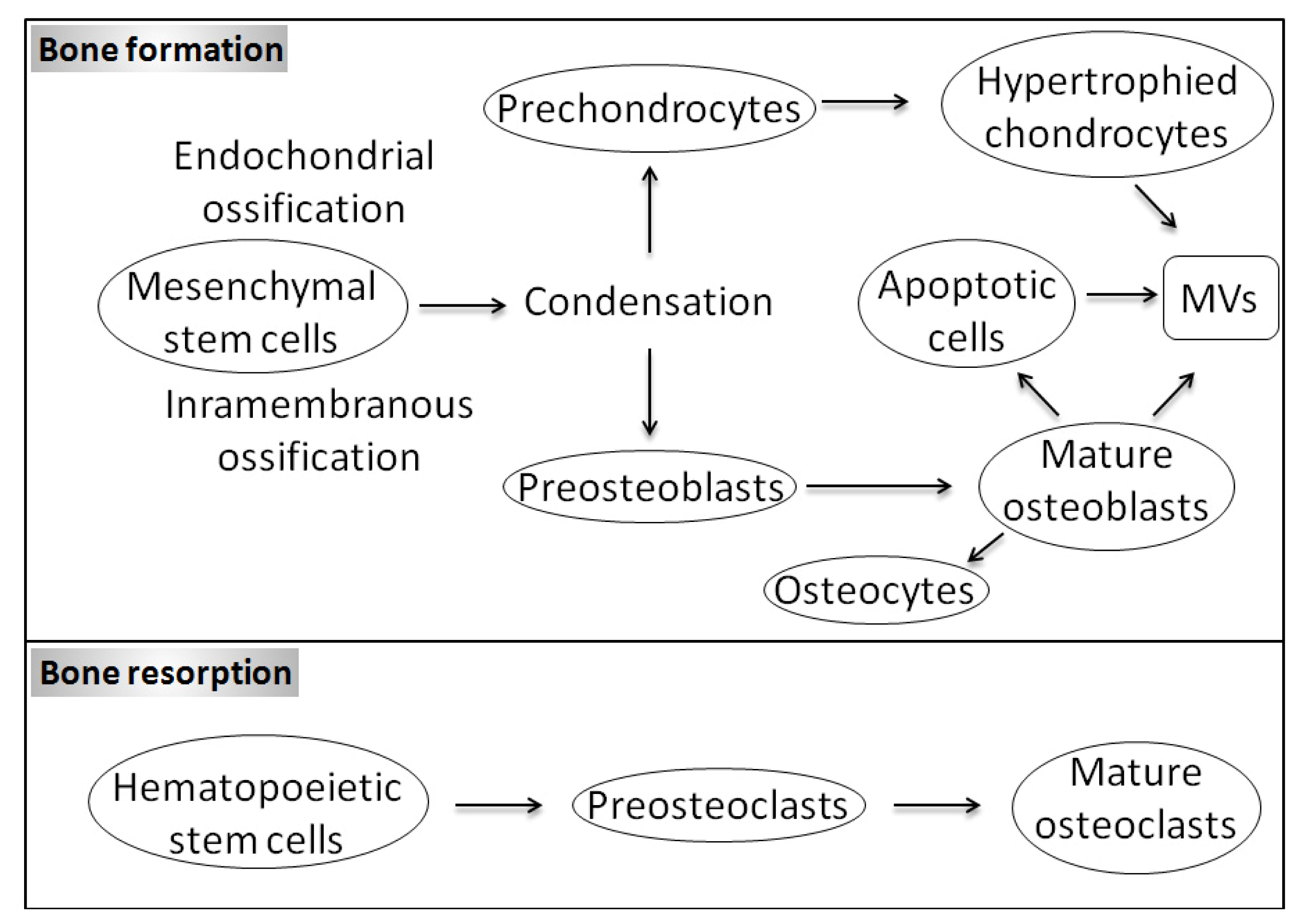{kind=link}
{kind=link}
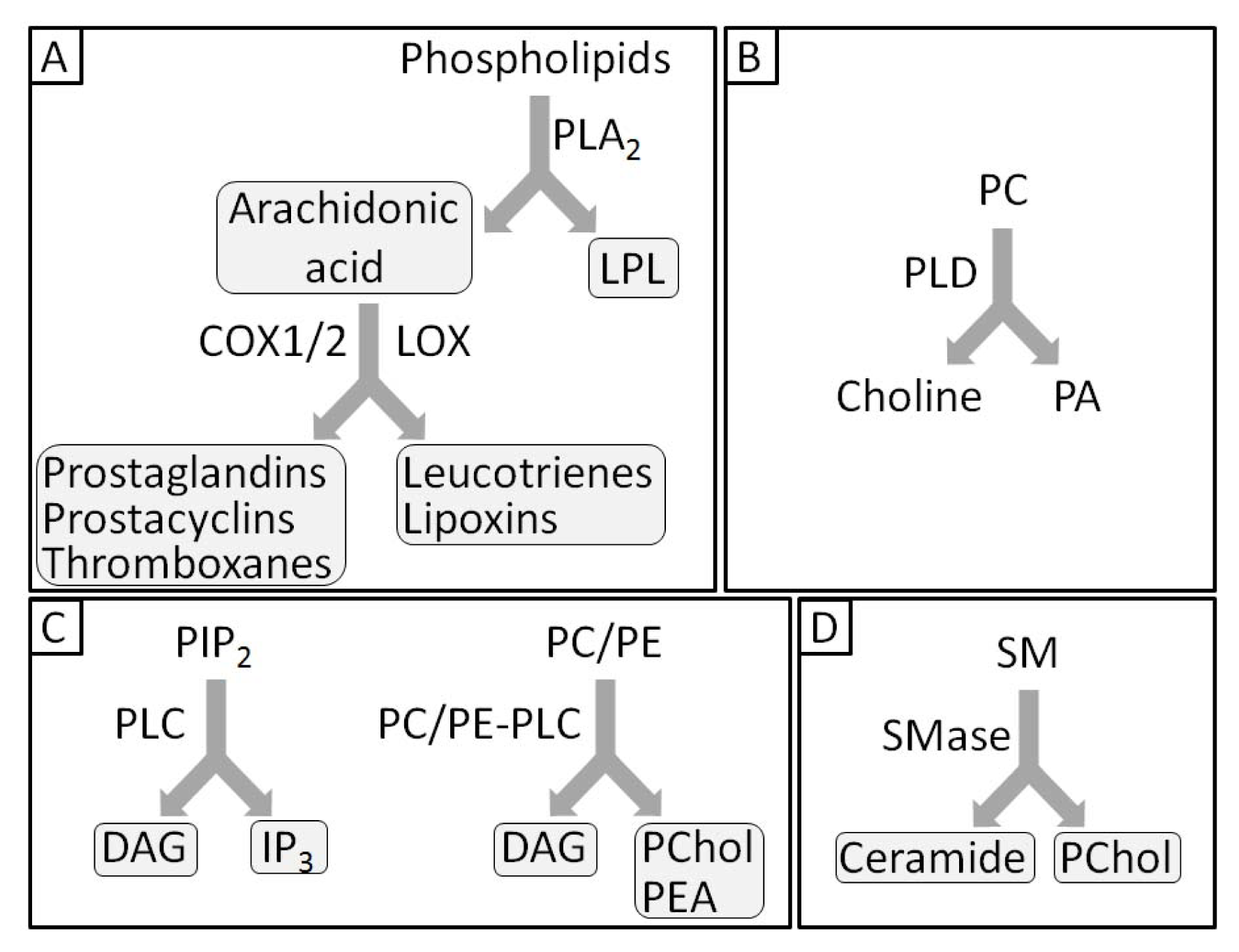{kind=link}
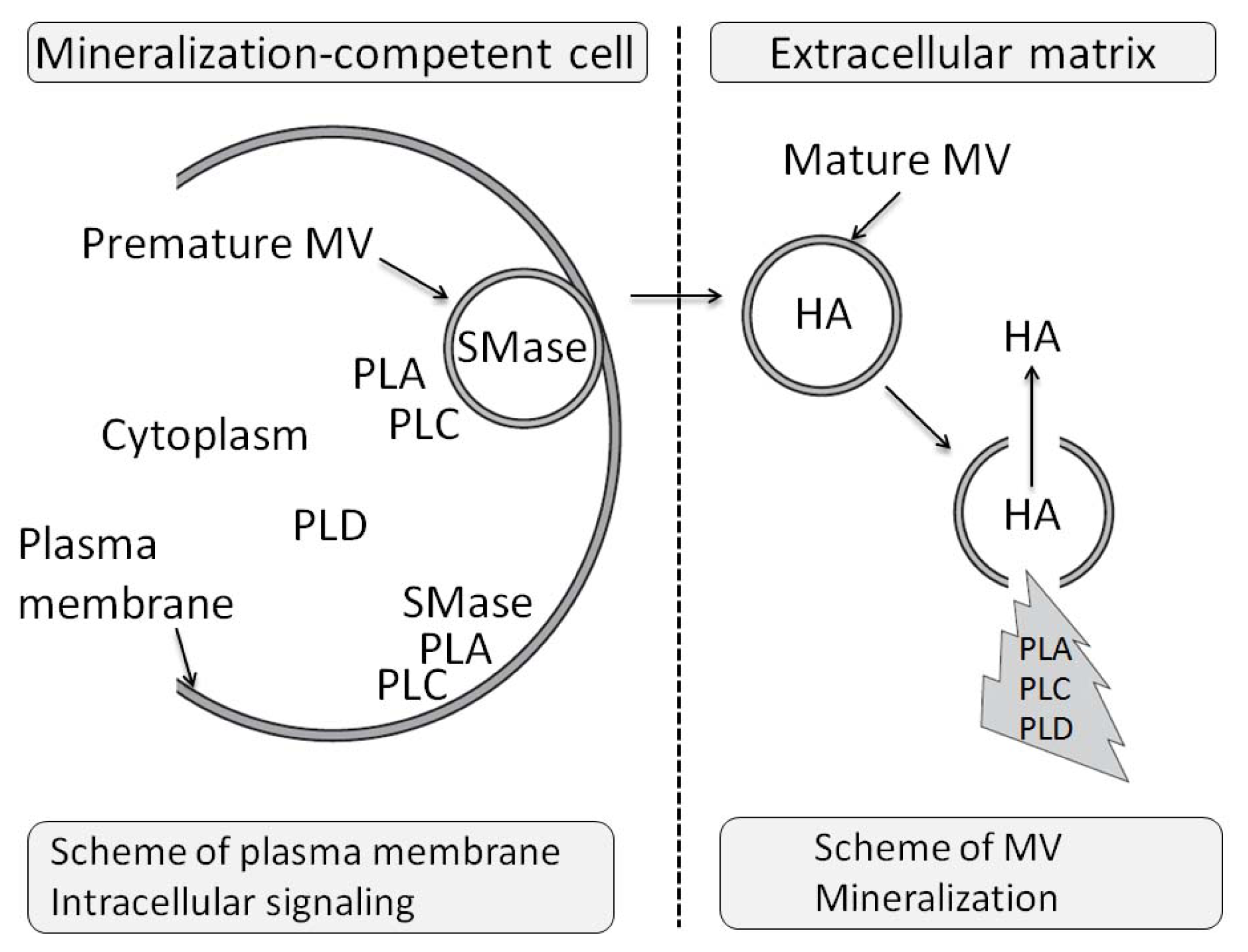{kind=link}
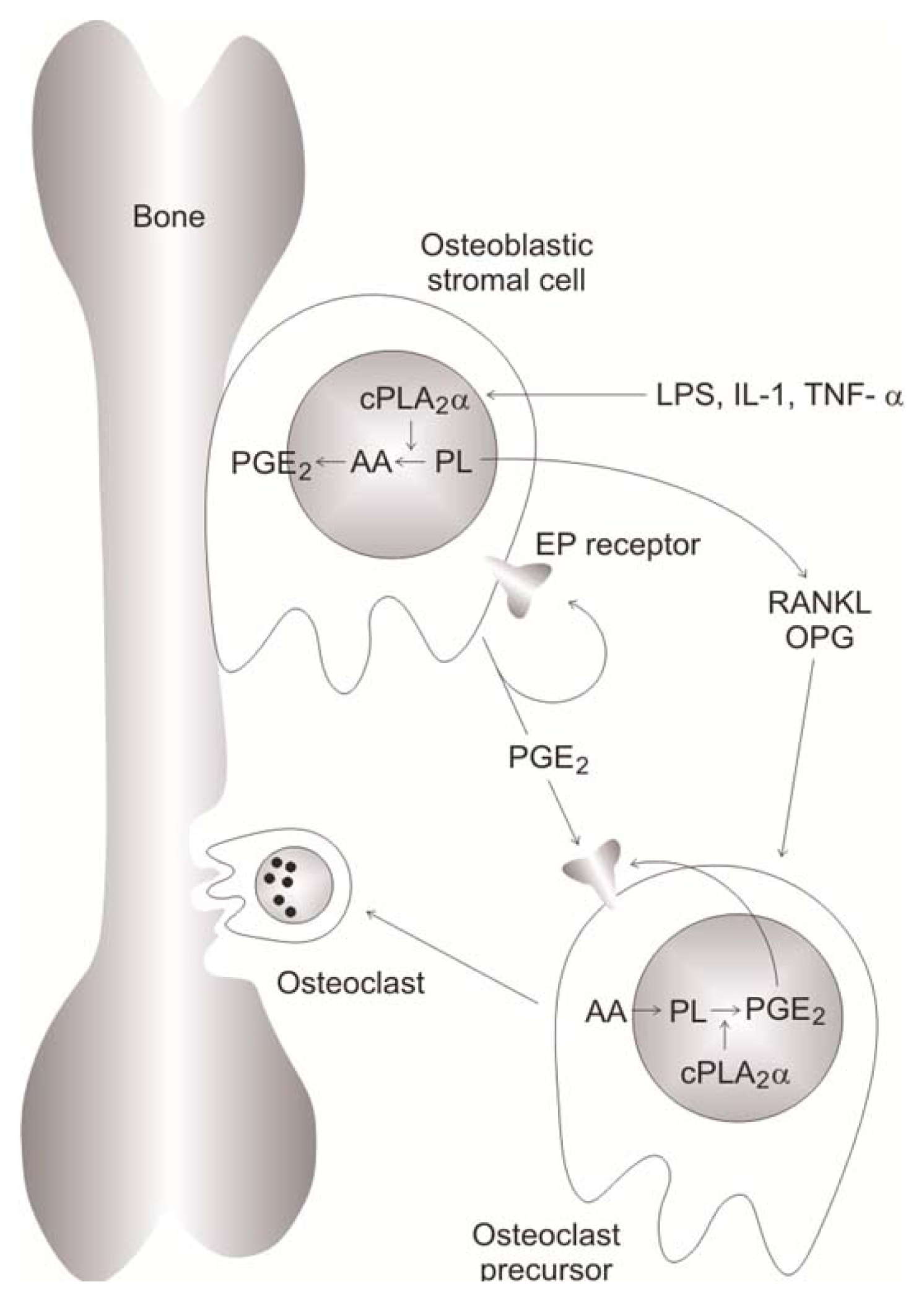{kind=link}
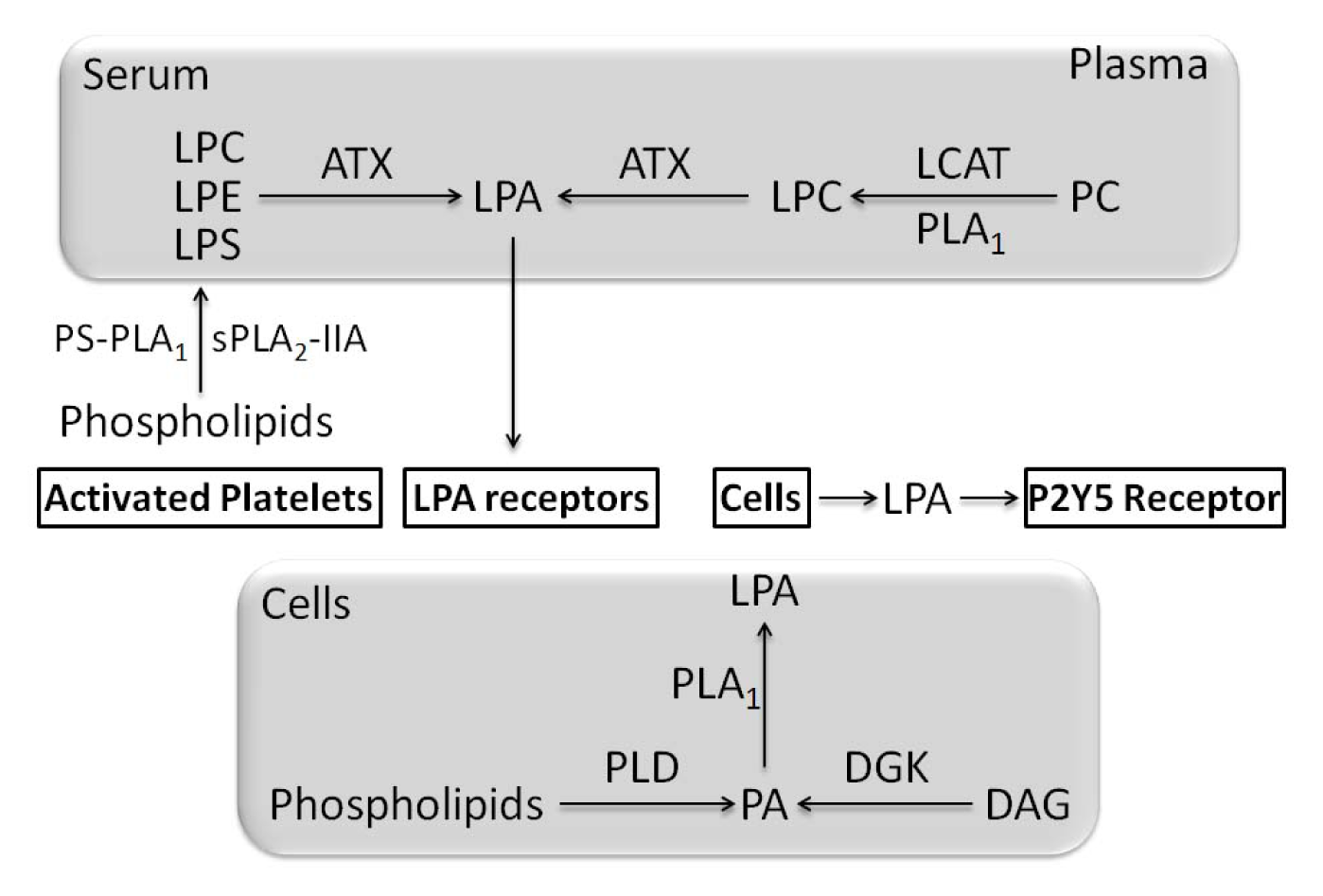{kind=link}
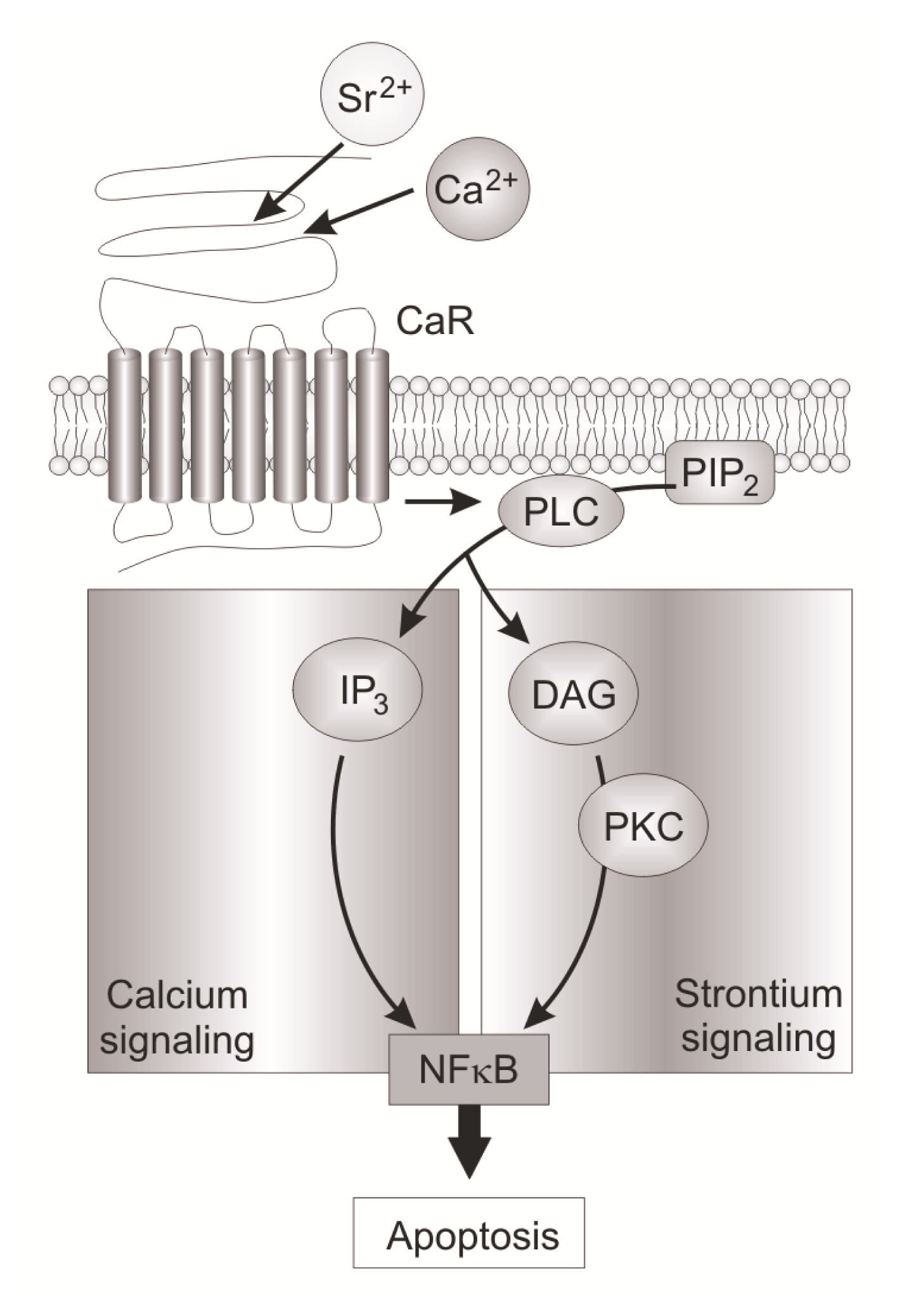{kind=link}
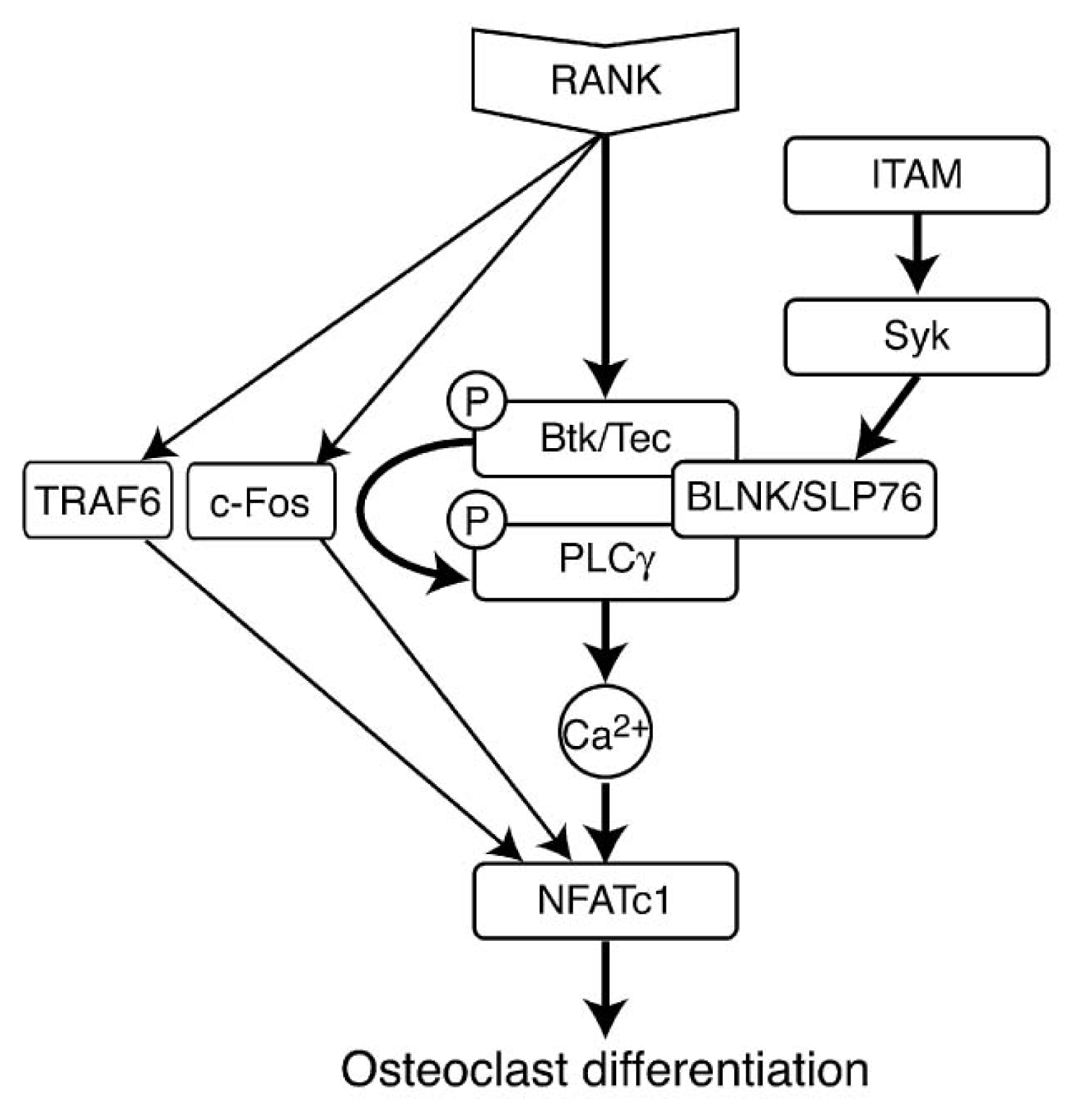{kind=link}
| % of Total lipid | |||||
|---|---|---|---|---|---|
| Chondrocytes | |||||
| Lipid | Whole cartilage | Proliferating | Hypertrophic | Cell membranes | MVs |
| SM | 8.6 ± 0.7 | 5.8 ± 0.4 | 8.0 ± 0.8 | 8.1 ± 0.8 | 13.4 ± 1.8 |
| PC | 45.2 ± 1.9 | 47.6 ± 1.5 | 38.0 ± 1.5 | 53.2 ± 2.2 | 41.8 ± 2.5 |
| LPC | 2.0 ± 0.6 | 1.9 ± 0.4 | 1.8 ± 0.4 | 3.5 ± 0.8 | 3.4 ± 0.8 |
| PE | 17.6 ± 1.0 | 16.9 ± 0.7 | 14.7 ± 0.8 | 14.6 ± 1.8 | 14.9 ± 1.8 |
| LPE | 2.0 ± 0.4 | 3.3 ± 0.7 | 2.4 ± 0.4 | 4.9 ± 1.2 | 6.5 ± 1.2 |
| PS | 5.1 ± 0.8 | 3.3 ± 0.3 | 5.0 ± 1.0 | 5.4 ± 0.7 | 9.3 ± 1.1 |
| LPS | 0.5 ± 0.2 | 0.2 ± 0.1 | 0.3 ± 0.2 | 2.2 ± 0.7 | 2.4 ± 0.8 |
| PI | 7.2 ± 0.8 | 6.2 ± 0.8 | 6.4 ± 0.8 | 6.1 ± 0.8 | 6.6 ± 0.6 |
| LPI | 1.1 ± 0.7 | 1.0 ± 0.6 | 0.5 ± 0.4 | 0.3 ± 0.2 | 1.1 ± 0.3 |
| PA | 2.0 ± 0.5 | 0.8 ± 0.3 | 1.6 ± 0.5 | 1.1 ± 0.2 | 0.9 ± 0.3 |
| PG | 1.2 ± 0.6 | 0.7 ± 0.3 | 1.2 ± 0.6 | 0.9 ± 0.2 | 1.3 ± 0.3 |
| di-PG | 3.0 ± 0.6 | 2.5 ± 0.4 | 2.9 ± 0.6 | 1.7 ± 0.3 | 1.5 ± 1.4 |
| Types of PLA1 | Groups | Origin |
|---|---|---|
| Extracellular PLA1 | PS-PLA1 | Human |
| mPLA1α | Human | |
| mPLA1β | Human | |
| Hepatic lipase | Human | |
| Endothelial lipase | Human | |
| Pancreatic lipase-related protein 2 | Human | |
| Intracellular PLA1 | iPLA1α | Human |
| iPLA1β | Human | |
| iPLA1γ | Human |
| Type | Group | Subgroup | Origin or commun source |
|---|---|---|---|
| sPLA2 | I | A | Cobras and kraits |
| I | B | Human/porcine pancreas | |
| II | A | Rattlesnake/human synovial | |
| II | B | Gaboon viper | |
| II | C | Rat/murine testis | |
| II | D | Human/murine pancreas/spleen | |
| II | E | Human/murine brain/heart/uterus | |
| II | F | Human/murine testis/embryo | |
| III | Lizard/bee | ||
| V | Human/murine heart/lung/macrophage | ||
| IX | Snail venom | ||
| X | Human spleen/thymus/leucocyte | ||
| XI | A | Green rice shoots (PLA2-I) | |
| XI | B | Green rice shoots (PLA2-II) | |
| XII | A | Human/murine | |
| XII | B | Human/murine | |
| XIII | Parvovirus | ||
| XIV | Symbiotic fungus/bacteria | ||
| cPLA2 | IV | A(α) | Human macrophage-like U937 cells/Platelets/Raw 264.7/rat kidney, ubiquitous |
| IV | B(β) | Human pancreas/liver/heart/brain/ubiquitous | |
| IV | C(γ) | Human heart/skeletal muscle | |
| IV | D(δ) | Murine placenta | |
| IV | E(ɛ) | Murine heart/skeletal muscle/testis/thyroid | |
| IV | F(η) | Murine thyroid/stomach | |
| iPLA2 | VI | A(β) | Human/murine |
| VI | B(γ) | Human/murine | |
| VI | C(δ) | Human/murine | |
| VI | D(ɛ) | Human | |
| VI | E(ζ) | Human | |
| VI | F(η) | Human | |
| PAF-AH | VII | A(lipoprotein-associated-PLA2) | Human, murine, porcine, bovine |
| VII | B(PAF-AH II) | Human, bovine | |
| VIII | A(α1) | Human | |
| VIII | B(α2) | Human | |
| Lysosomal PLA2 | XV | Human, murine, bovine | |
| Adipose PLA | XVI | Human,mouse |
| Types of PLA2 | Expression levels | Diseases | References |
|---|---|---|---|
| sPLA2-IIA | Highly expressed in synovial fluid | RA | [123,126,165,166,169] |
| sPLA2-IIA | Highly expressed in chondrocytes | RA | [126] |
| sPLA2-IID | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-IIE | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-V | Overexpressed in synovial fluid | RA | [124] |
| sPLA2-X | More or less expressed in synovial fluid | Active and inactive RA | [124] |
| sPLA2-IIA | Overexpressed in synovial fluid | OA | [126,167,168] |
| sPLA2-IIA | Overexpressed in VSMC | Infarctus heart | [164] |
| sPLA2-V | Overexpressed in VSMC | Infarctus Heart | [164] |
| cPLA2-α | cPLA2-α−/− mice loss in function | Prevention in collagen-induced arthritis | [174] |
| iPLA2β | iPLA2-β−/− mice loss in function | Low bone mass | [121] |
| Enzymes or products or animal models | Expression level or concentration | Physiological effects | Pathological effects | References |
|---|---|---|---|---|
| COX-2 | Increase in synovial fluid | RA | [38] | |
| mPGES-1 | Increase in synovial fluid | RA | [38] | |
| Mice deficient in COX2 | Null-COX | CIA reduction | [215] | |
| Mice deficient in mPGES-1 | Null-PGES-1 | CIA reduction | [217] | |
| PGE2 | High level in synovial fluid | RA | [239,240] | |
| Prostaglandin | Stimulate bone formation | [233] | ||
| Prostaglandin | Activate bone resorption in osteoporosis, RA, OA or in periodontis | [238] | ||
| PGD2 | Stimulate osteoblast calcification | [249] | ||
| PGF2α | Promote osteogenic differentiation | [251] | ||
| 15-Deoxy-Δ12,14- prostaglandin J2 | Prostaglandin D2 metabolite | Activates PPARγ and TNAP expression | [249] | |
| n-3 PUFA or conjugated linoleic acid | Exogenous addition | Beneficial effects due to modulation of COX-2 | [59] |
| Cox inhibitors | Physiological effects | Pathological effects | References |
|---|---|---|---|
| NSAIDS Ibuprofen Indomethacin | Inhibit fracture healing | [221–223] | |
| Indomethacin | Decrease TNAP activity | [222] | |
| NSAIDS | Decrease heterotopic calcification | [224–226] | |
| Keterolac | Decrease in spinal fusion | [228] | |
| COX-2 inhibitor | Decrease pain in RA | [231,241] |
| Type | Group | Origin |
|---|---|---|
| Non-specific PLC | Mammalian | |
| PI-specific | PLC-β PLCB1, PLCB2, PLCB3, PLCB4 | Mammalian |
| PI-specific | PLC-γ PLCG1, PLCG2 | Mammalian |
| PI-specific | PLC-δ PLCD1, PLCD3, PLCD4 | Mammalian |
| PI-specific | PLC-ɛ PLCE1 | Mammalian |
| PI-specific | PLC-η PLCH1, PLCH2 | Mammalian |
| PI-specific | PLC-ζ PLCZ1 | Mammalian |
| Phospholipase C-like | PLCL1, PLCL2 | Mammalian |
| Zinc-dependent prokaryotic PLC | Bacterial | |
| PI-DAG-lyase | Trypanosome | |
| SMase | Neutral SMase1 | Mammalian |
| Neutral SMase2 (SMPD3) | Mammalian | |
| Neutral SMase3 | Mammalian | |
| Lysosmal acid SMase | Mammalian | |
| Secreted zinc-dependent acid SMase | Mammalian | |
| Alkaline SMase | Mammalian |
| Type | Variants | Origin |
|---|---|---|
| PLDs with HKD motif | ||
| PLD1 | PLD1a, PLD1b, PLD1c, PLD1d | Mammalian |
| PLD2 | PLD2a, PLD2b, PLD2c | Mammalian |
| PLD3 | Mammalian | |
| Endonuclease-like mitochondrial PLD | Mammalian | |
| Non-HKD PLDs | ||
| GPI-PLD | Mammalian | |
| N-acyl PE-PLD | Mammalian | |
| cytochrome P450 1A2 | Mammalian | |
| cytochtome P450 2E1 | Mammalian | |
| ATX | Mammalian |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mebarek, S.; Abousalham, A.; Magne, D.; Do, L.D.; Bandorowicz-Pikula, J.; Pikula, S.; Buchet, R. Phospholipases of Mineralization Competent Cells and Matrix Vesicles: Roles in Physiological and Pathological Mineralizations. Int. J. Mol. Sci. 2013, 14, 5036-5129. https://doi.org/10.3390/ijms14035036
Mebarek S, Abousalham A, Magne D, Do LD, Bandorowicz-Pikula J, Pikula S, Buchet R. Phospholipases of Mineralization Competent Cells and Matrix Vesicles: Roles in Physiological and Pathological Mineralizations. International Journal of Molecular Sciences. 2013; 14(3):5036-5129. https://doi.org/10.3390/ijms14035036
Chicago/Turabian StyleMebarek, Saida, Abdelkarim Abousalham, David Magne, Le Duy Do, Joanna Bandorowicz-Pikula, Slawomir Pikula, and René Buchet. 2013. "Phospholipases of Mineralization Competent Cells and Matrix Vesicles: Roles in Physiological and Pathological Mineralizations" International Journal of Molecular Sciences 14, no. 3: 5036-5129. https://doi.org/10.3390/ijms14035036
APA StyleMebarek, S., Abousalham, A., Magne, D., Do, L. D., Bandorowicz-Pikula, J., Pikula, S., & Buchet, R. (2013). Phospholipases of Mineralization Competent Cells and Matrix Vesicles: Roles in Physiological and Pathological Mineralizations. International Journal of Molecular Sciences, 14(3), 5036-5129. https://doi.org/10.3390/ijms14035036